

Abstract
Guanylyl cyclase C (GCC) has been detected only in intestinal mucosa and colon carcinoma cells of placental mammals. However, this receptor has been identified in several tissues in marsupials, and its expression has been suggested in tissues other than intestine in placental mammals. Selective expression of GCC by colorectal tumor cells in extraintestinal tissues would permit this receptor to be employed as a selective marker for metastatic disease. Thus, expression of GCC was examined in human tissues and tumors, correlating receptor function with detection by PCR. GCC was detected by ligand binding and catalytic activation in normal intestine and primary and metastatic colorectal tumors, but not in extraintestinal tissues or tumors. Similarly, PCR yielded GCC-specific amplification products with specimens from normal intestine and primary and metastatic colorectal tumors, but not from extraintestinal tissues or tumors. Northern blot analysis employing GCC-specific probes revealed an ≈4-kb transcript, corresponding to recombinant GCC, in normal intestine and primary and metastatic colorectal tumors, but not in extraintestinal tissues. Thus, GCC is selectively expressed in intestine and colorectal tumors in humans and appears to be a relatively specific marker for metastatic cancer cells in normal tissues. Indeed, PCR of GCC detected tumor cells in blood from some patients with Dukes B colorectal cancer and all patients examined with Dukes C and D colorectal cancer, but not in that from normal subjects or patients with Dukes A colon carcinoma or other nonmalignant intestinal pathologies.
Guanylyl cyclase C (GCC) is a member of the family of receptor guanylyl cyclases, of which six members have been identified in mammals (see ref. 1 for review; refs. 2–4). Peptides that specifically bind to GCC include the Escherichia coli heat-stable enterotoxin (ST), guanylin, and uroguanylin (1, 5). Ligand binding to GCC activates guanylyl cyclase and elevates intracellular cGMP, resulting in phosphorylation of the cystic fibrosis transmembrane conductance regulator, increases in chloride flux, and, ultimately, fluid secretion (6–8).
In normal adult placental mammals, functional GCC has been identified only on intestinal mucosa cells, but not in normal extraintestinal tissues (9–11). Also, these receptors have been identified on human colon carcinoma cell lines in vitro (12–14). However, in marsupials, GCC is expressed in several organs, including lung, gall bladder, kidney, and testes (5, 15). Similarly, GCC has been demonstrated in regenerating adult rat liver (16). In addition, ST may alter electrolyte excretion in rabbit kidney (17). Finally, studies suggested that this receptor could be detected by molecular techniques in normal rat brain and adrenal gland and human and bovine airway epithelia (18). The anatomical pattern of expression of GCC in normal adult humans remains undefined.
Recently, ST binding activity and guanylyl cyclase activation was identified in human primary and metastatic colorectal tumors (19, 20). Thus, GCC may represent a selective marker of colorectal tumor metastases in extraintestinal tissues. Selective expression of GCC in normal intestinal mucosa and colorectal tumor cells might be exploited to develop diagnostic tests to detect tumor cells in normal tissues and blood for staging and surveillance of this disease. Thus, the present studies examine the selectivity of functional and molecular expression of GCC in normal human intestine, primary and metastatic colorectal tumors, extraintestinal tissues and tumors, and blood samples from normal subjects, patients with colon cancer, and those with other pathologies.
MATERIALS AND METHODS
Clinical Specimens.
Tissues and blood were obtained under an Institutional Review Board-approved protocol from Thomas Jefferson University Hospital, the National Disease Research Interchange (Philadelphia), and the Cooperative Human Tissue Network (Philadelphia) (19, 20). More than 280 human tissue samples were examined for ST binding and activation of guanylyl cyclase activity, and more than 260 tissue samples were examined for expression of GCC by reverse transcription–PCR (RT-PCR). Blood samples were collected in tubes containing EDTA, separated into plasma and cells by centrifugation, and either processed within 2 h of receipt or frozen at −80°C until use.
Cell Culture.
T84 and HT29 human colon carcinoma cells (American Type Culture Collection), human airway epithelial cells (Clonetics, San Diego), and 293 human kidney cells expressing rat GCC were grown to confluence and used as described (4, 12, 13, 21).
Receptor Binding and Enzyme Assays.
Membranes were prepared from tissues as described (19, 20). ST was iodinated (125I-labeled Tyr-4-ST) to a final specific activity of ≈2000 Ci/mmol (1 Ci = 37 GBq; ref. 22). Quantification of ST–receptor binding and activation of guanylyl cyclase were performed as described (19, 20, 23). Assays were performed at least in duplicate and the intraexperimental variability was ≤12% (SEM).
Nucleic Acid Extraction.
Total RNA was extracted from samples using a modified version of the acid guanidinium thiocyanate/phenol/chloroform method employing a single reagent (TRIzol reagent; GIBCO/BRL–Life Technologies, Gaithersburg, MD; refs. 24 and 25). Only samples exhibiting intact 28S and 18S ribosomal RNA were analyzed. mRNA was purified by Oligotex poly(A)+ mRNA affinity latex beads (Qiagen, Chatsworth, CA) using the manufacturer’s protocol. Greater than 90% of the mRNA was recovered in purified preparations. RNA preparations were stored in diethylpyrocarbonate-treated water (RNase-free) at −80°C. To remove contaminating genomic DNA, the RNA was treated with 1 unit of RQ1 RNase-free DNase per μl (Promega) for 15 min at 37°C, followed by a 30-min incubation at 95°C with 1 μl of RNase inhibitor (Panvera, Madison, WI).
RT-PCR.
Reverse transcription of mRNA (≤1 μg) was performed with 0.25 unit of avian myeloblastosis virus (AMV) reverse transcriptase XL per μl (Panvera) containing 10 mM Tris·HCl (pH 8.3), 50 mM KCl, 4 mM MgCl2, 1 mM each of dATP, dCTP, dGTP, and dTTP, 1 unit of RNase inhibitor per μl, and 1 μM GCC-specific antisense primer (nucleotides 325–345) in a total volume of 20 μl (26, 27). Thermal cycling proceeded for one cycle at 55°C for 30 min, 99°C for 5 min, and 4°C for 5 min. The resultant cDNA was subjected to PCR in the same reaction tube and included 2.5 units of TaKaRa Taq polymerase (Panvera) in 100 μl of: 10 mM Tris·HCl, 50 mM KCl, 2.5 mM MgCl, and 0.2 μM GCC-specific sense primer (nucleotides 119–140; refs. 26 and 28). Incubation and thermal cycling conditions were: 95°C for 2 min, one cycle; 94°C for 30 sec, 58°C for 30 sec, and 72°C for 90 sec, 35 cycles; and 72°C for 7 min, one cycle. Some tissues were analyzed as described above using GCC-specific nested primers. In these studies, first round amplification reactions (35 cycles) employed alternate antisense (nucleotides 688–709) and sense (nucleotides 13–36) primers (26). Second round amplification reactions (35 cycles) were initiated using 1–10 μl of the amplified cDNA solution from first-round reactions in a final volume of 100 μl under the same PCR conditions, using nested antisense (nucleotides 267–290) and sense (nucleotides 13–36) primers (26). Blood samples were analyzed by nested primer RT-PCR as described above, using antisense (nucleotides 1197–1218) and sense (nucleotides 685–708) primers for first-round amplification reactions and antisense (nucleotides 1000–1021) and sense (nucleotides 759–781) primers for second-round amplification reactions (26). Where indicated, PCR products were radiolabeled by performing PCR in the presence of 50 μM dCTP supplemented with [α-32P]dCTP (10 Ci/mmol). Following RT-PCR, samples were stored at 4°C until analysis, within 24 h of amplification. Primers specific for human β-actin (CLONTECH) were used as a positive control (29). In some studies of intestinal and airway epithelial cells, primers specific for the human cystic fibrosis transmembrane conductance regulator (antisense primers, GCCATCAGTTTACAGACACAG; sense primers, TTGCTGGATCCACTGGAGCAGG) were also used as a positive control (30, 31).
Ribonuclease Protection Assay.
A modification of the ribonuclease protection assay was used to confirm the presence of GCC-specific amplification products in PCRs (26, 32–34). T84 cell mRNA (≤50 ng) was reverse transcribed (antisense primer, nucleotides 325–345) and the resultant first-strand cDNA was used in a PCR with a GCC-specific sense primer (nucleotides 119–140), as described above (26). Reaction products were subjected to further amplification using identical primers but possessing a T7 transcription initiation start site on the antisense primer. The resultant antisense oligonucleotide (nucleotides 119–345) possessing a 5′ T7 transcription initiation start site was used as a template to generate a [32P]UTP-labeled antisense riboprobe using the MAXIscript T7 In Vitro Transcription Kit (Ambion, Austin, TX) according to the manufacturer’s instructions, to a final specific activity of 109 cpm/μg. Ribonuclease protection assays were performed using the 32P-labeled GCC-specific antisense riboprobe, amplification products of RT-PCRs, and the ribonuclease protection assay II kit (Ambion), according to the manufacturer’s instructions.
Northern Blot Analysis.
Messenger RNA was subjected to Northern blot analysis using a cDNA probe spanning nucleotides 13–702, corresponding to portions of the extracellular domain of human GCC (4, 26). This probe was generated from T84 cell mRNA by RT-PCR using GCC-specific primers (antisense, nucleotides 688–709; sense, nucleotides 12–33) corresponding to the extracellular domain (12, 26). The PCR fragment was gel-purified and ligated into pNoTA/T7 shuttle vector (Prime PCR CLONER Cloning System; 5 Prime → 3 Prime). The vector was transformed and grown in competent antibiotic-resistant E. coli. The plasmid containing the GCC probe was labeled with T4 DNA kinase (Pharmacia) and separated from unreacted components by spin column chromatography on Sephadex 50 (Millipore), and 200 ng was used for detection of RNA on blots. A probe specific for glyceraldehyde dehydrogenase was employed as a positive control in these studies (American Type Culture Collection).
Miscellaneous.
Protein was measured using bovine serum albumin (Bio-Rad) as standard (35). All reagents commercially obtained were of the highest analytical grade.
RESULTS
Binding of 125I-labeled ST and ST activation of guanylyl cyclase were examined in membranes prepared from human tissues (Table 1). All tissues of intestinal origin specifically bound ST in a concentration-dependent and saturable fashion (data not shown). Binding was detected in all anatomical divisions of the intestine and in all colorectal tumors examined, regardless of the metastatic location (Table 1). Analysis of these data by the method of Scatchard revealed curvilinear isotherms, demonstrating the presence of high (pM) and low (nM) affinity binding sites for ST in all tissues of intestinal origin (data not shown), in close agreement with previous studies (19, 20, 23). The KD and Bmax values for high and low affinity sites were similar in small intestine, cecum, and colon (Table 1). In contrast, Bmax values for high and low affinity sites in the rectum were lower than in other anatomical divisions of the intestine, in close agreement with previous studies (20). Specific ST binding was not detected in >40 different types of extraintestinal tissues and tumors (>100 total extraintestinal specimens) examined. Similarly, ST activated guanylyl cyclase in all colorectal tumors, regardless of their metastatic location, but not in any extraintestinal tissue or tumor examined (Table 1; refs. 19, 20, and 36). Activation was similar in small intestine, cecum, and colon, but lower in rectum, in close agreement with the reduced number of ST binding sites in that tissue (19, 20).
Table 1.
ST binding parameters and activation of guanylyl cyclase in membranes from normal human intestine and primary and metastatic colorectal tumors
| n | High affinity ST
binding
|
Low affinity ST binding
|
Guanylyl cyclase, fold | |||
|---|---|---|---|---|---|---|
| Bmax | KD, pM | Bmax | KD, nM | |||
| Normal | ||||||
| Small intestine | 9 | 38 ± 22.1 | 23.8 ± 13.4 | 320 ± 77 | 1.7 ± 0.8 | 2.11 ± 0.21 |
| Cecum | 3 | 25 ± 14.3 | 26.2 ± 10.1 | 300 ± 82 | 2.0 ± 0.7 | 1.95 ± 0.12 |
| Colon | 46 | 20 ± 12.2 | 34.4 ± 11.8 | 220 ± 49 | 2.7 ± 1.1 | 1.57 ± 0.22 |
| Rectum | 4 | 12 ± 10.1 | 47.0 ± 14.1 | 125 ± 34 | 2.5 ± 1.5 | 1.39 ± 0.08 |
| Primary CRC | ||||||
| Cecum | 7 | 20 ± 9.91 | 19.3 ± 9.2 | 260 ± 66 | 2.6 ± 0.2 | 1.81 ± 0.09 |
| Colon | 60 | 16 ± 11.4 | 28.3 ± 6.1 | 180 ± 36 | 3.1 ± 0.4 | 1.69 ± 0.10 |
| Rectum | 6 | 10 ± 7.72 | 31.0 ± 4.2 | 95 ± 44 | 4.9 ± 0.3 | 1.35 ± 0.12 |
| Metastatic CRC | ||||||
| Liver | 12 | 10 ± 4.45 | 28.1 ± 6.10 | 110 ± 19 | 2.1 ± 0.3 | 1.81 ± 0.27 |
| Lung | 5 | 15 ± 8.50 | 19.4 ± 4.12 | 195 ± 80 | 5.8 ± 0.1 | 1.92 ± 0.23 |
| Lymph node | 8 | 10 ± 5.21 | 29.3 ± 9.20 | 138 ± 27 | 4.8 ± 1.0 | 1.55 ± 0.26 |
| Ovary | 3 | 12 ± 4.82 | 44.7 ± 12.1 | 160 ± 38 | 6.9 ± 3.3 | 1.38 ± 0.21 |
| Peritoneum | 7 | 18 ± 7.86 | 35.5 ± 6.01 | 209 ± 17 | 5.6 ± 0.2 | 1.44 ± 0.19 |
| Mesentery | 1 | 27 ± 16.1 | 36.2 ± 6.10 | 295 ± 33 | 3.1 ± 1.2 | 1.41 ± 0.20 |
| Stomach | 1 | 17 ± 10.2 | 20.1 ± 9.21 | 100 ± 25 | 1.7 ± 0.4 | 1.50 ± 0.21 |
Equilibrium binding was conducted as described, and activation of guanylyl cyclase was quantified employing 1 μM ST as described. n = number of tissue specimens; Bmax = fmol of ST bound per mg of protein; fold = (cGMP produced in the presence of 1 μM ST) (cGMP produced in the absence of ST). For tissues where n = 1, the SEM reflects at least three determinations using the same tissue. For all other tissues, the SEM reflects results obtained with n tissues. CRC, colorectal cancer.
The specificity of expression of GCC was further examined by determining if mRNA encoding this protein could be detected in extraintestinal sites in humans using RT-PCR. GCC-specific primers corresponding to portions of the extracellular domain and mRNA extracted from intestinal tissues yielded an amplification product of ≈250 bases, the size predicted from the defined sequence of that protein (Fig. 1; ref. 26). This amplification product was identified in all segments of the colon and rectum (>20 specimens) and all primary and metastatic colorectal tumors examined (>55 specimens; Fig. 1). Restriction enzyme digestion of that amplification product with SmaI or AatII yielded cDNA fragments of the size predicted by the defined sequence for human GCC (data not shown; ref. 26). Similarly, sequencing of amplification products confirmed identity with GCC in all cases. However, GCC-specific amplification products were not detected with mRNA extracted from >40 different types of extra-intestinal tissues and tumors (>140 total extraintestinal specimens).
Figure 1.
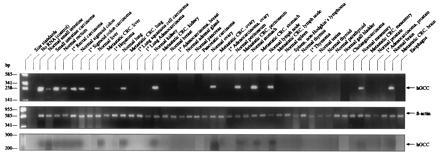
Expression of GCC in tissues detected by RT-PCR. (Top and Middle) RT-PCR was performed employing mRNA purified from indicated tissues and primers for GCC (Top) or β-actin (control; Middle), as described. (Bottom) Aliquots of the above RT-PCRs were subjected to the ribonuclease protection assay as described. Left lane indicates size markers. Arrows indicate the size of human GCC (hGCC; ≈250 bp) and β-actin (≈600 bp) predicted from their defined sequences.
The inability to detect GCC by RT-PCR in extraintestinal sites might reflect expression of that protein below the limits of detection of the methods used. The ribonuclease protection assay is a sensitive technique that can detect femtogram quantities of complementary oligonucleotides (32, 33). Thus, a GCC-specific riboprobe was labeled with [32P]nucleotide to high specific activity (109 cpm/μg), hybridized with products of RT-PCR incubations, digested with ribonuclease, and analyzed by electrophoresis. Protection of the labeled riboprobe against digestion and its detection by autoradiography indicates the presence of a GCC-specific RT-PCR amplification product. Riboprobe protection was observed with amplification products from normal colon and rectum and primary and metastatic colorectal tumors, confirming that these products were specific for GCC (Fig. 1). In contrast, amplification products from extraintestinal tissues and tumors did not protect the labeled riboprobe, supporting the suggestion that GCC is not expressed in those specimens.
Previous studies suggested that GCC expression might be detected in rat adrenal gland and brain and human and bovine airway epithelia (18). However, those studies used primers corresponding to portions of the cytoplasmic region, which included the catalytic domain of GCC (18). The catalytic domains of receptor guanylyl cyclases exhibit the highest degree of homology, >50%, compared with other signature domains of this family (1). Also, these earlier studies used hybridization conditions of relatively low stringency (4, 18). Thus, previous results might reflect hybridization with sequences other than GCC (18). Alternatively, earlier results may reflect expression of GCC transcripts expressed in relatively low abundance, below the limit of detection of RT-PCR employed in the present studies. Thus, select extraintestinal tissues were subjected to RT-PCR using nested primers corresponding to portions of the extracellular domain (16–18). These conditions permit amplifications of up to 10- to 20-fold, and should identify specific transcripts expressed in low abundance. A GCC-specific amplification product was detected in intestine, but not in liver, kidney, brain, adrenal gland, or human airway epithelial cells (data not shown). In contrast, an amplification product specific for the cystic fibrosis transmembrane conductance regulator, expressed in intestinal and airway mucosa, was detected in human intestine and airway epithelial cells (data not shown; refs. 30 and 31). These data further confirm that native GCC is not expressed in human extraintestinal tissues.
Northern blot analysis was conducted using a GCC-specific cDNA probe to determine if the transcript of this protein expressed by colorectal tumors was similar to that in normal intestinal mucosa. Indeed, a specific band of ≈4.4 kb was detected with normal colon and primary colonic tumors, which corresponded precisely to a band detected with cells expressing recombinant GCC and HT29 human colon carcinoma cells (data not shown). A similar band was detected with colorectal tumors metastatic to lung and liver, but not in normal specimens of those tissues (data not shown). These data demonstrate that the transcript of the gene encoding GCC expressed in primary and metastatic colorectal tumors is similar to that expressed in normal intestinal mucosa.
The above studies suggest that RT-PCR using GCC-specific primers might represents a specific and sensitive method for detecting metastatic colorectal tumor cells in blood. Thus, T84 human colon carcinoma cells were diluted with normal human blood and a 1-ml aliquot containing 106 normal blood cells and the indicated number of tumor cells assessed by ST binding, guanylyl cyclase activation, and RT-PCR employing GCC-specific nested primers (Fig. 2). Guanylyl cyclase activation by ST was least sensitive, requiring at least 106 tumor cells per ml for detection. By comparison, 125I-labeled ST binding could detect 1 cancer cell in 1000 normal blood cells, requiring at least 103 tumor cells per ml. However, RT-PCR was the most sensitive detection method, detecting 1 cancer cell in 106 normal blood cells.
Figure 2.
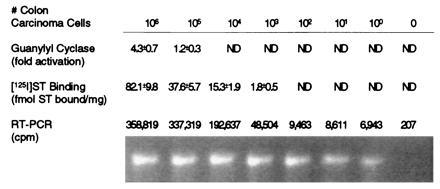
Sensitivity of enzyme activation, receptor binding, and RT-PCR to detect colon carcinoma cells in blood. The indicated number of T84 human colon carcinoma cells were diluted with 106 normal human blood cells in 1 ml and examined for ST (1 μM) activation of guanylyl cyclase and 125I-labeled ST (2.5 × 10−9 M) binding and by RT-PCR using GCC-specific nested primers, as described. PCR was conducted with 32P-labeled nucleotides, and the resultant amplification products resolved by electrophoresis were stained with ethidium bromide (lower row) or directly quantified (upper row). Fold activation was calculated as (cGMP produced with ST)/cGMP produced without ST). Values represent the mean ± SEM. ND, not detectable.
These studies demonstrating that colon carcinoma cells could be detected in blood were extended by comparing blood from patients with Dukes A, B, C, and D colorectal cancer, patients with nonmalignant pathology of the intestine, patients with extraintestinal malignancies, and normal male subjects using RT-PCR with GCC-specific nested primers (Fig. 3). Amplification products were not obtained with blood from normal subjects, nor from patients with adenomatous polyp disease, acute exacerbating inflammatory bowel disease, extraintestinal malignancies, or Dukes A colon carcinoma, or from some patients with Dukes B colon carcinoma. In contrast, specific amplification products were obtained with blood from some patients with Dukes B colorectal cancer and all patients examined with Dukes C and D colorectal cancer. The identity of those products with GCC was confirmed by restriction enzyme digestion and sequencing (data not shown).
Figure 3.
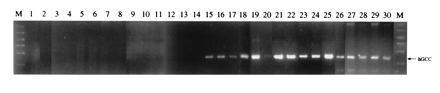
Detection of colon carcinoma cells in blood of patients using RT-PCR and GCC-specific nested primers. Total RNA was extracted from a 1-ml aliquot of blood cells, mRNA-purified, and used for RT-PCR, using GCC-specific nested primers, as described. Border lanes (M) indicate size markers (200–1000 bp). Arrow indicates the predicted size of the amplification product for human GCC (hGCC; ≈250 bp). Lane 1, normal volunteers (n = 10); lane 2, patients with a colonic adenomatous polyp (n = 6); lane 3, patient with an acute exacerbation of inflammatory bowel disease (n = 1); lane 4, patient with adenocarcinoma of the lung (n = 1); lane 5, patient with adenocarcinoma of the breast (n = 1); lane 6, patient with adenocarcinoma of the ovary (n = 1); lane 7, patient with lymphoma (n = 1); lane 8, patients with pancreatic carcinoma (n = 6); lanes 9–11, patients with Dukes A colorectal cancer; lanes 12–16, patients with Dukes B colorectal cancer; lanes 17–20, patients with Dukes C colorectal cancer; and lanes 21–30, patients with Dukes D colorectal cancer.
DISCUSSION
The present studies suggest that human GCC is selectively expressed in intestinal mucosa and primary and metastatic colorectal tumors, but not in extraintestinal tissues or tumors (19, 20). Selective tissue expression was detected by ligand binding, activation of guanylyl cyclase, RT-PCR, and Northern blot analysis. These results are consistent with earlier studies in animals demonstrating that ST binding and activation of guanylyl cyclase could be detected only in intestinal cells, but not in extraintestinal tissues (9–11). Also, they agree with those demonstrating expression of functional ST receptors in human colon carcinoma cells in vitro and in vivo (12–14, 19, 20). They suggest that GCC is similar to members of the family of receptor guanylyl cyclases that exhibit tissue-specific expression, such as guanylyl cyclases D, E, and F, rather than those expressed in many tissues, such as guanylyl cyclases A and B (1–4). It should be noted that these studies did not examine every tissue type, under all possible normal and pathological conditions, for expression of GCC. Thus, determining whether GCC is specifically expressed exclusively by intestinal mucosal and colorectal tumor cells, under all circumstances, will require further studies.
More recently, studies suggested that GCC might be expressed in rat adrenal gland and brain and bovine and human airway epithelia (18). These conclusions were based, in part, on results obtained by PCR using primers that included sequences corresponding to the catalytic domain and hybridization conditions of relatively low stringency (18). Receptor guanylyl cyclases share >50% sequence homology in their cognate catalytic domains (1–4, 26). In contrast, primers used in the present studies corresponded to the extracellular domain of GCC, which exhibits <10% homology with other members of the family (1–4, 26). The present studies demonstrated that amplification products were not detected in human adrenal gland, brain, and airway epithelial cells following RT-PCR employing GCC-specific primers corresponding to portions of the extracellular domain, hybridization conditions of high stringency, and high levels of amplification. Results from earlier studies may reflect amplification of sequences unrelated to guanylyl cyclases, since conditions of relatively low stringency were employed. Thus, computerized database analysis of sequence homology demonstrated >50 potential hybridization candidates for the primers used previously (18). Alternatively, those results could reflect the presence of a novel member of the receptor guanylyl cyclase family in those tissues. Indeed, a novel receptor guanylyl cyclase not yet fully characterized was identified previously in the intestine (4). Furthermore, previous results might reflect the expression of splice variants of GCC that possess only the intracellular, but not the extracellular, domain. Splice variants of the heterodimeric cytosolic form of guanylyl cyclase have been identified previously (37).
Recent studies demonstrated that ST binding and cyclase activation were detected in several tissues in opossum, including intestine, kidney, testes, lung, and gall bladder, and in adult regenerating rat liver (5, 15, 16). In addition, perfusion of isolated rabbit kidneys with ST altered the clearance of sodium and potassium (17). These results contrast sharply with those presented herein. That functional ST receptors can be detected in multiple tissues in opossum suggest that there are significant differences in the expression of GCC in marsupial compared with placental mammals (5, 15). Similarly, the relationship of expression of GCC in regenerating rat liver to that in normal human liver remains unclear (16). It is notable that GCC is expressed in several extraintestinal tissues, including liver, in the developing rat fetus (38). Thus, the expression of GCC in regenerating rat liver may be related to the expression of this protein in that tissue during ontogeny (16, 38).
Alternatively, a novel form of receptor guanylyl cyclase may be expressed in rabbit kidney (17). Indeed, guanylin and uroguanylin are low molecular weight, heat-stable peptides that share homology with ST. These peptides bind to and activate GCC in the intestine (5). Both peptides are synthesized as high molecular weight precursors and have been identified in mammalian urine (5). Interestingly, guanylin and uroguanylin have lower affinity for binding to and potency for activating GCC compared with ST (5). In fact, it has been suggested that GCC may not be the native receptor for these ligands. That suggestion is supported by the present results, demonstrating that expression of native GCC cannot be detected in human extraintestinal tissues or tumors. Rather, other receptor guanylyl cyclases or other proteins may function as receptors for guanylin and uroguanylin in extraintestinal tissues (5). Possibly, these other receptors mediate the observed changes in electrolyte transport when rabbit kidneys are perfused with high concentrations of ST (17).
About 150,000 new cases of colorectal cancer occur in the United States each year (39). The mainstay of staging and treatment continues to be surgery (40). However, about one-third of the patients undergoing surgery with curative intent develop recurrent disease, due to the presence of undetected metastatic tumor (39). In addition, delays in tumor recurrence and improvement in patient survival can be achieved with adjuvant therapy in patients with Dukes C disease (41). Thus, accurate staging to detect extraintestinal tumor could have a significant impact on the morbidity and mortality associated with colorectal cancer. Staging is dependent upon the ability to detect colorectal tumor cells in otherwise normal extraintestinal tissues. Currently, there are no specific and sensitive markers for staging colorectal cancer. The selective expression of GCC in metastatic colorectal cancer cells, but not in other extraintestinal tissues to which these cells typically metastasize, suggest that detection of the expression of this protein by RT-PCR may be useful for staging patients with this disease.
Similarly, there are no specific and sensitive markers to detect recurrent colorectal cancer following presumably curative surgery. Indeed, carcinoembryonic antigen (CEA), the most frequently employed marker for postoperative surveillance, detects <60% of recurrences, most after development of terminal disease (42). However, the selective expression of GCC by metastatic colorectal tumors, but not other extraintestinal tissues, suggests that detection of the expression of this protein by RT-PCR in blood might have use for postoperative surveillance for recurrent disease. In the present studies, GCC expression was not detected in blood from normal volunteers, patients with nonmalignant pathology of the intestine, and patients with extraintestinal malignancies. As anticipated, GCC was not detected in blood from patients with Dukes A colon carcinoma, since these tumors are confined to the epithelial layer of the intestine (43). Detection of GCC expression in blood from patients with Dukes B disease was heterogeneous, reflecting the variable penetration of tumor into the bowel wall characteristic of this stage (44, 45). GCC expression was detected in blood from Dukes C and D patients. In Dukes C patients, tumor cells have metastasized outside the bowel wall and into regional lymph nodes (43). It has been suggested that metastasis of tumor cells into the lymphoid system is contemporaneous with that into the circulation (46). Metastasis of tumor cells to distant tissues, such as liver and lung, as in Dukes D patients, requires hematogenous spread (43, 46). It should be noted that the prognostic significance, if any, of the detection of GCC by RT-PCR in patients with Dukes B, C, and D disease remains undefined. However, these studies suggest that the value of GCC as a marker for the diagnosis, staging, and postoperative surveillance of patients with colorectal cancer deserves further examination.
Acknowledgments
We gratefully acknowledge the following from Thomas Jefferson University for their contributions: members of the Departments of Surgery and Pathology; Lisa L. Liu of the Medical Records Department and Dawna Powell of the Blood Bank; and the Division of Clinical Pharmacology, Department of Medicine. We thank D. C. Robertson, Ph.D., University of Idaho, for providing ST and D. L. Garbers, Ph.D., and S. Schulz, Ph.D., for providing 293 cells expressing rat GCC. This research was supported by the National Science Foundation (IBN-9205717), the National Institutes of Health (1 R55 DK43805), W.W. Smith Charitable Trust, the Elsa U. Pardee Foundation, and Targeted Diagnostics and Therapeutics, Inc. Stephen L. Carrithers was the recipient of an National Institutes of Health Postdoctoral Fellowship (1 F32 CA63764-01).
Footnotes
Abbreviations: GCC, guanylyl cyclase C; ST, E. coli heat-stable enterotoxin; RT-PCR, reverse transcription–PCR.
References
- 1.Fulle H-J, Garbers D L. Cell Biochem Funct. 1994;12:157–165. doi: 10.1002/cbf.290120303. [DOI] [PubMed] [Google Scholar]
- 2.Yang R-B, Foster D C, Garbers D L, Fulle H-J. Proc Natl Acad Sci USA. 1995;92:602–606. doi: 10.1073/pnas.92.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulle H-J, Vassar R, Foster D C, Yang R B, Garbers D L. Proc Natl Acad Sci USA. 1995;92:3571–3575. doi: 10.1073/pnas.92.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz S, Green C K, Yuen P T S, Garbers D L. Cell. 1990;63:941–948. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 5.Forte L R, Currie M G. FASEB J. 1995;9:643–650. doi: 10.1096/fasebj.9.8.7768356. [DOI] [PubMed] [Google Scholar]
- 6.Field M, Graf L H, Jr, Laird W J, Smith P L. Proc Natl Acad Sci USA. 1978;75:2800–2804. doi: 10.1073/pnas.75.6.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chao A C, de Sauvage F J, Dong Y-J, Wagner J A, Goeddel D V, Gardner P. EMBO J. 1994;13:1065–1072. doi: 10.1002/j.1460-2075.1994.tb06355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markert T, Vaandrager A B, Gambaryan S, Pohler D, Hausler C, Walter U, De Jonge H R, Jarchau T, Lohmann S M. J Clin Invest. 1995;96:822–830. doi: 10.1172/JCI118128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes J M, Murad F, Chang B, Guerrant R L. Nature (London) 1978;271:755–756. doi: 10.1038/271755a0. [DOI] [PubMed] [Google Scholar]
- 10.Guerrant R L, Hughes J M, Chang B, Robertson D C, Murad F. J Infect Dis. 1980;142:220–228. doi: 10.1093/infdis/142.2.220. [DOI] [PubMed] [Google Scholar]
- 11.Rao M C, Guandolini S, Smith P L, Field M. Biochim Biophys Acta. 1980;632:35–46. doi: 10.1016/0304-4165(80)90247-0. [DOI] [PubMed] [Google Scholar]
- 12.Huott P A, Liu W, McRoberts J A, Giannella R A, Dharmsathaporn K. J Clin Invest. 1988;82:514–523. doi: 10.1172/JCI113626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaandrager A B, Bot A G M, De Vente J, De Jonge H R. Gastroenterology. 1992;102:1161–1169. [PubMed] [Google Scholar]
- 14.Cohen M, Jensen N, Hawkins J, Mann E, Thompson M, Lenze M, Giannella R A. J Cell Physiol. 1993;156:138–144. doi: 10.1002/jcp.1041560119. [DOI] [PubMed] [Google Scholar]
- 15.Forte L R, Krause W J, Freeman R H. Am J Physiol. 1989;257:F874–F881. doi: 10.1152/ajprenal.1989.257.5.F874. [DOI] [PubMed] [Google Scholar]
- 16.Laney D W, Jr, Bezerra J A, Kosiba J L, Degen S J F, Cohen M B. Am J Physiol. 1994;266:G899–G906. doi: 10.1152/ajpgi.1994.266.5.G899. [DOI] [PubMed] [Google Scholar]
- 17.Lima A A M, Montiero H S A, Fonteles M C. Pharmacol Toxicol (Copenhagen) 1992;70:163–167. doi: 10.1111/j.1600-0773.1992.tb00449.x. [DOI] [PubMed] [Google Scholar]
- 18.Schulz S, Chrisman T D, Garbers D L. J Biol Chem. 1992;267:16019–16021. [PubMed] [Google Scholar]
- 19.Carrithers S L, Parkinson S J, Goldstein S, Park P, Robertson D C, Waldman S A. Gastroenterology. 1994;107:1653–1661. doi: 10.1016/0016-5085(94)90804-4. [DOI] [PubMed] [Google Scholar]
- 20.Carrithers S L, Parkinson S J, Goldstein S D, Park P K, Urbanski R W, Waldman S A. Dis Colon Rectum. 1995;39:171–181. doi: 10.1007/BF02068072. [DOI] [PubMed] [Google Scholar]
- 21.Vaandrager A B, Schulz S, De Jonge H R, Garbers D L. J Biol Chem. 1993;268:2174–2179. [PubMed] [Google Scholar]
- 22.Thompson M R, Luttrell M, Overmann G, Giannella R A. Anal Biochem. 1985;148:26–36. doi: 10.1016/0003-2697(85)90623-2. [DOI] [PubMed] [Google Scholar]
- 23.Hugues M, Crane M R, O’Hanley P, Waldman S A. Biochemistry. 1991;30:10738–10745. doi: 10.1021/bi00108a019. [DOI] [PubMed] [Google Scholar]
- 24.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 25.Puissant C, Houdebine L M. BioTechniques. 1990;8:148–149. [PubMed] [Google Scholar]
- 26.de Sauvage F J, Camerato T R, Goeddel D V. J Biol Chem. 1991;266:17912–17918. [PubMed] [Google Scholar]
- 27.Beverley S M. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Mith J A, Struhl K, editors. New York: Wiley; 1994. pp. 15.4.1–15.1.6. [Google Scholar]
- 28.Kramer M, Coen D M. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Mith J A, Struhl K, editors. New York: Wiley; 1994. pp. 15.1.1–15.1.8. [Google Scholar]
- 29.Walther W, Stein U, Eder C. BioTechniques. 1994;17:674–675. [PubMed] [Google Scholar]
- 30.Trapnell B C, Chu C-S, Paako P K, Banks T C, Yoshimura K, Ferrans V J, Chernick M S, Crystal R G. Proc Natl Acad Sci USA. 1991;88:6565–6569. doi: 10.1073/pnas.88.15.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crawford I, Maloney P C, Zeitlin P L, Guggino W B, Hyde S C, Turley H, Gatter K C, Harris A, Higgins C F. Proc Natl Acad Sci USA. 1991;88:9262–9266. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melton D A, Krieg P A, Rebagliati M R, Maniatis T, Zinn K, Green M R. Nucleic Acids Res. 1984;12:7035–7056. doi: 10.1093/nar/12.18.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch F F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. pp. 7.71–7.78. [Google Scholar]
- 34.Gilman M. In: Current Protocols in Molecular Biology. Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Mith J A, Struhl K, editors. New York: Wiley; 1994. pp. 4.7.1–4.7.8. [Google Scholar]
- 35.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Krause G, Bayeri A, Heim J-M, Singh S, Gerzer R. Gut. 1994;35:1250–1257. doi: 10.1136/gut.35.9.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cchajlani V, Franberg P-A, Ahlner J, Axelsson K L, Wikberg J E S. FEBS Lett. 1991;290:157–158. doi: 10.1016/0014-5793(91)81248-7. [DOI] [PubMed] [Google Scholar]
- 38.Laney D W, Jr, Mann E A, Dellon S C, Perkins D R, Giannella R A, Cohen M B. Am J Physiol. 1992;263:G816–G821. doi: 10.1152/ajpgi.1992.263.5.G816. [DOI] [PubMed] [Google Scholar]
- 39.Silverberg E, Boring C C, Squires T S. Cancer. 1990;40:9–26. [PubMed] [Google Scholar]
- 40.Pihl E, Hughes E S R, McDermott F T, Milne B J, Price A B. J Surg Oncol. 1981;16:333–341. doi: 10.1002/jso.2930160406. [DOI] [PubMed] [Google Scholar]
- 41.Moertel C G, Fleming T R, Macdonald J S, Haller D G, Laurie J A, Goodman P J, Ungerleider J S, Emerson W A, Tormey D C, Glick J H, Veeder M H, Mailliard J A. N Engl J Med. 1990;322:352–358. doi: 10.1056/NEJM199002083220602. [DOI] [PubMed] [Google Scholar]
- 42.Moertel C G, Fleming T R, Macdonald J S, Haller D G, Laurie J A, Tangen C. J Am Med Assoc. 1993;270:943–947. [PubMed] [Google Scholar]
- 43.Wolmark N, Fisher B, Wieand H S. Ann Surg. 1986;203:115–121. doi: 10.1097/00000658-198602000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubia C A, Emas S, Nylander G. Surg Gynecol Obstet. 1977;145:682–684. [PubMed] [Google Scholar]
- 45.Jass J R, Love S, Northover J M A. Lancet. 1987;i:1303–1306. doi: 10.1016/s0140-6736(87)90552-6. [DOI] [PubMed] [Google Scholar]
- 46.Cady B. Arch Surg (Chicago) 1984;119:1067–1072. doi: 10.1001/archsurg.1984.01390210063014. [DOI] [PubMed] [Google Scholar]