

Abstract
Recognition of self is emerging as a theme for the immune recognition of human cancer. One question is whether the immune system can actively respond to normal tissue autoantigens expressed by cancer cells. A second but related question is whether immune recognition of tissue autoantigens can actually induce tumor rejection. To address these issues, a mouse model was developed to investigate immune responses to a melanocyte differentiation antigen, tyrosinase-related protein 1 (or gp75), which is the product of the brown locus. In mice, immunization with purified syngeneic gp75 or syngeneic cells expressing gp75 failed to elicit antibody or cytotoxic T-cell responses to gp75, even when different immune adjuvants and cytokines were included. However, immunization with altered sources of gp75 antigen, in the form of either syngeneic gp75 expressed in insect cells or human gp75, elicited autoantibodies to gp75. Immunized mice rejected metastatic melanomas and developed patchy depigmentation in their coats. These studies support a model of tolerance maintained to a melanocyte differentiation antigen where tolerance can be broken by presenting sources of altered antigen (e.g., homologous xenogeneic protein or protein expressed in insect cells). Immune responses induced with these sources of altered antigen reacted with various processed forms of native, syngeneic protein and could induce both tumor rejection and autoimmunity.
Most antigens defined on human cancers are expressed both by malignant and normal cells (1–4). Studies of immune recognition of human cancer have shown that differentiation antigens (5), expressed by malignant cells and their normal cell counterparts, comprise a major group of tumor antigens recognized by the host (3). Thus, immunity against cancer in humans might be directed against self molecules. The question then arises how a host might convert from a state of immune tolerance or ignorance to immune response, to differentiation antigens on cancer, and whether such an immune response would be capable of rejecting tumors.
The central work in the immune response to human cancer has been done in melanoma. A set of melanoma antigens is expressed both on malignant cells and normal melanocytes or related neuroectodermal cells (6–9). Three of these antigens are melanosomal membrane glycoproteins [tyrosinase/albino protein, tyrosinase-related protein 1 (or gp75/brown protein), and the gp100/pMel 17/silver protein], and one is an uncharacterized melanocyte-specific protein (MelanA/MART-1 antigen) (10–23). Thus, one dominant set of antigens recognized on human melanoma are melanocyte differentiation antigens.
Products of the brown locus expressed by melanocytes and melanoma are recognized by autoantibodies and T cells of persons with melanoma and are relevant tumor autoantigens (21, 23). We have established a syngeneic model in C57BL/6 mice to investigate immunogenicity of the brown locus protein and potential sequelae of autoimmunity (24). We show that (i) there is apparent tolerance to syngeneic gp75, supporting previous studies in allophenic mice that demonstrate that mice are tolerant to cutaneous melanocytes (25); (ii) autoantibodies to gp75 can be actively induced by altering the source of antigen; (iii) active immunization against gp75 can lead to tumor protection; and (iv) active immunization against gp75 can induce manifestations of autoimmunity.
MATERIALS AND METHODS
Mice, Tumors, and Antibodies.
C57BL/6 (6- to 8-week-old females) were obtained from the Jackson Laboratory. B16F1 and B16F10 are mouse melanoma cell lines of C57BL/6 origin kindly provided by Isaiah Fidler (M. D. Anderson Cancer Center, Houston) (26). B78H.1 is a variant of B16 melanoma that does not express the gp75 antigen (from Anthony Albino, Memorial Sloan–Kettering). JBRH is a melanoma from C57BL/6 provided by P. Livingston (Memorial Sloan–Kettering), and TIB88 is a gp75− skin fibroblast cell line derived from C57BL/6 mice (American Type Tissue Collection). Sf9 insect cells from Spodoptera frugiperda were obtained from Invitrogen. The human melanoma cell lines have been described previously (7). All cell lines were tested routinely for mycoplasma contamination. Rabbit polyclonal peptide antibodies PEP-1, PEP-7, and PEP-8, which recognize gp75, tyrosinase, and TRP-2, respectively, were provided by Vincent Hearing (National Cancer Institute; ref. 27). The mAb TA99 specifically recognizes gp75 (21).
Plasmid and Baculovirus Constructs.
For construction of plasma of murine gp75, a 1.8-kb EcoRI fragment from the plasmid pHOMERB2 (kindly provided by S. Shibahara, Sendai, Japan), which contains the full-length cDNA for murine gp75 (28), was cloned into the expression vector pcEXV-3 (29). 3.27 cells are B78.H1 melanoma cells, and TIB88/gp75 are TIB88 fibroblasts transfected with the gp75 cDNA and selected for high expression of gp75 based on immunofluorescence staining and immunoprecipitation with mAb TA99. For construction of murine gp75 in baculovirus expression vector, a recombinant baculovirus containing full-length murine gp75 protein was constructed and isolated in collaboration with Charles Tackney (Imclone, New York) using methods reported in ref. 30.
Purification of gp75.
The gp75 antigen was purified from B16F1 melanoma grown in C57BL/6 mice or from gp75/Sf9 insect cell lysates using a protocol described previously (31). Briefly, TA99 was conjugated to Affi-Gel-10 (Bio-Rad). Purified gp75 protein was eluted with 0.1 M glycine (pH 2.5) and collected into 1 M Tris (pH 8.3). Purified gp75 protein was analyzed by gel electrophoresis and silver staining and was >90% pure. Lysates of 1 × 106 and 5 × 106 gp75/Sf9 cells contained 14 μg and 70 μg of gp75, respectively. Protein content of gp75 in B16 melanoma cells and Sf9 cells expressing gp75 was determined by Western blots against standard purified gp75, measured using digitization with a CCD-72 series video camera (Dage–MTI, Michigan City, IN).
Peptides of Murine gp75.
Mouse gp75 peptide sequences representing potential B-cell determinants were selected by using a hydrophobicity plot of the amino acid sequence (32). Three strongly hydrophilic peptides were chosen and synthesized using an Applied Biosystems model 431A peptide synthesizer and standard fluorenylmethoxycarbonyl chemistries. The peptides span amino acids 69–89, 221–245, and 517–537 of mouse gp75 (28). Bovine serum albumin was conjugated with glutaraldehyde to murine gp75 peptides in a one-step procedure using a standard methodology (33).
Immunization and Tumor Protection Experiment.
Mice were injected intraperitoneally (i.p.), intravenously (i.v.), intradermally (i.d.), or subcutaneously (s.c.) with cells, lysates, or purified protein or peptide as indicated. Protein or cells were injected in PBS or with one of the following adjuvants: complete and incomplete Freund’s adjuvant (Sigma), QS21 (Cambridge Biotech), or Detox (Ribi Immunochem). Mouse gp75 purified from melanoma cells or baculovirus-infected insect cells was emulsified in complete Freund’s adjuvant for the initial injection and incomplete Freund’s adjuvant for subsequent immunizations. Mice were injected with 10–12 μg of purified gp75 per injection. Sf9 cells infected with wild-type or recombinant gp75 baculovirus were harvested by scraping and then were freeze–thawed three times prior to injection. Human melanoma cell lines freeze–thawed three times were injected i.p. in PBS or in Freund’s adjuvant. Irradiated melanoma cells were treated with 900 cGy. B16F10 melanoma cells transduced with the cytokines interleukin 2 and interferon γ (IFN-γ) have been described (24). B16F10 melanoma cells that secrete mouse granulocyte/macrophage colony-stimulating factor (GM-CSF) were stably selected after transfection of plasmid pcEXV-3 that expresses cDNA for mouse GM-CSF (courtesy of David Golde, Memorial Sloan–Kettering Cancer Center), and secreted 200–500 units in 24 h. Booster injections were given at 10- to 14-day intervals, and mice were tail bled ≈1 week after each immunization. For B16F10 melanoma lung metastases, C57BL/6 mice were injected i.v. through the tail vein with B16F10 melanoma cells in sterile saline. To assess lung metastases, mice were killed at 14–20 days after tumor challenge, and surface lung metastases were scored and counted as black nodules under a dissecting microscope. Surface lung metastases were detected by day 4–5 under a dissecting microscope and by day 5–8 by eye. For histologic evaluation, tissues and tumors were fixed in formalin solution, blocked in paraffin, and stained with hematoxylin/eosin. Statistical analysis of tumor growth was performed using the Bonferroni two-sided t test, a conservative analysis to allow for multiple comparisons. (All animal experiments were in accordance with institutional and National Institutes of Health guidelines.)
Immunoprecipitation with [35S]Methionine, Glycosidase Digestion, and Peptide Mapping.
Cell lines were labeled with trans-[35S]methionine and lysed as described (31, 33). For pulse–chase experiments, cells were labeled at 5–10 min and chased in media containing cold methionine. For each immunoprecipitate, 3–10 × 106 cpm trichloroacetic acid-insoluble precipitate in 200 μl lysis buffer was incubated with mouse sera or control antibody, followed by the addition of 50 μl protein A-Sepharose. Proteins were analyzed by 9% SDS/PAGE N-glycosidase F (N-glycanase) and endo β-N-acetylglucosaminidase H (Endo H) were obtained from Genzyme. Glycosidase digestions were performed as reported (33). Peptide mapping was performed as described by limited proteolysis with Staphylococcus aureus V8 protease (34).
ELISA, Western Blot Analysis, and Cytotoxic T Lymphocyte (CTL) Assays.
For ELISA, B16F10 melanoma cells (gp75+) or B78H.1 melanoma cells (gp75−) were used as target cells as described (25). Serially diluted serum or positive control mAb TA99 was added for 1 h, and a 1:500 dilution of alkaline phosphatase-conjugated goat anti-human Ig or anti-mouse IgG (Sigma) was subsequently added. p-Nitrophenyl phosphate (1 μg/ml; Sigma) in 0.25 M MgCl2 and 10% diethanolamine (pH 9.8) was added. Optical density at 405 nm was measured on a FisherBiotech microkinetics reader (Fisher Scientific). Western blots were performed as described (35). Horseradish peroxidase-conjugated goat anti-mouse Ig (Sigma) was used for detection and visualized using ECL detection reagents (Amersham). Cell-mediated cytotoxicity was assessed in vitro by a 51Cr release assay. Spleen mononuclear cells from immunized mice were stimulated for 5–7 days with irradiated B16F10 melanoma cells treated with IFN-γ at 200 units/ml to induce class I and II major histocompatibility antigens. Approximately 1 × 105 to 1 × 106 IFN-γ-treated B16F10 target cells were labeled with 100 μCi (1 Ci = 37 GBq) of 51Cr (New England Nuclear). Stimulated splenic effector cells were added at various effector-to-target ratios up to 100:1, and lysis assays were performed in triplicate. Spontaneous release was measured by incubating target cells in medium alone, and maximum release was obtained by adding 1% (vol/vol) Nonidet P-40 to target cells. The spontaneous release of target cells was <20% of maximum release.
RESULTS
Immunization with Syngeneic gp75 Does Not Induce Autoantibodies to gp75.
The brown locus encodes the type I membrane glycoprotein gp75, known as tyrosinase-related protein 1. This protein is a melanosomal protein, but is also expressed at the cell surface (24). The b allele of the brown locus is expressed in melanocytes and melanomas of C57BL/6 mice. Autoantibody and CTL responses to the b locus protein were assessed in syngeneic C57BL/6 mice after immunization with gp75 antigen in both cell-associated and purified forms (Table 1).
Table 1.
Immunization against mouse gp75
| Source of gp75 | Adjuvant | No of injections | Route | Dose | Ab/CTL Response |
|---|---|---|---|---|---|
| B16 melanoma | None | 1 | s.c., i.v. | 50, 00 | No response |
| Irradiated B16 | None | 5 | i.p. | 8 × 106 | No response |
| Irradiated B16 | F, D, Q | 5 | i.p. | 8 × 106 | No response |
| 3.27 tx | None | 5 | i.p., s.c. | 8 × 106 | No response |
| 3.27 tx | F, D, Q | 5 | i.p., s.c. | 8 × 106 | No response |
| TIB88/gp75 tx | F | 4 | i.p., s.c. | 5 × 106 | No response |
| B16–IFN-γ | None | 5 | s.c., i.v. | 8 × 106 | No response |
| B16–IL-2 | None | 5 | s.c., i.v. | 8 × 106 | No response |
| B16–IL-2/IFNγ | None | 5 | s.c., i.v. | 8 × 106 | No response |
| B16–GM-CSF | None | 5 | s.c., i.v. | 8 × 106 | No response |
| Purified gp75 | F, Q | 5 | s.c., i.v. | 10 μg | No response |
| gp75 peptides | F, Q | 5 | id | 100 μg | No response |
C57BL/6 mice were immunized with gp75 sources as indicated and described in the Materials and Methods. Dose refers to individual dose per injection, either number of cells per injection or amount of protein or peptide. One week following the last immunization, sera were tested by immunoprecipitation and Western blots against lysates of syngeneic B16F10 melanoma and ELISA for antibodies (Ab) to gp75 and splenocytes were tested for CTL responses as described. Under adjuvant, F = Freund’s adjuvant; D = Detox adjuvant; and Q = QS21 adjuvant. No of injections = number of immunizations at 7- to 14-day intervals. B16–IFN-γ, –IL-2 (interleukin 2), and –GM-CSF refer to B16F16 melanoma cells expressing the designated cytokine. tx, Transfectant. 3.27 are B78 H.1 melanoma cells transfected with gp75, and TIB88/gp75 are syngeneic fibroblasts transfected with gp75.
C57BL/6 mice were immunized with (i) syngeneic gp75+ B16 melanoma cells (which express a nonmutant b locus protein); (ii) syngeneic B16 cells expressing interleukin 2, GM-CSF, and IFN-γ; (iii) syngeneic gp75− B16 melanoma variant, B78H.1, and syngeneic fibroblasts transfected with cDNA expressing the mouse b allele; (iv) hydrophilic peptides of gp75 conjugated to carrier protein; and (v) full-length gp75 glycoprotein purified from syngeneic melanoma cells (Table 1). Cells, purified glycoprotein, or peptides were combined with adjuvants, including Freund’s, a mixture of bacterial cell wall skeletons and an endotoxin derivative (Detox), and a saponin component (QS21). Immunizations were tested by i.p., s.c., and i.d. routes. After immunizations, mice were assessed for antibodies against gp75 by ELISA, immunoprecipitation. and Western blots and for CTL to B16 as described. No antibodies or CTL against gp75 were detected after any of these immunization strategies, supporting the notion that C57BL/6 mice maintain tolerance to the gp75 glycoprotein.
gp75 Expressed in Insect Cells Induces Autoantibodies.
C57BL/6 mice were immunized with lysates of insect Sf9 cells expressing either syngeneic gp75 in a baculovirus vector (gp75/Sf9) or wild-type baculovirus (wt/Sf9). Mice immunized with gp75/Sf9, with or without Freund’s adjuvant, developed autoantibodies to gp75 (145/148 mice), but none after immunization with wt/Sf9 lysates (0/46 mice) (Fig. 1). No CTL response was observed in five mice immunized with gp75/Sf9. Autoantibodies appeared after two to four immunizations, lasted greater than 4 months after the last immunization, and reacted with gp75 expressed in syngeneic melanocytic cells (B16F10 and JBRH melanomas). Although autoantibodies were detected after immunization with gp75/Sf9 lysates alone (25/28 mice), antibody responses were consistently stronger when Freund’s adjuvant was included (120/120 mice). Autoantibodies were observed consistently after immunization with lysates of 1 × 106 (Fig. 2) and 5 × 106 gp75/Sf9 cells (Fig. 1) per immunization, both with and without Freund’s adjuvant. Antibodies were IgG class, based on reactivity with rabbit anti-mouse IgG and protein A and copurification of antibody reactivity with IgG fractions from sera (data not shown).
Figure 1.
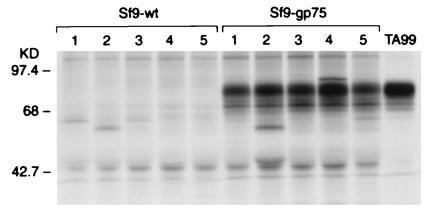
Mice immunized with gp75/Sf9 produced antibodies against gp75 in syngeneic melanocytic cells. C57BL/6 mice were immunized with lysates of insect Sf9 cells infected with either recombinant baculovirus expressing syngeneic mouse gp75 (gp75/Sf9) or wt baculovirus (wt/Sf9). Mice were immunized four times subcutaneously with freeze–thawed Sf9 cells, 5 × 106 cells per immunization. Sera from immunized mice were used to immunoprecipitate syngeneic gp75 from [35S]methionine-labeled B16 melanoma lysates. The mAb TA99 was used as a positive control (right lane). Results for five mice in each group are shown (lanes 1–5).
Figure 2.
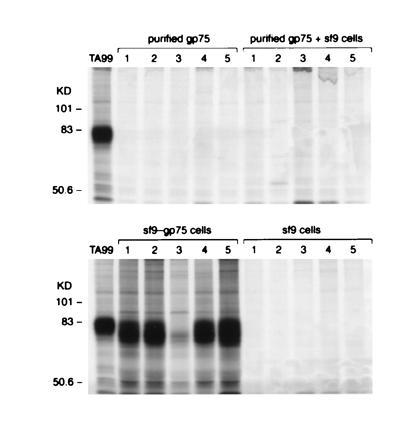
Purified mouse gp75 mixed with lysates of wt/Sf9 cells does not induce autoantibodies to gp75. C57BL/6 mice were immunized with purified gp75 from B16 melanoma cells (purified gp75), purified gp75 from B16 melanoma cells mixed with lysates from 1 × 106 Sf9 cells (purified gp75 + sf9), lysates of 1 × 106 insect Sf9 cells infected with either recombinant baculovirus expressing syngeneic mouse gp75 (gp75/sf9 cells), or wt baculovirus (sf9 cells). Purified gp75 from B16 melanoma contained 10 μg of gp75, and gp75/Sf9 contained 14 μg of gp75. All immunizations included Freund’s adjuvant. Mice were immunized four times s.c. Sera from immunized mice were used to immunoprecipitate syngeneic gp75 from [35S]methionine-labeled B16 melanoma lysates. The mAb TA99 was used as a positive control (left lane). Results for five mice in each group are shown (lanes 1–5).
The difference in immunogenicity between gp75/Sf9 and mouse gp75 was not due simply to quantitative differences in the amount of gp75 in the two preparations; 8 × 106 B16 melanoma cells contained 20 μg of gp75 (see Table 1) compared with only 14 μg in 1 × 106 gp75/Sf9 cells. Also, 10 μg of purified mouse gp75 mixed with Sf9 lysates did not induce autoantibodies (Fig. 2). Although Sf9 cells can apparently provide an adjuvant effect (36, 37), these results suggested that other differences between gp75 produced in mouse cells versus insect cells were necessary to induce autoantibodies.
Induction of Immunity Against an Intracellular, Early Processed Form of gp75.
In contrast to immunization with gp75/Sf9 lysates, immunization with purified gp75 (12 μg) produced in gp75/Sf9 insect cells plus Freund’s adjuvants induced autoantibodies that recognized 68–70 kDa of early processed forms of gp75 (Fig. 3). The identification of the 68- to 70-kDa bands as gp75 was confirmed by preclearing of these bands by mAb TA99 before immunoprecipitation with immune sera and showing identity of peptide maps of these bands and mAb TA99 immunoprecipitates of gp75 (data not shown). These autoantibodies recognized an early processed form of gp75 and did not recognize mature gp75. This form of gp75 contained only immature, high mannose N-linked carbohydrates that were sensitive to the glycosidase Endo H and was detected within a 5-min pulse with [35S]methionine (data not shown). These results localize this gp75 form to the endoplasmic reticulum (or pre-Golgi compartment). There was no evidence for response against immature gp75 after immunization with Sf9/gp75 at early time points (e.g., after one or two immunizations), suggesting that the antibody response to gp75 did not simply evolve from a response to immature gp75 to recognition of fully mature gp75. Thus, purified gp75 produced in insect cells induced autoantibodies that only recognized a sequestered, early posttranslational form, although one mouse eventually developed autoantibodies against mature gp75 after five immunizations (mouse 2 in Fig. 3). These results led us to mix purified gp75 from Sf9/gp75 cells with lysates of wt/Sf9 and compare immunogenicity with whole lysates of Sf9/gp75 cells, predicting that the Sf9 lysate would enhance the antibody response to mature gp75. Surprisingly, the mixture of exogenous gp75 from Sf9/gp75 cells mixed with Sf9 lysates did not induce any detectable autoantibodies to gp75 at 10 μg (0/5 mice) or 50 μg (0/3 mice) individual doses (data not shown). These results suggest that endogenous expression of gp75 in Sf9 cells was required for autoantibody responses to fully mature gp75. Thus, the “adjuvant” effect of Sf9 lysates depended on endogenous expression of gp75; Sf9 lysates actually inhibited antibody response to purified gp75 from insect cells.
Figure 3.
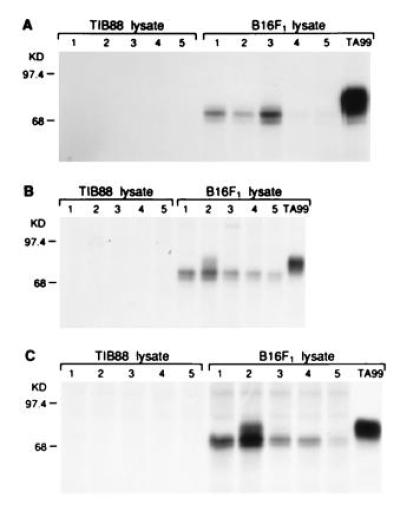
Mice immunized with gp75 purified from Sf9 cells recognized an early processed form of gp75. Sera from C57BL/6 mice immunized with purified mouse gp75 (12 mg) produced in baculovirus were assessed for antibody responses to syngeneic gp75 by immunoprecipitation of lysates from [35S]methionine-labeled syngeneic B16 melanoma cells or syngeneic gp75-negative TIB88 fibroblasts. The mAb TA99 was used as a positive control (right lane). Results for five mice are shown (lanes 1–5) after 3 immunizations (A), after 4 immunizations (B), and after 5 immunizations (C). All mice developed antibodies to syngeneic gp75 after four immunizations. These autoantibodies recognized an earlier processed form of gp75 (see Results), but mouse 2 developed autoantibodies that recognized mature, fully processed gp75 after four immunizations.
Human gp75 Expressed in Human Melanoma Induces Autoantibodies in Mice.
All mice immunized with the gp75+ human melanoma cell line SK-MEL-19 with Freund’s adjuvants (20/20 immunized mice) developed autoantibodies to gp75 (Fig. 4). There was no response without adjuvant (0 of 5 mice) (Fig. 4). Preclearing of immunoprecipitates with mAb TA99 against gp75 showed that the autoantigen was gp75, and identity of the gp75 55-kDa core polypeptides was supported by N-glycanase digestions (data not shown). No antibodies to gp75 were detected in sera of 12 mice immunized with the gp75− human melanomas SK-MEL-131 or SK-MEL-37 plus Freund’s adjuvant. Three of five mice immunized with purified human gp75 (10 μg/dose for five immunizations) with Freund’s adjuvant developed autoantibodies to gp75, although the antibody responses were generally weaker possibly due to the lower amount of purified gp75 used compared with the amount of gp75 in SK-MEL-19 lysates (data not shown). Thus, human gp75 broke apparent tolerance to gp75 in C57BL/6 mice (Fig. 4).
Figure 4.
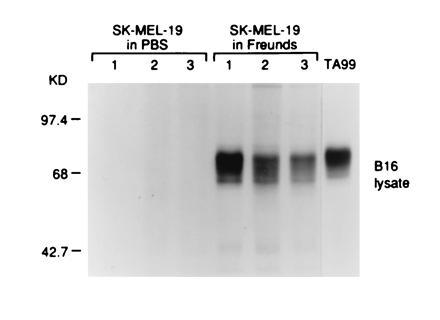
Mice immunized with human gp75 expressed in SK-MEL-19 melanoma cells produced antibodies against gp75 in syngeneic melanocytic cells. C57BL/6 mice were immunized with lysates of SK-MEL-19 melanoma cells with either Freund’s adjuvant or PBS. Mice were immunized four times s.c. with freeze–thawed cells, 5 × 106 cells per immunization. Sera from immunized mice were used to immunoprecipitate syngeneic gp75 from [35S]methionine-labeled B16 melanoma lysates. The mAb TA99 was used as a positive control (right lane). Results for three mice in each group are shown (lanes 1–3).
Tumor Protection and Autoimmunity.
The in vivo effects of immune recognition of gp75 were investigated using a syngeneic tumor model. B16 melanoma cells and normal melanocytes in C57BL/6 mice express the wt b allele of the brown locus. As described above, the product of this allele is recognized by sera from syngeneic mice immunized with mouse gp75 in gp75/Sf9 cells and human gp75, but purified gp75 from gp75/Sf9 cells induces autoantibodies preferentially against a sequestered, early processed form of gp75. We have previously shown that passive transfer of mouse mAb against gp75 into mice bearing B16F10 tumors leads to tumor rejection (38). Mice immunized with gp75/Sf9 lysates, starting immunization concomitantly with tumor challenge, were protected from lung metastases of B16F10 melanoma (Fig. 5). There was even significant protection when immunizations were started 4 days after tumor challenge as metastases became established, although these effects were modest (53% decrease in lung metastases; P = 0.01). There was no significant protection in mice immunized with wt/Sf9 lysates compared with unimmunized control animals (P > 0.40). Passive transfer of sera from mice immunized with gp75/Sf9 to five unimmunized mice produced a 68% decrease in lung metastases compared with mice treated with equivalent amount of normal mouse sera (P = 0.02), supporting the notion that tumor protection was at least partly mediated by humoral mechanisms.
Figure 5.
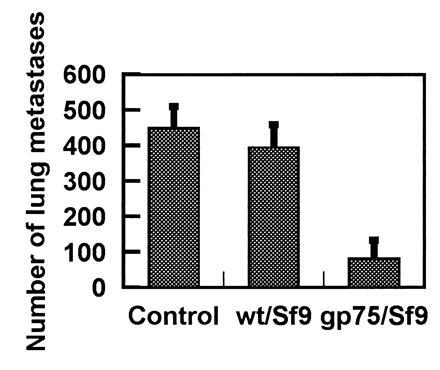
Immunization with gp75/Sf9 protects against melanoma lung metastases. C57BL/6 mice, five per group, were immunized with 5 × 106 of gp75/Sf9 or control wt/Sf9 or were not immunized (Control). Lung metastases of B16F10 melanoma were assessed at day 14 following challenge with 100,000 melanoma cells intravenously. The mean number of lung colonies ± SD (error bars) is shown.
Mice immunized against the immature, early processed form of gp75 (using purified gp75 from gp75/Sf9 cells shown in Fig. 3) were not significantly protected against B16F10 metastases (366 ± 78 metastases in four immunized mice versus 412 ± 94 metastases in five unimmunized control mice in one experiment, which is representative of the three experiments which were performed). It is worth noting that the one mouse in this group that went on to develop autoantibodies against mature gp75 (mouse 2 in Fig. 3) was protected against lung metastasis (only 21 metastases). Overall, these results suggest that antibody recognition of mature gp75, but not early forms of gp75 in the processing pathway, can lead to tumor rejection.
Mice immunized with human gp75+ SK-MEL-19 were also markedly protected against B16F10 melanoma compared with unimmunized mice (4 ± 7 metastases in immunized mice versus 275 ± 77 lung metastases in control mice; six mice per group). Although immunization with the gp75− melanoma SK-MEL-131 did not induce any tumor protection (data not shown), recognition of other xenogeneic antigens other than gp75 could not be critically assessed.
Mice immunized with gp75/Sf9 developed depigmentation over an 8-week observation (5/5 mice), while no depigmentation was observed in mice immunized with wt/Sf9 over 5 months of observation (0 of 5 mice). Depigmentation appeared as patchy white areas with map-like borders against a black coat background (Fig. 6). The depigmentation developed after four immunizations (after 40 days) without any manipulation of coat hairs (e.g., depilation). No changes in weight, behavior, or feeding were noted, and otherwise mice appeared healthy. Histologic sections of depigmented skin at 1 and 3 months showed depigmented hairs, hairbulbs without pigment, and follicles that lacked pigmented melanocytes. No infiltrates of mononuclear cells or other inflammatory cells were observed in or around hair follicles. There were no signs in the eyes (choroid and retina) of changes in pigmentation or pigmented granules, inflammation, or alterations in cellular morphologies or tissue architecture.
Figure 6.
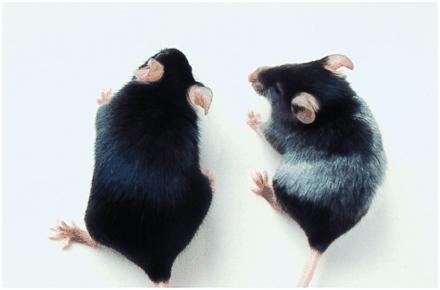
Alterations in coat color of C57BL/6 mice immunized with Sf9/gp75 but not Sf9/wt cell lysates. Black C57BL/6 mice, five per group, were immunized with lysates from 5 × 106 Sf9/gp75 or Sf9/wt cells s.c. with Freund’s adjuvant every 10–14 days for five immunizations. Depigmentation was observed in coats of all five mice immunized with Sf9/gp75 but no mice immunized with Sf9/wt. Depigmentation appeared without dipilation of the coats. (Right) A representative mouse immunized with Sf9/gp75. (Left) A control mouse immunized with Sf9/wt.
DISCUSSION
T cells and antibodies against autologous melanoma recognize autoantigens expressed on normal tissues. The most prevalent autoantigens are differentiation antigens, expressed by melanomas and melanocytes or other neuroectoderm-derived normal tissues. A second class of antigens, represented by the MAGE, BAGE, and GAGE families, are expressed on melanomas and other cancers, and in normal testes (4). Little is known about the function of this set of antigens, but they may represent developmentally controlled molecules that are up-regulated in certain activation settings. A third set of antigens are prototypes for unique tumor antigens and are detected in melanomas from individual patients. These include proteins with point mutations and mutations leading to alternative reading frames (20, 39, 40). These mutations can lead to new epitopes or aggretopes [capacity to bind major histocompatibility complex (MHC) molecules].
In mouse models using tumors induced by potent carcinogens, unique antigens are dominant epitopes that are recognized by the cellular arm of the immune system. These unique antigens presumably reflect the effects of carcinogens inducing widespread mutations, a few which will be presented to the immune system. Tumor rejection in these models appears to be primarily mediated by immune responses to these unique antigens. However, immune recognition of human melanoma is directed also against self molecules. It is unclear at this point whether immune recognition of these self antigens can induce tumor rejection. It is not even clear whether these autoreactive B and T cells are the result of active immune responses to melanoma, are activated by other host events unrelated to cancer, or simply reflect the immune repertoire of natural immunity. For instance, autoantibodies to ganglioside antigens can be found in ≈5% of persons with metastatic melanoma, but are also detected in a similar proportion of healthy, nontransfused males (refs. 6 and 41; A.N.H., unpublished observations). Presence of serum autoantibodies to gangliosides correlates with improved prognosis in persons with metastatic melanoma, suggesting but not proving that these autoantibodies provide some protection against progression of melanoma (42, 43). Despite the suggestion of an immunological effect against melanoma, there are no autoimmune manifestations, even though gangliosides are expressed on normal neuroectoderm-derived tissues (43).
Given the prevalence of autoantigens on human cancers, tolerance is emerging as a central theme for immune recognition of human cancer. There was apparent tolerance to gp75 in C57BL/6 mice, and tolerance for antibody responses was broken by immunization with human gp75 or by insect cells expressing mouse gp75. These results suggest that immunization with altered forms of the self protein may be one strategy to break tolerance. These observations are reminiscent of the phenomenon of molecular mimicry, where immune responses against self antigens can be triggered by structurally homologous molecules expressed by infectious pathogens.
Immunization with gp75/Sf9 lysates induced autoantibodies against mature gp75 and broke apparent tolerance, while purified gp75 from gp75/Sf9 cells induced selective recognition of a sequestered, early intracellular form of gp75. Mouse gp75 expressed in insect cells contains branched high mannose sugar chains, reminiscent of the endoplasmic reticulum form of mammalian gp75, compared with the complex sugars on mature mouse gp75 (33, 44–46). These two glycosylated forms have different surface charges (e.g., sialic acid residues on mouse gp75) and conformations (based on recognition by mAbs). The preferential recognition of early processed forms of gp75 after immunization with gp75/Sf9 may reflect the presence of mannose-rich residues on both the immunogen, gp75/Sf9, and the target antigen in the endoplasmic reticulum. Although these results suggest that Sf9 lysates contained an adjuvant effect for antibody responses, this possible adjuvant effect was not observed when purified native mouse gp75 or gp75 purified from Sf9 cells was mixed with Sf9 lysates or a variety of other adjuvants (Table 1 and Fig. 2). In fact, Sf9 lysates actually appeared to inhibit the antibody response to purified gp75 produced by baculovirus in Sf9 cells. Thus, endogenous expression of gp75 by Sf9 cells was required to induce autoantibodies, and depended presumably on how gp75 was packaged by the Sf9 cells. Because proteins expressed in Sf9 cells can form insoluble complexes, it is possible that the “adjuvant” effect observed in these studies was due to the insoluble nature of the antigen produced endogenously in Sf9 cells.
Autoantibodies against the sequestered intracellular form predictably did not provide tumor protection. However, immunization with endogenous gp75 in lysates of Sf9 cells induced an autoantibody response to mature self gp75, leading to tumor protection. The mature form of gp75 is expressed in melanosomes, but a proportion of gp75 also reaches the surface of melanoma cells, providing one explanation for antibody mediated tumor rejection (47). The induction of IgG antibodies suggests that helper T-cell responses were involved. Recently, Sun et al. (48) have shown that DNA released from Drosophila cells can stimulate B lymphocytes and that these activated B cells can costimulate T lymphocytes, providing one explanation for an adjuvant effect of insect cells. Also, it is predicted that gp75 may be presented through the class II MHC pathway to CD4 T cells, since gp75 contains an endosomal sorting signal (shared by other melanosomal membrane proteins) (49) that could traffic it near or through the MHC II compartment within the endocytic pathway. There is a precedent for this notion since tyrosinase can be presented through the class II MHC pathway (14). However, preliminary experiments show that immunization with Sf9/gp75 does not induce proliferative T-cell responses against gp75 peptides that bind class II MHC (data not shown), suggesting that induction of autoantibodies by Sf9/gp75 may not simply be due to recruitment of T-cell help against gp75.
These findings also confirm that autoimmunity directed against tyrosinase-related proteins expressed by melanocytes can influence coat color in mice. The induction of concomitant tumor rejection and autoimmunity is relevant to a clinical observation in persons with metastatic melanoma who develop vitiligo; vitiligo is associated with an improved prognosis (50, 51). This mouse model has recapitulated this association, showing that an active response to a member of the tyrosinase family of proteins can mediate melanoma rejection and induce vitiligo.
Acknowledgments
We thank Shakantula Tiwari for expert technical assistance in these studies. This work was supported by grants from the National Institutes of Health (CA56821, AR41465, and CA33049), the Louis and Anne Abrons Foundation, and Swim Across America.
Footnotes
Abbreviations: GM-CSF, granulocyte/macrophage colony-stimulating factor; CTL, cytotoxic T lymphocyte; IFN-γ, interferon γ; wt, wild type; MHC, major histocompatibility complex.
References
- 1.Pardoll D M. Nature (London) 1994;369:357. doi: 10.1038/369357a0. [DOI] [PubMed] [Google Scholar]
- 2.Tsomides T J, Eisen H N. Proc Natl Acad Sci USA. 1994;91:3487–3489. doi: 10.1073/pnas.91.9.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houghton A N. J Exp Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boon T, van der Bruggen P. J Exp Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyse E A, Old L J. Annu Rev Genet. 1969;3:269–285. [Google Scholar]
- 6.Houghton A N, Taormina M C, Ikeda H, Watanabe T, Oettgen H F, Old L J. Proc Natl Acad Sci USA. 1980;77:4260–4264. doi: 10.1073/pnas.77.7.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houghton A N, Eisinger M, Albino A P, Cairncross J G, Old L J. J Exp Med. 1982;156:1755–1766. doi: 10.1084/jem.156.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irie R F, Irie K, Morton D L. Cancer Res. 1976;36:3510–3517. [PubMed] [Google Scholar]
- 9.Livingston P O, Natoli E J, Calves M J, Stockert E, Oettgen H F, Old L J. Proc Natl Acad Sci USA. 1987;84:2911–2915. doi: 10.1073/pnas.84.9.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brichard V, Van Pel A, Wolfel T, Wolfel C, De Plaen E, Lethe B, Coulie P, Boon T. J Exp Med. 1993;178:489–495. doi: 10.1084/jem.178.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brichard V G, Herman J, Van Pel A, Wildmann C, Gaugler B, Wolfel T, Boon T, Lethe B. Eur J Immunol. 1996;26:224–230. doi: 10.1002/eji.1830260135. [DOI] [PubMed] [Google Scholar]
- 12.Robbins P F, El-Gamil M, Kawakami Y, Stevens E, Yannelli J R, Rosenberg S A. Cancer Res. 1994;54:3124–3126. [PubMed] [Google Scholar]
- 13.Wolfel T, Van Pel A, Brichard V, Schneider J, Seliger B, Meyer zum Buschenfelde K H, Boon T. Eur J Immunol. 1994;24:759–764. doi: 10.1002/eji.1830240340. [DOI] [PubMed] [Google Scholar]
- 14.Topalian S L, Rivoltini L, Mancini M, Markus N R, Robbins P F, Kawakami Y, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:9461–9465. doi: 10.1073/pnas.91.20.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawakami Y, Eliyahu S, Delgado C H, Robbins P F, Rivoltini L, Topalian S L, Miki T, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:3515–3519. doi: 10.1073/pnas.91.9.3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins P F, Rivoltini L, Yannelli J R, Appella E, Rosenberg S A. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox A L, Skipper J, Chen Y, Henderson R A, Darrow T L, Shabanowitz J, Engelhard V H, Hunt D F, Slingluff C., Jr Science. 1994;264:716–719. doi: 10.1126/science.7513441. [DOI] [PubMed] [Google Scholar]
- 18.Coulie P G, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, Mattei S, De Plaen E, Lurquin C, Szikora J P, Renauld J C, Boon T. J Exp Med. 1994;180:35–42. doi: 10.1084/jem.180.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sensi M, Traversari C, Radrizzani M, Salvi S, Maccalli C, Mortarini R, Rivoltini L, Farina C, Nicolini G, Wolfel T, Brichard V, Boon T, Bordignon C, Anichini A, Parmiani G. Proc Natl Acad Sci USA. 1995;92:5674–5678. doi: 10.1073/pnas.92.12.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde K H, Beach D. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 21.Vijayasaradhi S, Bouchard B, Houghton A N. J Exp Med. 1990;171:1375–1380. doi: 10.1084/jem.171.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Pel A, van der Bruggen P, Coulie P G, Brichard V G, Lethe B, van den Eynde B, Uyttenhove C, Renauld J C, Boon T. Immunol Rev. 1995;145:229–250. doi: 10.1111/j.1600-065x.1995.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 23.Wang R, Robbins P, Kawakami Y, Kang X-Q, Rosenberg S. J Exp Med. 1996;183:799–804. doi: 10.1084/jem.181.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara I, Takechi Y, Houghton A N. J Exp Med. 1995;182:1609–1614. doi: 10.1084/jem.182.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mintz B, Silvers W K. Science. 1967;158:1484–1486. doi: 10.1126/science.158.3807.1484. [DOI] [PubMed] [Google Scholar]
- 26.Fidler I J. Nat New Biol. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- 27.Jimenez M, Tsukamoto K, Hearing V J. J Biol Chem. 1991;266:1147–1156. [PubMed] [Google Scholar]
- 28.Shibahara S, Tomita Y, Sakakura T, Nager C, Chaudhuri B, Muller R. Nucleic Acids Res. 1986;14:2413–2427. doi: 10.1093/nar/14.6.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller J, Germain R N. J Exp Med. 1986;164:1478–1489. doi: 10.1084/jem.164.5.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lucklow V A, Summers M D. Biotechnology. 1988;6:47–55. [Google Scholar]
- 31.Vijayasaradhi S, Houghton A N. Int J Cancer. 1991;47:298–303. doi: 10.1002/ijc.2910470221. [DOI] [PubMed] [Google Scholar]
- 32.Kyte J, Doolittle R. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 33.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. pp. 1–726. [Google Scholar]
- 34.Cleveland D W, Fischer S G, Kirshner M W, Laemmli U K. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 35.Cote R J, Morrissey D M, Houghton A N, Thomson T M, Daly M E, Oettgen H F, Old L J. Proc Natl Acad Sci USA. 1986;83:2959–2963. doi: 10.1073/pnas.83.9.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prehaud C, Takehara K, Flamand A, Bishop D H. Virology. 1989;173:390–399. doi: 10.1016/0042-6822(89)90551-5. [DOI] [PubMed] [Google Scholar]
- 37.Ghiasi H, Kaiwar R, Nesburn A B, Wechsler S L. J Gen Virol. 1992;73:719–722. doi: 10.1099/0022-1317-73-3-719. [DOI] [PubMed] [Google Scholar]
- 38.Hara I, Nguyen H, Takechi Y, Gansbacher B, Chapman P B, Houghton A N. Int J Cancer. 1995;61:253–260. doi: 10.1002/ijc.2910610219. [DOI] [PubMed] [Google Scholar]
- 39.Coulie P G, Lehmann F, Lethe B, Herman J, Lurquin C, Andrawiss M, Boon T. Proc Natl Acad Sci USA. 1995;92:7976–7980. doi: 10.1073/pnas.92.17.7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robbins P, El-Gamil M, Li Y, Kawakami Y, Loftus D, Appella E, Rosenberg S. J Exp Med. 1996;183:1185–1192. doi: 10.1084/jem.183.3.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Livingston P O. Ann NY Acad Sci. 1993;690:204–213. doi: 10.1111/j.1749-6632.1993.tb44009.x. [DOI] [PubMed] [Google Scholar]
- 42.Jones P C, Sze L L, Morton D L, Irie R F. J Natl Cancer Inst. 1981;66:249–254. [PubMed] [Google Scholar]
- 43.Livingston P, Wong G, Adluri S, Too Y, Padavan M, Parente R, Hanlon C, Calves M, Helling F, Ritter G, Oettgen H, Old L. J Clin Oncol. 1994;12:1036–1044. doi: 10.1200/JCO.1994.12.5.1036. [DOI] [PubMed] [Google Scholar]
- 44.Jarvis D L, Summers M D. Mol Cell Biol. 1989;9:214–223. doi: 10.1128/mcb.9.1.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuroda K, Geyer H, Geyer R, Doerfler W, Klenk H. Virology. 1990;174:418–429. doi: 10.1016/0042-6822(90)90095-9. [DOI] [PubMed] [Google Scholar]
- 46.Roux L, Lloyd K O. Arch Biochem Biophys. 1986;251:87–96. doi: 10.1016/0003-9861(86)90054-8. [DOI] [PubMed] [Google Scholar]
- 47.Takechi, Y., Hara, I. Naftzger, C., Xu, Y. & Houghton, A. N. (1996) Clin. Cancer Res., in press. [PubMed]
- 48.Sun S, Cai Z, Langlade-Demoyen P, Kosaka H, Brunmark A, Jackson M R, Peterson P A, Sprent J. Immunity. 1996;4:555–564. doi: 10.1016/s1074-7613(00)80482-3. [DOI] [PubMed] [Google Scholar]
- 49.Vijayasaradhi S, Xu Y, Bouchard B, Houghton A N. J Cell Biol. 1995;130:807–820. doi: 10.1083/jcb.130.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duhra P, Ilchyshyn A. Clin Exp Dermatol. 1991;16:303–305. doi: 10.1111/j.1365-2230.1991.tb00383.x. [DOI] [PubMed] [Google Scholar]
- 51.Nordlund J J, Kirkwood J M, B M, Forget, Milton G, Albert D M, Lerner A B. J Am Acad Dermatol. 1983;9:689–696. doi: 10.1016/s0190-9622(83)70182-9. [DOI] [PubMed] [Google Scholar]