

Abstract
We have previously identified a 94- to 97-kDa oxidized low density lipoprotein (LDL)-binding protein in mouse macrophages as macrosialin (MS), a member of the lamp family. Earlier immunostaining studies have shown that MS and its human homolog, CD68, are predominantly intracellular proteins. However, using sensitive techniques such as flow cytometry (FACS) and cell-surface-specific biotinylation, we now show that there is significant surface expression of these proteins. FACS analysis of intact cells using mAb FA/11 showed small but definite surface expression of MS in resident mouse peritoneal macrophages but this was greatly enhanced with thioglycollate elicitation. Biotinylation of intact cells and detergent-solubilized cell preparations followed by immunoprecipitation revealed 10–15% of the total MS content of elicited macrophages on the plasma membrane. Similar results were obtained with untreated RAW 264.7 cells. FACS analysis of intact THP-1 monocytic cells showed minimal surface expression of CD68 on unactivated cells (4% of total cell content). Stimulation with phorbol 12-myristate 13-acetate increased both surface and total CD68 expression considerably. Furthermore, the specific binding at 4°C and uptake at 37°C of 125I-labeled oxidized LDL by activated THP-1 cells was inhibited by 30–50% by CD68 mAbs KP-1 and EBM-11. Thus, although the surface expression of MS/CD68 at steady-state represents only a small percentage of their total cellular content, these proteins can play a significant role in oxidized LDL uptake by activated macrophages in vitro and could contribute to foam cell formation in atherosclerotic lesions.
The conversion of monocyte/macrophages to foam cells depends upon the uptake not of native low density lipoprotein (LDL) but of one or more modified forms of LDL (1). The first modification of LDL shown to lead to foam cell formation was chemical acetylation, as described by Goldstein et al. (2). They showed that this was due to uptake by a new receptor, the acetyl-LDL receptor or scavenger receptor A, later cloned by Kodama et al. (3). However, the generation of acetyl-LDL in vivo has not been demonstrated and seems unlikely to occur at a rate sufficient to give it an important role. Oxidized LDL (OxLDL), on the other hand, is also taken up more avidly than native LDL by macrophages (4–7), in part by way of scavenger receptor A, and oxidative modification of LDL does occur in vivo (8).
That oxidative modification plays an important role in atherogenesis is strongly implied by a large number of studies in animal models (cholesterol-fed rabbits, LDL-receptor-deficient rabbits, cholesterol-fed hamsters, cholesterol-fed nonhuman primates, and transgenic apolipoprotein E-deficient or LDL-receptor-deficient mice), showing that the appropriate use of one or another antioxidant compound slows the progression of the fatty streak lesion (9, 10). The enhanced uptake of OxLDL by monocyte/macrophages occurs in part by way of the same receptor that recognizes acetyl-LDL but additional receptors must be involved (11–14). Endemann et al. (15) showed that CD36 can bind and take up OxLDL, a finding that has been confirmed by others (14, 16, 17) and OxLDL uptake by human monocyte/macrophages clearly occurs in part by way of this receptor. Hamster SR-BI can bind acetyl-LDL and OxLDL (14) but the tissue distribution of this receptor and its high affinity for high density lipoprotein (HDL) have suggested quite a different role for it, namely, in the selective uptake of cholesterol esters in steroidogenic tissues (18). Finally, studies in this laboratory, using ligand blotting, identified a distinct 94-to 97-kDa protein expressed in mouse peritoneal macrophages (MPMs) that had a higher affinity for OxLDL than for acetyl-LDL (19) and DeRijke and van Berkel (20) identified a similar protein in rat Kupffer cells. We have recently purified this 94- to 97-kDa protein from MPMs and identified it as macrosialin (MS) (21). MS was first identified as a macrophage glycoprotein antigen by Smith and Koch (22) and later characterized and cloned in the laboratories of Gordon and Simmons (23). Holness and Simmons (24) also cloned the human homolog CD68. MS and CD68 are closely related to the family of lysosomal-associated membrane proteins (lamps) (25). They are predominantly intracellular proteins, found mainly in the late endosomal compartment (22, 26, 27). Electron microscopic studies failed to show more than a trace of MS on the plasma membrane of activated MPMs (28). The function of MS/CD68 has yet to be established but its predominantly intracellular localization and its extensive glycosylation has suggested a role in relationship to endosomal function such as antigen processing or presentation and protection of the lysosomal membrane from hydrolytic enzymes (23, 24). The present studies were undertaken to determine whether there is surface expression of MS/CD68 on macrophages and to determine whether or not these molecules play a receptor role in the binding and uptake of OxLDL.
MATERIALS AND METHODS
Materials.
Phorbol 12-myristate 13-acetate (PMA), saponin, ethidium bromide, and propidium iodide were purchased from Sigma. Triton X-114, octyl glucoside, and streptavidin conjugated to alkaline phosphatase were from Boehringer Mannheim. Sulfo-N-hydroxysuccinimide-biotin was from Pierce.
Antibodies.
DS4 guinea pig antiserum against the C-terminal cytoplasmic tail of MS was obtained as described (21). mAbs used were FA/11, a rat anti-mouse IgG2a directed against MS (a gift from R. P. daSilva and S. Gordon, Oxford University); KP-1 and EBM-11, both mouse anti-human CD68 IgG1 mAb hybridoma supernatants (Dako); and MB47, a mouse anti-human apolipoprotein B-100 mAb (a kind gift from L. Curtiss, Scripps Research Institute, La Jolla, CA). Control antibodies used were a mouse anti-human pancytokeratin–fluorescein isothiocyanate-conjugated polyclonal IgG (Sigma); a mouse IgG1 (PharMingen and Sigma); a mouse monoclonal anti-human CD4 and a mouse anti-human CD45 (both IgG1 hybridoma supernatants from Dako); and a rat IgG2a (BioSource International, Camarillo, CA). Secondary antibodies used were goat anti-rat IgG F(ab)2 and goat anti-mouse F(ab)2 (Accurate Chemicals); and goat anti-mouse immunoglobin (Fab specific) and sheep anti-mouse IgG (Sigma). They were purchased as conjugates of fluorescein isothiocyanate, phycoerythrin, or alkaline phosphatase.
Cells and Cell Culture.
Resident and thioglycollate-elicited MPMs were prepared as described (19). The human monocytic cell line THP-1 (American Type Culture Collection) was grown in suspension in RPMI 1640 medium with 10% fetal calf serum, 2 mM glutamine, 100 mg of streptomycin and 100 units of penicillin per ml, and 5 × 10−5 M 2-mercaptoethanol. THP-1 monocytes were plated and differentiated to adherent macrophages with 10−7 M PMA and grown in the same complete medium for 3 to 4 days.
Flow Cytometry.
For flow cytometry, 106 cells in suspension (or PMA-differentiated THP-1 cells harvested by gentle scraping from 100-mm plates) were washed with ice-cold buffer A (PBS/0.1% BSA/0.01% NaN3) and incubated for 20 min at 4°C with 1–2 μg of primary antibody in 100 μl of buffer A, washed twice, and incubated for another 20 min with 50 μl of a 1:100 diluted fluorescent-labeled secondary antibody. Cells were washed twice and immediately analyzed on a flow cytometer (Becton Dickinson FACS). All incubations and washes were performed at 4°C with ice-cold buffer A. Nonspecific binding to Fc receptors on THP-1 cells was blocked by using an Fc receptor blocking solution (Accurate Chemicals) per the instructions of the manufacturer. To distinguish between surface and intracellular expression of MS/CD68, cell membranes were permeabilized for 5 min with 0.5% saponin in PBS and washed with buffer A prior to incubation with antibodies at 4°C. The integrated mean fluorescence per cell before and after permeabilization, corrected for values with control IgG, were taken as proportional to surface and total receptors, respectively. To assess cell viability and control for cell death, cells were briefly incubated with ethidium bromide, propidium iodide, or anti-pancytokeratin-fluorescein isothiocyanate. Data were analyzed with cellquest (Becton Dickinson).
Ligand and Western Blot Analysis.
OxLDL was prepared as described (7). Detergent extracts of THP-1 monocytes and fractions from different stages of purification were electrophoresed on SDS/8% polyacrylamide gels, the proteins were electrotransferred to nitrocellulose membranes and probed with unlabeled OxLDL as described (19, 21). Bound ligand was detected by incubating with anti-human apolipoprotein B (MB47) followed by sheep anti-mouse IgG conjugated to alkaline phosphatase and the phosphatase substrates, 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium (Bio-Rad). Western blot analysis was carried out by first blocking the nitrocellulose strips with Super Block (Pierce) for 1 hr. The blots were rinsed with PBS/0.1% Tween 20, incubated with a 1:50 dilution of KP-1 in PBS/Tween 20 for 3 to 4 hr, and then washed three times. The bound antibodies were detected using alkaline phosphatase-conjugated sheep anti-mouse IgG. Biotinylated proteins were detected using a 1:300 dilution of streptavidin conjugated to alkaline phosphatase.
Purification of CD68.
CD68 was purified from THP-1 monocytes starting with 56 mg of membrane protein from Triton X-114 detergent extracts. The 100,000 × g membranes were prepared from suspension cultures essentially as described (29) and extracted with 1% Triton X-114. After removal of the insoluble material, 16 ml of the detergent extract was diluted to 50 ml with buffer B [50 mM Tris·HCl/0.15 M NaCl/0.1 mM EDTA, pH 8, containing the protease inhibitor mixture (50 units of aprotinin per ml/5 mM benzamidine/0.1 mM phenylmethlysulfonyl fluoride/14.5 μM pepstatin A/0.1 mM leupeptin)] and applied without any phase separation to a 10-ml wheat germ agglutinin-Sepharose column preequilibrated with buffer B containing 0.3% Triton X-114 (buffer C). The column was washed with 50 ml of buffer C and then detergent exchange was performed using 50 ml of buffer B containing 40 mM octyl glucoside (buffer D). Bound proteins were eluted with 0.5 M N-acetylglucosamine in buffer D and subjected to further purification on a delipidated OxLDL column as described for MS (21).
Cell Surface Biotinylation and Immunoprecipitation.
Freshly isolated MPMs (30 × 106 cells) in suspension were cooled on ice and washed three times with 10 ml of ice-cold PBS. The cells were then reacted with 10 ml of water soluble sulfo-N-hydroxysuccinimide-biotin (100 μg/ml) in ice-cold PBS at 4°C for 1 hr on a rotary shaker. The cells were washed three times with ice-cold PBS and pelleted at 300 × g. Whole-cell octyl glucoside extracts were prepared by treatment with 0.1–0.5 ml of buffer D for 1–1.5 hr at 4°C followed by sedimentation of the insoluble material at 100,000 × g. MS was immunoprecipitated with antiserum DS4 as described (21). To assess the total cellular content of MS, whole-cell detergent extracts were first prepared and then proteins were biotinylated at 4°C using sulfo-N-hydroxysuccinimide-biotin at 4 mg/ml followed by immunoprecipitation. Immunoprecipitates were solubilized by boiling with 40 μl of 5× SDS sample buffer for 5 min, subjected to SDS/PAGE, and electroblotting on to nitrocellulose, and the biotinylated proteins were analyzed by streptavidin blotting as described above.
OxLDL Cellular Binding and Uptake.
Differentiated THP-1 cells were cultured for 4 days with PMA in 12-well tissue culture dishes. Cell surface binding and uptake were measured as described (12). Cells were washed three times with DMEM complete medium and incubated with 125I-labeled OxLDL (5 μg/ml; specific activity, ≈600 cpm/ng) in the presence or absence of an excess of unlabeled OxLDL or with indicated concentrations of the CD68 mabs EBM-11 and KP-1 or an isotype-matched IgG1 control. Binding at 4°C was analyzed after 5–6 hr. Uptake at 37°C was analyzed after 4–5 hr.
RESULTS
Surface Expression of MS.
As shown in Fig. 1A, FACS analysis of resident MPMs using mAb FA11 showed minimal surface expression of MS, in agreement with the finding of Rabinowitz et al. (28). On the other hand, thioglycollate-elicited MPMs, which show more than a 10-fold increase in total cellular MS expression (21, 30), clearly expressed MS on the plasma membrane (Fig. 1B). Surface expression was confirmed and quantified by biotinylation of thioglycollate-elicited intact MPMs followed by immunoprecipitation of MS from a detergent extract of the membranes by using a polyvalent antiserum against the C-terminal tridecapeptide of MS as described (21). As shown in Fig. 2, lane 1, biotinylated MS was readily detected on intact cells both in the monomeric form (94–97 kDa) and in the dimeric form (185–200 kDa) that is generated during purification (21). On the other hand, α-actinin, an intracellular cytoskeletal marker, was not biotinylated under these conditions (data not shown). Another aliquot of cells was biotinylated after detergent disruption so that the total cellular content of MS could be assessed. As shown in Fig. 2, lane 2, the amount of intracellular MS exceeded considerably the amount expressed on the cell surface. Densitometric scanning, summing the monomer and dimer bands, indicated that at steady state, 10–15% of the total MS cellular pool is expressed on the plasma membrane. We have applied the same methods to the murine macrophage cell line RAW 264.7 and observed that it also expresses 5–10% of its total MS on the surface (data not shown). However, biotinylation was not sufficiently sensitive to detect MS on the surface of resident MPMs.
Figure 1.
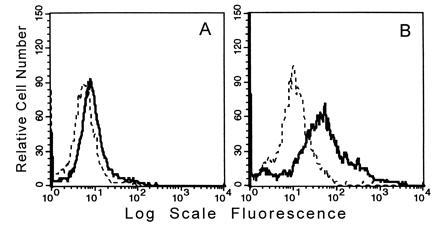
FACS analysis of MS surface expression on MPMs. Binding of FA/11 (bold line) compared with that of a rat isotype control IgG (dashed line) to intact resident MPMs (A) or thioglycollate-elicited MPMs (B) is shown.
Figure 2.
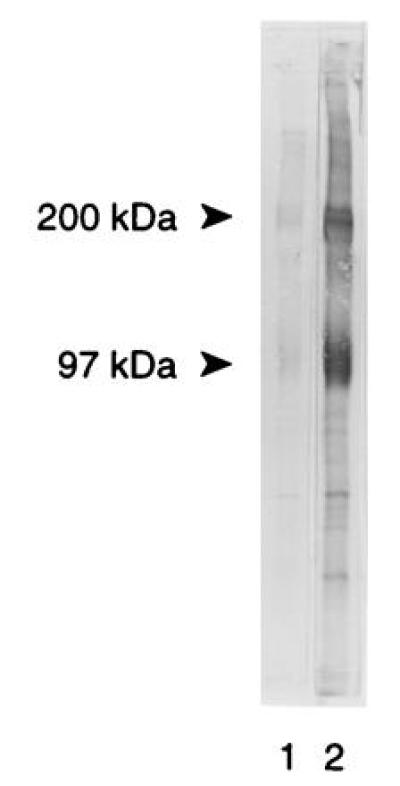
Cell surface expression of MS on elicited MPMs. Thioglycollate-elicited macrophages were harvested and subjected to cell-surface-specific biotinylation at 4°C followed by detergent extraction of cells and immunoadsorption with DS4 antiserum (lane 1). Alternatively, cell extracts were biotinylated and then processed for immunoadsorption (lane 2) to quantify total cellular expression of MS. The immunoprecipitates were subjected to SDS/PAGE and electroblot transfer, and the nitrocellulose strips were probed with streptavidin-alkaline phosphatase.
Purification of CD68.
Earlier we described (19) a 120-kDa protein present in THP-1 monocyte/macrophages and human peritoneal macrophages that binds OxLDL and is induced by PMA. We have now further characterized this protein and demonstrated that it is CD68, the human homolog of MS, which was reported by Micklem et al. (31) to have a molecular mass of 110 kDa. Western blot analysis of membrane preparation from THP-1 monocytes and differentiated (PMA treated) THP-1 macrophages with three different mAbs against CD68 (KP-1, EBM-11, and PGM-1) revealed diffuse bands that comigrated with 130- to 150-kDa and 110- to 125-kDa OxLDL-ligand-binding bands, respectively (data not shown). We subjected membrane detergent extracts of THP-1 monocytes to a purification scheme similar to that reported for MS (21), including affinity chromatography first on wheat germ agglutinin and then on delipidated OxLDL columns. Fig. 3 shows that the CD68 protein detected by Western blot analysis with KP-1 (Upper) and the OxLDL ligand binding activity (Lower) of the 130- to 150-kDa band coisolated in the same fractions. The elution profile of CD68 on the OxLDL column was identical to that of MS. A prominent 80-kDa band in the crude membrane extracts that binds KP-1 (Upper, lanes 1, 3, and 4) was retained on the lectin column but did not bind OxLDL on ligand blots and was not retained on the OxLDL column. This could represent a degradation product of CD68 that lacks the OxLDL-binding domain or a protein of related structure. The sharp 97-kDa OxLDL-binding band in the crude extracts (Lower, lane 1) is absent in the purified preparation.
Figure 3.
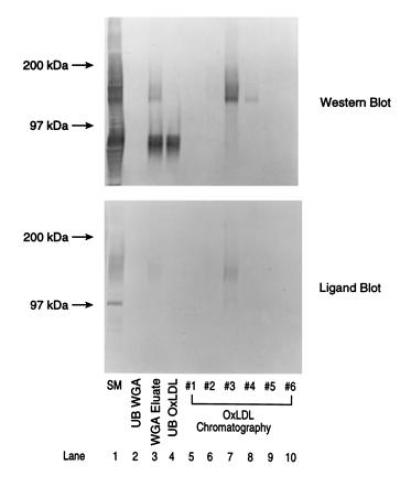
Copurification of CD68 and the 130- to 150-kDa OxLDL binding activity from THP-1 monocytes. Membrane detergent extracts of THP-1 monocytes were subjected to purification on wheat germ agglutinin lectin and OxLDL columns, and 20-μl aliquots of fractions (20–80 μg of protein) were examined by Western blot analysis with KP-1 monoclonal CD68 antibody and for OxLDL-ligand blotting activities.
Surface Expression of CD68 on THP-1 Cells.
FACS analysis using mAbs EBM-11 and KP-1 showed very low levels of surface expression of CD68 on unstimulated THP-1 human monocytic cells (Fig. 4A). Comparison of mean fluorescence before and after permeabilization of THP-1 monocytes showed that 4.1% of the total cellular CD68 was on the plasma membrane at steady state (data not shown). Other monocytic cell lines (U937 and Monomac 6) and peripheral blood human monocytes also showed only limited expression on the cell surface (data not shown), consistent with earlier findings (32). Treatment with PMA, shown to increase the mRNA levels for CD68 in U937 cells (24), greatly increased both total expression of CD68 (15-fold) and relative (22.4% instead of 4.1%) surface expression (Fig. 4B). In all of the FACS studies reported herein, cells at all permeable to ethidium bromide or propidium iodide were omitted from analysis. As another negative control, we used an antibody against cytoskeletal proteins (anti-pancytokeratin). Fig. 5A shows surface expression of CD68 detected with KP-1; none of the cells were positive for the cytoskeletal antibody. After permeabilization with saponin, the signal intensity for CD68 increased markedly (Fig. 5B), as expected, and all the cells were now positive with anti-pancytokeratin. Comparison of the mean fluorescence of nonpermeabilized and permeabilized cells before and after PMA stimulation showed that both total cellular CD68 and relative surface expression increased considerably. The binding of the IgG isotype control also increased after permeabilization, although to a lesser degree than KP-1, perhaps reflecting nonspecific binding to intracellular proteins.
Figure 4.
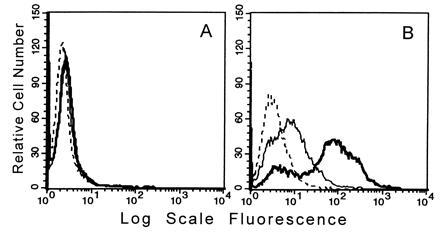
FACS analysis of CD68 surface expression on intact THP-1 cells. Binding of KP-1 (bold line) and EBM-11 (normal line) compared with that of a mouse isotype control IgG (dashed line) to intact unstimulated THP-1 monocytes (A) or to THP-1 macrophages after 3 days of stimulation with 100 nM PMA (B). In A the curves for KP-1 and EBM-11 are superimposed on one another. Only cells negative for binding of pancytokeratin were included in the analysis.
Figure 5.
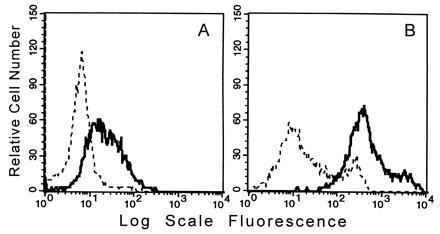
FACS analysis of CD68 cell surface and total cellular expression in PMA-stimulated THP-1 macrophages. Binding of KP-1 (bold line) and mouse isotype control IgG (dashed line) to THP-1 cells treated 3 days with PMA before permeabilization (A) and after permeabilization (B). In A, as in Fig. 4, the cells selected for analysis were those totally negative for pancytokeratin but after permeabilization (B), the entire cell population was positive for pancytokeratin.
Functional Role of CD68 in OxLDL Binding and Uptake by Macrophages.
To directly test the contribution of CD68 to OxLDL binding and uptake by THP-1 macrophages, we carried out competition studies with the mAbs EBM-11 and KP-1 (Fig. 6). Nonspecific binding of labeled ligand was determined in the presence of a 40- to 50-fold excess of unlabeled OxLDL and this was subtracted from all total binding values to yield specific binding values. Since isotype-matched control antibodies sometimes showed some nonspecific inhibition of total 125I-labeled OxLDL binding (5–30%), the data are expressed as percentage of binding or uptake remaining relative to that of control IgG (100%). The apparent Kd value for binding of OxLDL to THP-1 cells treated for 4 days with PMA was ≈5 μg/ml (data not shown) and the competition with the antibodies was performed at this concentration of ligand. Fig. 6 Left shows the effects of the antibodies on specific 125I-labeled OxLDL binding at 4°C. EBM-11 at 20 μg/ml inhibited binding of ligand by ≈50% and KP-1 had an even more dramatic effect, inhibiting OxLDL binding by ≈75%. EBM-11 and KP-1 at 30 μg/ml also inhibited the cell-associated radioactivity at 37°C (Fig. 6 Right) by ≈50%. The siginificant inhibition of OxLDL binding by several IgG isotype controls or irrelevant monoclonal IgG1 hybridoma supernatants was unexpected. The reason for this is not known but might reflect the fact that OxLDL can bind to Fc receptors (33). In some studies, instead of using a nonspecific IgG-matched isotype as control, we used a mAb against CD45, a surface leukocyte antigen expressed on THP-1 cells (34) as the control. The CD45 mab inhibited OxLDL uptake by close to 30%. Using this value in the presence of anti-CD45 as control, EBM-11 inhibited by an additional 19% and KP-1 inhibited by an additional 36%. Thus, CD68 contributes significantly to OxLDL binding and uptake by PMA-stimulated THP-1 macrophages.
Figure 6.
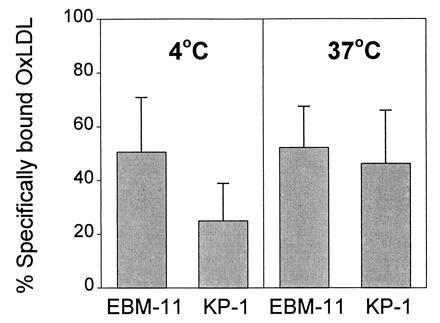
Inhibition by CD68 mAbs of specific binding and uptake of 125I-labeled OxLDL (5 μg/ml) by THP-cells. THP-1 cells were treated with PMA for 4 days after which binding at 4°C (Left) and uptake at 37°C (Right) were measured in the absence or presence of a 40- to 50-fold excess unlabeled OxLDL, EBM-11 (n = 5), KP-1 (n = 4), or an isotype-matched control IgG. Cell-bound and cell-associated radioactivity was assessed after 4–5 hr and were ≈260 and ≈800 ng of OxLDL per mg of cell protein, respectively. Nonspecific OxLDL binding and uptake (≈131 and 360 ng/mg of cell protein, respectively) were subtracted from all values to yield specific binding values. Activities in the presence of control IgG are taken as 100% (see Results). Data represent mean ± SD.
DISCUSSION
Our findings that little or no MS/CD68 can be demonstrated on the plasma membrane of resident MPMs and only a little over 4% on nonstimulated human monocytic THP-1 cells are consonant with the conclusions of others (26–28). However, we now provide clear evidence for significant increases in surface expression of MS in thioglycollate-elicited MPMs and of CD68 in PMA-stimulated THP-1 cells. The biotinylation studies, controlled by using α-actinin as a negative control, consistently demonstrated 10–15% of the total MS on the surface of the elicited mouse macrophages and FACS analysis showed more than 20% on the surface of PMA-treated THP-1 cells. Finally, the inhibition of OxLDL binding and uptake into THP-1 cells by two mAbs directed against CD68 establishes a functional role for the receptor expressed on the surface. Binding and uptake of 125I-labeled OxLDL were inhibited by 30–50%. Recent studies in this laboratory show that cells stably expressing CD68 cDNA show an increased binding and uptake of labeled OxLDL with a Kd value for the ligand binding of 5–6 μg/ml (T. Albert, D. Lamb, M.P.R., D.S., and O.Q., unpublished results), which is similar to that in THP-1 macrophages. This supports the present data demonstrating the role of CD68 as a macrophage receptor for OxLDL.
The limited surface expression of MS/CD68 (relative to the total cellular content) in the face of its appreciable contribution to uptake of OxLDL requires comment. MS and CD68 are considered to be members of the lamp family based on significant homologies of sequence and domain organization (23). All members of the family are highly glycosylated and all are predominantly found intracellularly. MS, however, differs from other members of the lamp family in that it is found predominantly in the late endosomal fraction, whereas the other members of the family are found predominantly in the lysosomal fraction (28, 25). MS/CD68 appears to have a rather limited tissue distribution, being found in macrophages, Langerhans cells, and dendritic cells almost exclusively (22–23, 32, 35), whereas other lamp family members are more widely distributed (25). Lippincott-Schwartz and Fambrough (36) have carefully studied the subcellular distribution of LEP-100, the chicken homolog of lamp-1. At steady state, they found that no more than 2–3% of the total cell content of LEP-100 was on the plasma membrane; most of it was found in lysosomes and endosomes. However, kinetic studies showed that the rate of movement of LEP-100 from plasma membrane to endosomes and on into lysosomes was extremely rapid. The rate constant for movement from endosomes to the plasma membrane was approximately 0.138 min−1 and the rate constant for the reverse process was about 0.34 min−1 (36). When transfer between membrane compartments was inhibited by chloroquine, the steady-state distribution shifted dramatically: now fully 20–25% of the total cell content of the receptor was at the plasma membrane.
Targeting of lamp proteins to the lysosome depends on a highly conserved sequence in their short cytoplasmic tail, especially on an AY or GY, six residues from the transmembrane region (37). Mutations deleting or shifting the tyrosine cause the lamps to accumulate on the plasma membrane. The cytoplasmic tail of MS/CD68 is highly homologous to that of the lamp family (23, 24).
The key point is that the lamp proteins may have functions relating to cell–cell interaction or cell–ligand interaction even though only a small fraction of the total protein is at any one time on the cell surface. One reason this is possible is that these proteins are abundantly expressed so that even as little as 5 or 10% on the surface corresponds to a large absolute number of copies on the surface (25, 34). For example, there can be as many as 50,000 lamp molecules on the surface at steady state (25). Furthermore, as long as the population at the cell surface can be renewed rapidly, the cell surface compartment can play a role in binding and uptake of ligands, adherence to matrix, etc. Indeed, Fukuda and coworkers (38) have shown that lamp-1 is involved in cell–cell adhesion and may be critically important with respect to metastasis of malignant cells. From our data on the inhibition of OxLDL binding by mAbs, at half-saturating concentrations of ligand, we calculate that there are at least 4000–8000 CD68 molecules on the surface of each PMA-stimulated THP-1 macrophage cell. Interestingly, Bottalico et al. (39) reported that THP-1 macrophages differentiated for 2 days with PMA expressed only about 3700 acetyl-LDL binding sites per cell and interpretation of the Scatchard analysis reported for 7-day differentiated THP-1 cells shows 6000 acetyl-LDL receptors per cell (40). We suggest that like the lamps, MS/CD68 can shuttle rapidly between the endosomes and the plasma membrane and participate in the rapid internalization of OxLDL. If the ligand is an aggregrate of OxLDL or an oxidatively damaged cell, the macrophage receptors involved in binding to the target cell would be “trapped” and the continuous movement of receptors between endosome and plasma membrane would lead to an increase in the total number of receptors on the plasma membrane. This would be true even if there was no increase in the rate of that movement since it is already fast. Additionally, there might be, as a result of a signaling mechanism, an actual increase in the rate of transport of receptor molecules to the surface. The recent demonstration that lamp-1 and lamp-2 are localized in mobilizable secretory granules in human neutrophils and appear on the cell surface when the secretory vesicles are mobilized (41) lends support to this hypothesis.
Sambrano et al. (42) showed that OxLDL could strongly inhibit the binding of oxidatively damaged red blood cells to MPMs. Later studies in the same laboratory showed that the same was true for the binding of apoptotic thymocytes (43). Sambrano and Steinberg (43) proposed that the biologically important function of the so-called scavenger receptors might lie in their ability to recognize damaged cells; the ability to recognize OxLDL is presumably a reflection of some similarities between an oxidized lipoprotein and an oxidized (or otherwise damaged) plasma membrane. This is most certainly true for CD36, which is known to be involved in recognition of apoptotic thymocytes and neutrophils, acting in concert with αvβ3 integrin and thrombospondin (44). The studies of Endemann et al. (15) and the studies of Nozaki et al. (17) on monocyte-derived macrophages from patients deficient in CD36 established that this receptor is also potentially involved in the recognition and uptake of OxLDL. Scavenger receptor A, which is responsible for a significant fraction of the uptake of OxLDL by macrophages (12), has recently been implicated by Platt et al. (45) in the macrophage recognition and uptake of apoptotic thymocytes. Whether or not MS/CD68 is similarly playing a dual role remains to be determined. The restricted distribution of MS/CD68 and their predominance in cell types specialized for phagocytosis coupled with their increased expression in the presence of phagocytic stimuli is suggestive, but further studies are needed. Furthermore, since nonactivated monocytes do express a small amount of CD68 on the cell surface, one can envisage a role for its carbohydrate moiety in cell adhesion and attachment to the endothelium mediated by selectins. The relative activities and contributions of various scavenger receptors at different stages of atherosclerotic lesion development and progression remains to be elucidated.
Acknowledgments
We are indebted to Dr. Monty Krieger, Massachusetts Institute of Techology, for his careful reading of the manuscript and for his helpful critique. We thank Richard Elam for his help with the graphics and Audrey Threlkeld for her skillful preparation of the manuscript. This research was supported in part by the National Heart, Lung, and Blood Institute (NIH 2P50 HL14197-23 SCOR on Arteriosclerosis) and by the Stein Institute for Research on Aging. M.P.R. is a recipient of an American Heart Association Grant-in-Aid, California affiliate.
Footnotes
Abbreviations: LDL, low density lipoprotein; MS, macrosialin; FACS, flow cytometry; OxLDL, oxidized LDL; MPM, mouse peritoneal macrophage; lamp, lysosomal-associated membrane protein; PMA, phorbol 12-myristate 13-acetate.
References
- 1.Brown M S, Goldstein J L. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 2.Goldstein J L, Ho Y K, Basu S K, Brown M S. Proc Natl Acad Sci USA. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kodama T, Freeman M, Rohrer L, Zabrecky J, Matsudaira P, Krieger M. Nature (London) 1990;343:531–535. doi: 10.1038/343531a0. [DOI] [PubMed] [Google Scholar]
- 4.Henriksen T, Mahoney E M, Steinberg D. Proc Natl Acad Sci USA. 1981;78:6499–6503. doi: 10.1073/pnas.78.10.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henriksen T, Mahoney E M, Steinberg D. Ann NY Acad Sci. 1982;401:102–116. doi: 10.1111/j.1749-6632.1982.tb25711.x. [DOI] [PubMed] [Google Scholar]
- 6.Henriksen T, Mahoney E, Steinberg D. Arteriosclerosis (Dallas) 1983;3:149–159. doi: 10.1161/01.atv.3.2.149. [DOI] [PubMed] [Google Scholar]
- 7.Steinbrecher U P, Parthasarathy S, Leake D S, Witztum J L, Steinberg D. Proc Natl Acad Sci USA. 1984;81:3883–3887. doi: 10.1073/pnas.81.12.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palinski W, Rosenfeld M E, Ylä-Herttuala S, Gurtner G C, Socher S S, Butler S W, Parthasarathy S, Carew T E, Steinberg D, Witztum J L. Proc Natl Acad Sci USA. 1989;86:1372–1376. doi: 10.1073/pnas.86.4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinberg D, Parthasarathy S, Carew T E, Khoo J C, Witztum J L. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg D. Adv Exp Med Biol. 1995;369:39–48. doi: 10.1007/978-1-4615-1957-7_5. [DOI] [PubMed] [Google Scholar]
- 11.Freeman M, Ekkel Y, Rohrer L, Penman M, Freedman N J, Chisolm G M, Krieger M. Proc Natl Acad Sci USA. 1991;88:4931–4935. doi: 10.1073/pnas.88.11.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sparrow C P, Parthasarathy S, Steinberg D. J Biol Chem. 1989;264:2599–2604. [PubMed] [Google Scholar]
- 13.Arai H, Kita T, Yokode M, Narumiya S, Kawai C. Biochem Biophys Res Commun. 1989;159:1375–1382. doi: 10.1016/0006-291x(89)92262-6. [DOI] [PubMed] [Google Scholar]
- 14.Acton S L, Schere P E, Lodish H F, Krieger M. J Biol Chem. 1994;269:21003–21009. [PubMed] [Google Scholar]
- 15.Endemann G, Stanton L W, Madden K S, Bryant C M, White R T, Protter A A. J Biol Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- 16.Nicholson A C, Frieda S, Pearce A, Silverstein R L. Arterioscler Thromb Vasc Biol. 1995;15:269–275. doi: 10.1161/01.atv.15.2.269. [DOI] [PubMed] [Google Scholar]
- 17.Nozaki S, Kashiwagi H, Yamashita S, Nakagawa T, Kostner B, Tomiyama Y, Nakata A, Ishigami M, Miyagawa J-I, Kameda-Takemura K, Kurata Y, Matsuzawa Y. J Clin Invest. 1995;96:1859–1865. doi: 10.1172/JCI118231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acton S, Rigotti A, Landschulz K T, Xu S, Hobbs H H, Krieger M. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 19.Ottnad E, Parthasarathy S, Sambrano G, Ramprasad M P, Quehenberger O, Kondratenko N, Green S, Steinberg D. Proc Natl Acad Sci USA. 1995;92:1391–1395. doi: 10.1073/pnas.92.5.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Rijke Y B, van Berkel T J C. J Biol Chem. 1994;269:824–827. [PubMed] [Google Scholar]
- 21.Ramprasad M P, Fischer W, Witztum J L, Sambrano G, Quehenberger O, Steinberg D. Proc Natl Acad Sci USA. 1995;92:9580–9584. doi: 10.1073/pnas.92.21.9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith M J, Koch G L E. J Cell Sci. 1987;87:113–119. doi: 10.1242/jcs.87.1.113. [DOI] [PubMed] [Google Scholar]
- 23.Holness C L, da Silva R P, Fawcett J, Gordon S, Simmons D L. J Biol Chem. 1993;268:9661–9666. [PubMed] [Google Scholar]
- 24.Holness C L, Simmons D L. Blood. 1993;81:1607–1613. [PubMed] [Google Scholar]
- 25.Fukuda M. J Biol Chem. 1991;266:21327–21330. [PubMed] [Google Scholar]
- 26.Saito N, Pulford K A F, Breton-Gorius J, Mason D Y, Cramer E M. Am J Pathol. 1991;139:1053–1059. [PMC free article] [PubMed] [Google Scholar]
- 27.Parwaresch M R, Radzun J, Kriepe H, Hansmann M L, Barth J. Am J Pathol. 1986;124:141–151. [PMC free article] [PubMed] [Google Scholar]
- 28.Rabinowitz S, Horstmann H, Gordon S, Griffiths G. J Cell Biol. 1992;116:95–112. doi: 10.1083/jcb.116.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramprasad M P, Li R, Bradley W A, Gianturco S H. Biochemistry. 1995;34:9126–9135. doi: 10.1021/bi00028a023. [DOI] [PubMed] [Google Scholar]
- 30.Rabinowitz S, Gordon S. J Exp Med. 1991;174:827–836. doi: 10.1084/jem.174.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Micklem K, Rigney E, Cordell J, Simmons D, Stross P, Turley H, Seed B, Mason D. Br J Haematol. 1989;73:6–11. doi: 10.1111/j.1365-2141.1989.tb00210.x. [DOI] [PubMed] [Google Scholar]
- 32.Pulford K A F, Sipos A, Cordell J L, Stross W P, Mason D Y. Int Immunol. 1990;2:973–980. doi: 10.1093/intimm/2.10.973. [DOI] [PubMed] [Google Scholar]
- 33.Stanton L W, White T R, Bryant C M, Protter A A, Endemann G. J Biol Chem. 1992;267:22446–22451. [PubMed] [Google Scholar]
- 34.Buzzi M, Lu L, Lombardi A J, Posner M R, Brautigan D L, Fast L D, Frackelton A R., Jr Cancer Res. 1992;52:4027–4035. [PubMed] [Google Scholar]
- 35.Betjes M G, Haks M C,, Tuk C W, Beelen R H. Immunobiology. 1991;183:79–87. doi: 10.1016/S0171-2985(11)80187-7. [DOI] [PubMed] [Google Scholar]
- 36.Lippincott-Schwartz J, Fambrough D M. Cell. 1987;49:669–677. doi: 10.1016/0092-8674(87)90543-5. [DOI] [PubMed] [Google Scholar]
- 37.Williams M A, Fukuda N. J Cell Biol. 1990;111:955–966. doi: 10.1083/jcb.111.3.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saitoh O, Wang W C, Lotan R, Fukuda M. J Biol Chem. 1992;267:5700–5711. [PubMed] [Google Scholar]
- 39.Bottalico L A, Wager R E, Agellon L B, Assoian R K, Tabas I. J Biol Chem. 1991;266:22866–22871. [PubMed] [Google Scholar]
- 40.Suematsu Y, Nishizawa Y, Shioi A, Hino M, Tahara H, Inaba M, Morli H, Otani S. J Cell Physiol. 1995;165:547–555. doi: 10.1002/jcp.1041650313. [DOI] [PubMed] [Google Scholar]
- 41.Dahlgren C, Carlsson S R, Karlsson A, Lundqvist H, Sjolin C. Biochem J. 1995;311:667–674. doi: 10.1042/bj3110667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrano G R, Parthasarathy S, Steinberg D. Proc Natl Acad Sci USA. 1994;91:3265–3269. doi: 10.1073/pnas.91.8.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sambrano G, Steinberg D. Proc Natl Acad Sci USA. 1995;92:1396–1400. doi: 10.1073/pnas.92.5.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savill J, Fadok V, Henson P, Haslett C. Immunol Today. 1993;14:131–136. doi: 10.1016/0167-5699(93)90215-7. [DOI] [PubMed] [Google Scholar]
- 45.Platt N, Suzuki H, Kurihara Y, Kodama T, Gordon S. Proc Natl Acad Sci USA. 1996;93:12456–12460. doi: 10.1073/pnas.93.22.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]