

Abstract
Moderate somatic stress inhibits gastric acid secretion. We have investigated the role of endogenously released NO in this phenomenon. Elevation of body temperature by 3°C or a reduction of 35 mmHg (1 mmHg = 133 Pa) in blood pressure for 10 min produced a rapid and long-lasting reduction of distension-stimulated acid secretion in the rat perfused stomach in vivo. A similar inhibitory effect on acid secretion was produced by the intracisternal (i.c.) administration of oxytocin, a peptide known to be released during stress. Intracisternal administration of the NO-synthase inhibitor, NG-nitro-l-arginine methyl ester (l-NAME) reversed the antisecretory effect induced by all these stimuli, an action prevented by intracisternal coadministration of the NO precursor, l-arginine. Furthermore, microinjection of l-NAME into the dorsal motor nucleus of the vagus nerve reversed the acid inhibitory effects of mild hyperthermia, i.v. endotoxin, or i.c. oxytocin, an action prevented by prior microinjection of l-arginine. By contrast, microinjection of l-NAME into the nucleus tractus solitarius failed to affect the inhibitory effects of hyperthermia, i.v. endotoxin, or i.c. oxytocin. Immunohistochemical techniques demonstrated that following hyperthermia there was a significant increase in immunoreactivity to neuronal NO synthase in different areas of the brain, including the dorsal motor nucleus of the vagus. Thus, our results suggest that the inhibition of gastric acid secretion, a defense mechanism during stress, is mediated by a nervous reflex involving a neuronal pathway that includes NO synthesis in the brain, specifically in the dorsal motor nucleus of the vagus.
Keywords: brain–gut axis, central nervous system, central neurotransmitters
The definition of the “stress syndrome” by Selye in 1936 (1) was based on his pioneer observations that exposure to various “noxious stimuli” elicited a body response characterized by the combination of gastrointestinal alterations along with adrenal hypertrophy and thymolymphatic involution. The acute inhibitory effects of stress on gastric acid secretion had been clinically observed in patients with a gastric fistula over 100 years before the characterization of this syndrome (2). However, the first experimental evidence of this was provided by Cannon in 1929 (3) when he described an emotionally related gastric hyposecretion in cats facing a barking dog.
More recent investigations have confirmed a reduction in gastric acid output following exposure to stress induced by a large variety of stimuli including restraint (4), rotation (5), high ambient temperature (6), unpredictable electrical shocks (7), endotoxin (8), or ionizing radiation (9). It has not, however, been apparent whether such an inhibitory response to stress is the consequence of a local mechanism or, as suggested more recently, has centrally mediated components involving the release of various neuropeptides (10–12). We have now established that the inhibitory effect on acid secretion in the stomach elicited by stress involves a nervous reflex in the central nervous system. In addition, we have found that NO released centrally is involved in this phenomenon. Furthermore, we have identified functionally in the brain an anatomical location of the nitrergic neurons involved in this process and confirmed by inmunohistochemical techniques their activation following the induction of stress.
MATERIALS AND METHODS
General Preparation.
Wistar rats of either sex (250–280 g) were fasted for 24 h before the experiments but were allowed drinking water. Under urethane anesthesia (1.5 g/kg, i.p.), the trachea was intubated, a jugular vein was cannulated, and systemic arterial blood pressure was measured from a cannula inserted into a carotid artery and attached to a pressure transducer. Two soft catheters were introduced in the stomach through incisions in the cervical esophagus and the duodenum, and the gastric lumen was flushed with 50–100 ml of saline to remove any solid content. Thereafter, the stomach lumen was perfused continuously (0.9 ml/min) via the esophageal catheter with saline at room temperature. After 1 h of stabilization, the gastric effluent was collected every 30 min, and the H+ output was determined by automatic titration of 8 ml aliquots of the perfusate to pH 7.0 with 0.01 M NaOH. A stable acid output for 60 min was considered as the basal rate of acid secretion.
To stimulate acid production, the stomach was distended by placing the tip of the antroduodenal cannula 20 cm above the perfused stomach, where it was maintained for the remaining 90 min of the experimental period. In all cases each preparation was used for a single response only, a procedure that gave a reproducible response, with treatment being allocated randomly between preparations. Values of acid production were expressed as μeq of H+ per 100 g body weight for 30 min (when referring to the acid output of a particular period of 30 min) or as the difference (Δ) between the total production of acid over the 90-min period of gastric distension and the basal acid output over a similar period, expressed as Δ μeq of H+ per 100 g of body weight for 90 min.
Experimental Procedure.
In the first series of studies, stress was induced by hyperthermia (+3°C), which was provoked by placing the animal under a heating lamp. Body temperature was constantly recorded by a rectal thermometer and maintained at 39°C for the 90-min experimental period. In a second group of experiments, stress was induced with mild hypotension by rapidly bleeding the animal from the arterial cannula into a reservoir, to a mean blood pressure of 70 mmHg (1 mmHg = 133 Pa). After maintaining this blood pressure for 10 min, the shed blood was slowly reinfused and acid secretion stimulated immediately afterwards. In a third series of studies, an acid inhibitory mediator released by stress, oxytocin (8 μg/kg), was injected i.v. or intracisternally (i.c.) 10 min before stimulation of acid production. Control animals received an i.c. administration of saline.
Further experiments were performed with the inhibitor of NO synthesis, NG-nitro-l-arginine methyl ester (l-NAME, 800 μg/kg, i.c.) to investigate the role of NO in the acid inhibitory effects of stress. To evaluate the specificity of the effects of l-NAME, l-arginine (12 mg/kg, i.c.) was coadministered with l-NAME, using doses of both compounds selected from preliminary dose-response studies.
Microinjections in the Dorsal Vagal Complex.
The dorsal vagal complex in the brainstem medulla was selected because it provides a major contribution to the nervous innervation of the stomach and plays a crucial role in the modulation of gastric secretory activity (13). Microinjections into the two nuclei comprising the dorsal vagal complex, the dorsal motor nucleus (DMN) and the nucleus tractus solitarius (NTS), were performed to identify the cerebral nucleus implicated in the acid-inhibitory responses to stress. Animals were placed into a stereotaxic instrument (Stoelting, Mod 51600), their heads were fixed in a nose-down position, and the skin and muscle overlying the occipital bone were retracted. Following the removal of a small segment of this bone a 1 μl Hamilton syringe, 26 gauge, was positioned accordingly to the atlas of Paxinos and Watson (14). The coordinates used for microinjection sites were: DMN (dorsoventral, 8.3 mm; anteroposterior from bregma, −13.8 mm; lateral from midline, 0.6 mm) and NTS (dorsoventral, 7.9 mm; anteroposterior from bregma, −13.8 mm; lateral from midline, 1 mm). l-NAME (80 μg/kg), l-arginine (800 μg/kg), or vehicle were microinjected in a volume of 0.1 μl delivered by pressure injection over 30 sec. The syringe was left in place for a further 3 min to allow diffusion of the injected solution. Ten minutes later, stress was induced by hyperthermia or animals received an i.v. administration of endotoxin (5 μg/kg) or an i.c. injection of oxytocin (8 μg/kg), and gastric acid secretion was stimulated by distension immediately afterwards.
At the end of the experimental period rats were killed and their brains were removed and fixed in phosphate buffer 0.1 M containing 4% paraformaldehyde at pH 7.4. Coronal sections (40 μm) were mounted, stained with Thionine Nissl, and examined microscopically. The location of microinjection sites was identified by the visualization of the tip of the needle track and marked on plates reproduced from the atlas of Paxinos and Watson (14).
Immunohistochemical Studies.
In separate experiments and after the standard 90-min period of hyperthermia (39°C), 50 ml of 0.9% saline were perfused through the left ventricle of the rat, followed by 500 ml of fixative solution containing 4% paraformaldehyde in 0.1 M phosphate buffer at pH 7.4. Nonstressed animals were used as controls. Brains were removed, cut into 4–5-mm coronal blocks, postfixed for 3 h in 4% buffered paraformaldehyde at room temperature, and rinsed by immersing them overnight at 4°C in 0.1 M phosphate buffer containing 30% saccharose. An antibody specific for the neuronal NO synthase (nNOS) was used (15, 16) for the identification of this enzyme in the areas of the brain studied.
Brain blocks were frozen and serial 40-μm frontal sections were obtained. Free-floating sections containing the whole forebrain were incubated with the nNOS antiserum and the avidin-biotin peroxidase complex procedure (17) was used to visualize immunoreactive sites. Briefly, all free-floating sections were incubated for 30 min in phosphate-buffered saline containing 3% normal goat serum and 0.2% Triton X-100. Subsequently, they were incubated overnight at 4°C with nNOS antiserum diluted 1:2500 in phosphate-buffered saline/Triton X-100. After several washes in phosphate-buffered saline, the sections were incubated for 1 h in biotinylated goat antirabbit immunoglobulin. After a further washing, the sections were incubated for a 90-min period in peroxidase-linked avidin-biotin complex. The peroxidase activity was demonstrated by the nickel-enhanced diaminobenzidine procedure (18).
Control procedures were performed on sham-operated animals. No immunolabeling was observed when the primary antibody was omitted or replaced with an equivalent concentration of preimmune normal rabbit serum. The specificity for nNOS was also shown after incubation of tissue sections with primary antiserum preabsorbed overnight at 4°C with 2 ng/ml of antigen extracted from rat brain (15).
Drugs.
Urethane, oxytocin, l-NAME, l-arginine, saccharose, and thionine were purchased from Sigma. Normal goat serum and avidin-biotin peroxidase complex were purchased from ICN and Vector Laboratories, respectively. Unless otherwise mentioned, all drugs were dissolved in saline immediately before use and administered in a volume of 1 ml/kg (i.v.), 10 μl per rat (i.c.), or 0.1 μl per rat (cerebral microinjection).
Statistical Analysis.
All data are expressed as mean ± SEM. Comparisons between groups were made by Student’s t test for unpaired data, except for the blood pressure studies in which Student’s t test for paired data was used. A probability of P < 0.05 or less was considered as statistically significant.
RESULTS
Effects of Hyperthermia, Hypotension, and Oxytocin on Gastric Acid Secretion.
In control rats, distension of the stomach with an intragastric pressure of 20 cm water produced a progressive increase in acid secretion that, after 90 min, reached a submaximal H+ production (Fig. 1). The total stimulated acid output in these animals was Δ 34 ± 8 μeq of H+ per 100 g for 90 min (n = 8).
Figure 1.
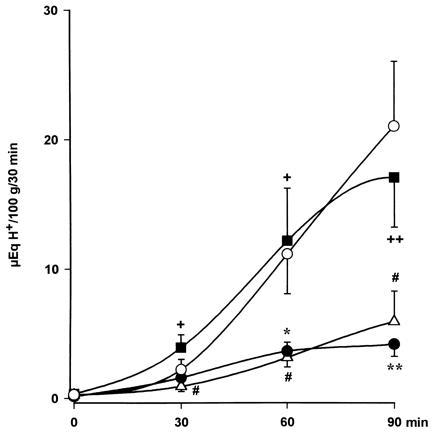
Acid secretion in the perfused stomach of the anesthetized rat. Distension (20 cm H2O, ○) resulted in increased output of H+ that was greatly reduced by hyperthermia (39°C, •). l-NAME (800 μg/kg i.c., ▪) prevented the acid-inhibitory effects of hyperthermia, an action that was abolished by administration of l-arginine (12 mg/kg, i.c., ▵). Each point represents mean ± SEM of at least six animals. Significance of difference from the control group is shown by ∗ (P < 0.05) and ∗∗ (P < 0.01), from the hyperthermia group by + (P < 0.05) and ++ (P < 0.01), and from the l-NAME-treated group by # (P < 0.05).
Increasing the body temperature from the control level of 36°C to 39°C induced a 74 ± 3% inhibition (P < 0.001) in the total production of acid to Δ 9 ± 1 μeq of H+ per 100 g for 90 min (n = 9). Similarly, a transient hypotension following reduction of blood pressure to 70 mmHg for a 10-min period inhibited (P < 0.01) acid production by 77 ± 5% (n = 13), as shown in Fig. 2.
Figure 2.
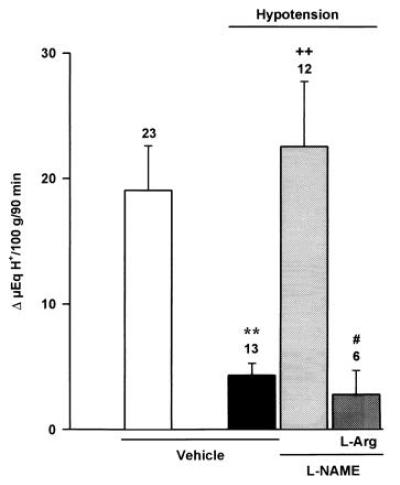
Effects of l-NAME (800 μg/kg, i.c.) or vehicle on the inhibitory actions of hypotension (70 mmHg, 10 min) on acid secretion induced by gastric distension (20 cm H2O) in the anesthetized rat. The administration of l-arginine (12 mg/kg, i.c.) restored the acid inhibitory effects of hypotension in animals treated with l-NAME. Results, expressed as the difference between basal and stimulated acid output, are mean ± SEM and the number above columns is the number of animals used. Significance from the control group is shown by ∗∗ (P < 0.01), from the hypotension only group by ++ (P < 0.01), and from the l-NAME-treated group by # (P < 0.05).
The i.c. injection of oxytocin (8 μg/kg, n = 14) significantly (P < 0.01) decreased stimulated acid secretion by 74 ± 8% (Fig. 3). However, when administered i.v. this dose of oxytocin did not modify acid output (Δ 45 ± 9 μeq of H+ per 100 g for 90 min, n = 6).
Figure 3.
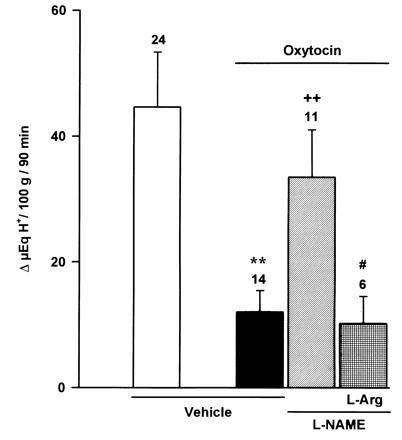
Effects of l-NAME (800 μg/kg, i.c.) or vehicle on the inhibitory actions of oxytocin (8 μg/kg, i.c.) on distension-stimulated acid secretion (20 cm H2O) in the anesthetized rat. The administration of l-arginine (12 mg/kg, i.c.) restored the acid inhibitory effects of oxytocin in animals treated with l-NAME. Results, expressed as the difference between basal and stimulated acid output, are mean ± SEM and the number above columns is the number of animals used. Significance from the control group is shown by ∗∗ (P < 0.01); from the oxytocin-treated group by ++ (P < 0.01); and from the l-NAME-treated group by # (P < 0.05).
Effects of Inhibition of NO Synthesis.
Pretreatment i.c. with l-NAME (800 μg/kg) prevented the acid-inhibitory effects induced by hyperthermia (Fig. 1). Prior i.c. administration of this dose (800 μg/kg) of l-NAME similarly reestablished acid production stimulated by distension in rats undergoing a 10-min period of hypotension (Fig. 2). Similarly, prior i.c. injection of l-NAME (800 μg/kg) restored acid secretory responses in rats treated i.c. with oxytocin (8 μg/kg) as shown in Fig. 3.
In control studies, the secretory response to distension (Δ 42 ± 8 μeq of H+ per 100 g for 90 min, n = 6) was not modified by i.c. administration of 800 μg/kg of l-NAME (Δ 50 ± 9 μeq of H+ per 100 g for 90 min, n = 10). This dose of l-NAME (800 μg/kg), if administered i.v., did not significantly influence the acid-inhibitory effects of hyperthermia (Δ 7 ± 4 μeq of H+ per 100 g for 90 min, n = 3), hypotension (Δ 3 ± 1 μeq of H+ per 100 g for 90 min, n = 4) or i.c. administration of oxytocin (Δ 8 ± 4 μeq of H+ per 100 g for 90 min, n = 5).
The reversal by i.c. l-NAME (800 μg/kg) of the inhibition of acid secretion by hyperthermia and hypotension was prevented by i.c. administration of l-arginine (12 mg/kg) as shown in Figs. 1 and 2. Furthermore, in animals pretreated with l-NAME (800 μg/kg, i.c.), the i.c. coadministration of l-arginine (12 mg/kg) reestablished the acid inhibitory effects of i.c. oxytocin (8 μg/kg) as shown in Fig. 3.
Location of Cerebral NO Synthesis.
Prior administration of l-NAME (80 μg/kg) into the DMN prevented the acid-inhibitory effects of hyperthermia (Fig. 4). Similarly, microinjection of l-NAME (80 μg/kg) in the same nucleus restored the production of acid in rats treated with i.v. endotoxin (5 μg/kg; Fig. 5), or i.c. oxytocin (8 μg/kg), an acid-inhibitory mediator released by stress (Fig. 6). In contrast, microinjection of l-NAME (80 μg/kg) into the NTS did not influence the acid-inhibitory effects of hyperthermia, i.v. endotoxin, or i.c. oxytocin, as shown, respectively, in Figs. 4, 5, 6. Furthermore, microinjections of vehicle into the DMN or the NTS did not significantly modify the gastric secretory response induced by distension.
Figure 4.
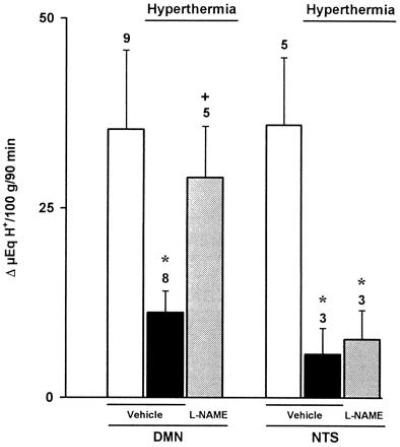
Effects of l-NAME (80 μg/kg) or vehicle microinjected into the DMN or the NTS on gastric acid secretion stimulated by distension (20 cm H2O) in rats undergoing hyperthermia (39°C). Results, expressed as the difference between basal and stimulated acid output, are mean ± SEM and the number above columns is the number of animals used. Significance from the respective control group is shown by ∗ (P < 0.05) and from the respective hyperthermic group by + (P < 0.05).
Figure 5.
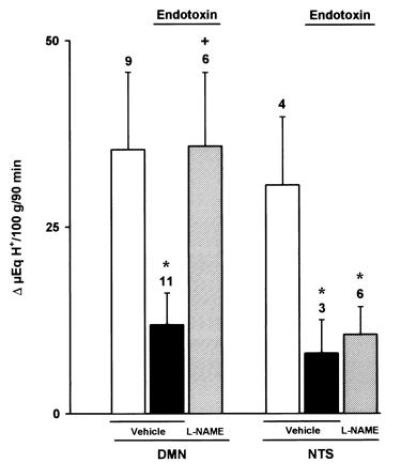
Effects of l-NAME (80 μg/kg) or vehicle microinjected into the DMN or the NTS on gastric acid secretion stimulated by distension (20 cm H2O) in endotoxin-treated rats (5 μg/kg, i.v.). Results, expressed as the difference between basal and stimulated acid output, are mean ± SEM and the number above columns is the number of animals used. Significance from the respective control group is shown by ∗ (P < 0.05) and from the respective endotoxin-treated group by + (P < 0.05).
Figure 6.
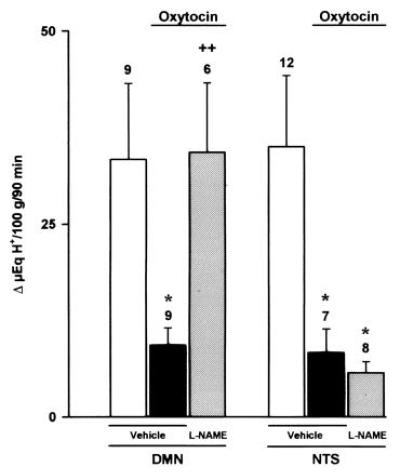
Effects of l-NAME (80 μg/kg) or vehicle microinjected into the DMN or the NTS on gastric acid secretion stimulated by distension (20 cm H2O) in oxytocin-treated rats (8 μg/kg, i.c.). Results, expressed as the difference between basal and stimulated acid output, are mean ± SEM and the number above columns is the number of animals used. Significance from the respective control group is shown by ∗ (P < 0.05) and from the respective oxytocin-treated group by ++ (P < 0.01).
This low dose of l-NAME (80 μg/kg), if administered i.c., did not modify the acid inhibitory effects of endotoxin (Δ 8 ± 6 μeq of H+ per 100 g for 90 min, n = 4). In animals pretreated with higher i.c. doses of l-NAME (800 μg/kg), microinjection of l-arginine (800 μg/kg) into the DMN reestablished the inhibition of gastric acid secretion by endotoxin (Δ19 ± 9 μeq of H+ per 100 g for 90 min, n = 8) or oxytocin (8 ± 4 μeq of H+ per 100 g for 90 min, n = 5). However the same dose of l-arginine (800 μg/kg) when administered i.c. did not influence the reversal induced by i.c. l-NAME (800 μg/kg).
Effects on Systemic Blood Pressure.
In animals undergoing hypotension, the reinfusion of the shed blood was accompanied by a rapid increase in systemic blood pressure to levels significantly higher (123 ± 5 mmHg, n = 6) than the resting values (105 ± 7 mmHg, n = 6), which remained for 20 min and then slowly decreased to resting values. Blood pressure was not modified by the increase in body temperature or the administration of oxytocin (8 μg/kg, i.c.) or endotoxin (5 μg/kg, i.v.). Pretreatment with either l-NAME (800 μg/kg i.c. or 80 μg/kg by microinjection) or l-arginine (12 mg/kg i.c. or 800 μg/kg by microinjection) did not influence systemic arterial blood pressure (n = 5 in both) during the 90-min evaluation period.
Immunohistochemical Studies.
As shown in Figs. 7 and 8, control animals (n = 4) exhibited a limited amount of nNOS-immunoreactivity located in the hippocampus and the rostral portion of the medulla oblongata. The DMN and NTS displayed a network of neurons and nerve fibers containing nNOS. Following a 90-min period of hyperthermia (n = 6) there was a substantial increase in the intensity of nNOS-immunoreactivity in both DMN and NTS, expressed in the cell bodies of neurons and the network of nerve fibers that pervade the two nuclei (Fig. 9). A limited amount of nNOS-immunoreactivity was also present in the cell bodies of neurons that form the hypoglossal nucleus and in different areas of the hippocampus.
Figure 7.
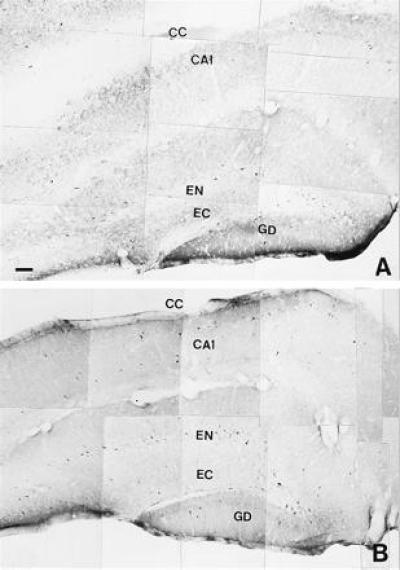
Distribution of nNOS immunoreactive neural structures forming different areas of the hippocampus from control (A) and hyperthermic (39°C) (B) animals. CA1, field of Ammon’s Horn; CC, corpus callosum; EN, part endal of the dentate gyrus; EC, part ectal of the dentate gyrus; GD, dentate gyrus. (Bars = 0.005 mm.)
Figure 8.
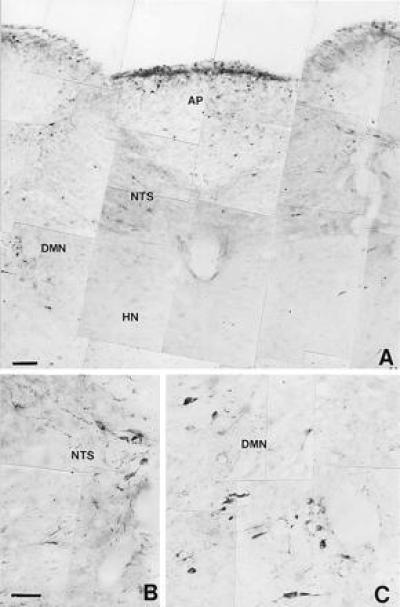
Microphotograph showing nNOS immunoreactive neural structures distributed in the most rostral portion of the medulla oblongata (A) from a control rat. HN, hypoglossal nucleus. (B and C) Large magnification of immunoreactive neurons in the NTS and the DMN, respectively. (Bars = 0.005 mm.)
Figure 9.
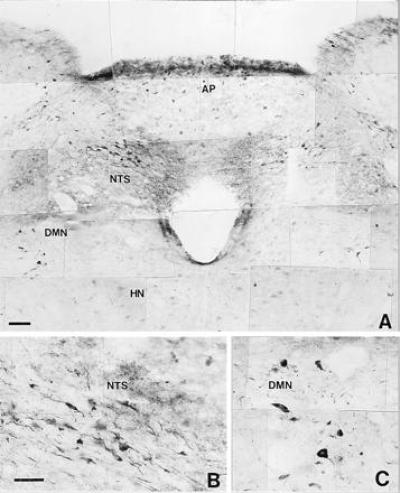
Microphotograph showing nNOS immunoreactive neural structures distributed in the most rostral portion of the medulla oblongata (A) from a hyperthermic rat (39°C). HN, hypoglossal nucleus. Note that the microphotograph shows an increase in the immunoreactivity in the NTS and DMN. Some neurons of the hypoglossal nucleus show light immunoreactivity. (B and C) Greater magnification of immunoreactive neurons in the NTS and DMN, respectively. (Bars = 0.005 mm.)
DISCUSSION
We have shown that induction of moderate stress by elevation of body temperature by 3°C or by a 35 mmHg reduction in blood pressure for 10 min produced a rapid and long-lasting inhibition of gastric acid secretion. This antisecretory response involves a reflex in which NO released in the brain plays a role, since i.c. administration of l-NAME reversed the acid inhibitory effects of both hyperthermia and hypotension. The low doses of l-NAME used had no such effect when administered i.v. and, furthermore, did not affect systemic arterial blood pressure when administered by either route, therefore excluding the possibility that systemic peripheral actions of this compound could be responsible for the reversal of acid inhibition.
In recent studies (19), the suppression of acid secretion by low doses of endotoxin has been shown to be prevented by l-NAME administered i.c. Endotoxin may be considered to be a challenge to which the body responds with stress and therefore the inducer of mechanisms similar to those elicited by hyperthermia and hypotension. Thus, diverse models of moderate stress indicate the existence of a common central pathway involving the acute generation of NO in the brain, which mediates the downregulation of gastric acid secretion.
Stress releases a number of mediators, some of which have been reported to inhibit gastric acid secretion through centrally mediated mechanisms (12, 20–22). In the current study i.c. administration of one of these mediators, oxytocin (23), abolished the distension-stimulated acid response at doses that, if administered i.v., had no such effect. Administration of l-NAME i.c. reversed the inhibitory action of oxytocin, thus implying the involvement of NO in the acid inhibitory actions of this peptide. Support for the specificity of these actions comes from the finding that l-arginine given i.c. reversed the effects of l-NAME in animals treated with oxytocin. Similarly, i.c. administration of l-arginine restored the acid inhibitory effects of both hyperthermia and hypotension in rats treated i.c. with l-NAME. These results not only provide further evidence that activation of a common l-arginine:NO pathway in the central nervous system mediates the acid inhibitory actions of stress, but also implicate its involvement in the actions of such mediators released during stress.
The DMN provides the major source of innervation to the stomach (24) and, in addition, it has been demonstrated that neurons within this nucleus play an important role in the control of gastric acid secretion (13, 25, 26). The DMN has been shown to contain a number of neurons and processes that exhibit inmunoreactivity to nNOS (16). Furthermore, there is recent in vitro evidence suggesting the involvement of NO in cell-to-cell signaling in the DMN (27). Although NO is now known to be a transmitter in many nonadrenergic/noncholinergic nerves in the peripheral nervous system (28, 29), and acts as an intercellular messenger in the brain (30–32), the biological functions of NO in the nervous system have not yet been fully elucidated. The present study demonstrates that microinjection of l-NAME into the DMN reversed the acid inhibitory actions of hyperthermia at doses that had no such effect when administered i.c. In addition, similar microinjections of l-NAME in the DMN prevented the inhibition of acid secretion induced by i.v. endotoxin or i.c. oxytocin, while microinjection of l-arginine in the DMN restored the acid-inhibitory effects of both endotoxin and oxytocin in rats treated with i.c. l-NAME, again suggesting that a common pathway is involved in the responses to various stressful situations. The NTS is located in the vicinity of the DMN (24), is also implicated in the regulation of gastrointestinal function (13) and, moreover, shows a greater density of nNOS immunoreactivity than the DMN (16). However, microinjection of l-NAME in the NTS failed to affect the acid inhibitory effects of hyperthermia, endotoxin or oxytocin, thus indicating that the NO-synthesizing neuronal structures involved in the abolition of acid responses induced by stress may be located more specifically within the DMN.
Immunohistochemical studies demonstrated an increase of nNOS immunoreactivity in the DMN following hyperthermia. This rapid augmentation in nNOS-immunoreactivity was not generalized and other areas of the medulla oblongata, such as the hypoglossal nucleus, or neural structures located in the telencephalon, such as the hippocampus, exhibited only minor increases in immunoreactivity following hyperthermia. The fact that the nNOS-immunoreactivity also increased in the NTS suggests that stress activates NO-dependent pathways in addition to the one we have investigated in the present study.
Thus, a nervous reflex involving the release of NO in the DMN mediates the stress-induced inhibition of gastric acid secretion. This centrally mediated antisecretory response should be considered as a physiological protective response against stress. It is clear that any disruption of the brain centers involved in such a neuronal pathway would affect this inhibitory reflex response and lead to the persistence, or even enhancement of the gastric secretion of acid during stress. Interestingly, stress induced by shock, massive burns, sepsis, or major trauma is associated with hyposecretion of gastric acid, whereas stress associated with brain injury is accompanied by an increase in acid secretion (33). In general, therefore, during stress, mechanisms leading to gastric ulceration as well as protective mechanisms such as the one described here, are triggered. Failure of the latter will lead to an increase in the severity of stress ulcerations with life-threatening consequences.
Acknowledgments
We thank Prof. J. Rodrigo for help in the immunohistochemical studies and Mrs. A. Higgs for helpful comments. This study was supported by Fondo de Investigaciones Sanitarios Grant FIS95/1343 and Direccion General de Investigacion Cientifica y Técnica Grants SAF95-0472, and PM95-145.
Footnotes
Abbreviations: l-NAME, NG-nitro-l-arginine methyl ester; NOS, NO synthase, nNOS, neuronal NOS; DMN, dorsal motor nucleus; NTS, nucleus tractus solitarius; i.c., intracisternally.
References
- 1.Selye H. Nature (London) 1936;138:32. [Google Scholar]
- 2.Beaumont W. In: Experiments and Observations on the Gastric Juice and the Physiology of Digestion. Osler W, editor. New York: Dover; 1833. [Google Scholar]
- 3.Cannon W B. Bodily Changes in Pain, Hunger, Fear and Rage. New York: Appleton; 1929. [Google Scholar]
- 4.Menguy R. Dig Dis. 1960;5:911–916. doi: 10.1007/BF02232858. [DOI] [PubMed] [Google Scholar]
- 5.Mine K, Noda T, Fujiwara M, Tsuruta N, Ueki S, Nakagawa T. Pharmacol Biochem Behav. 1983;19:359–364. doi: 10.1016/0091-3057(83)90065-5. [DOI] [PubMed] [Google Scholar]
- 6.Witty R T, Long J P. Am J Physiol. 1970;219:1359–1362. doi: 10.1152/ajplegacy.1970.219.5.1359. [DOI] [PubMed] [Google Scholar]
- 7.Paré W P, Livingston A., Jr Physiol Behav. 1973;11:521–526. doi: 10.1016/0031-9384(73)90039-5. [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Cuesta M A, Barrachina M D, Piqué J M, Whittle B J R, Esplugues J V. Eur J Pharmacol. 1992;218:351–354. doi: 10.1016/0014-2999(92)90191-6. [DOI] [PubMed] [Google Scholar]
- 9.Dorval E D, Meuller G P, Eng R R, Durakovic A, Conklin J J, Dubois A. Gastroenterology. 1985;89:374–380. doi: 10.1016/0016-5085(85)90339-7. [DOI] [PubMed] [Google Scholar]
- 10.Lenz H J, Raedler A, Greten H, Vale W W, Rivier J E. Gastroenterology. 1988;95:1510–1517. doi: 10.1016/s0016-5085(88)80070-2. [DOI] [PubMed] [Google Scholar]
- 11.Kauffman G L, Zhang L, Xing L, Seaton J, Colony P, Demers L. Ann NY Acad Sci. 1990;597:175–190. doi: 10.1111/j.1749-6632.1990.tb16166.x. [DOI] [PubMed] [Google Scholar]
- 12.Katoh H, Ohtake M, Sakaguchi T. Neuropeptides. 1991;20:169–173. doi: 10.1016/0143-4179(91)90127-5. [DOI] [PubMed] [Google Scholar]
- 13.Krowicki Z K, Hornby P J. In: Regulatory Mechanisms in Gastrointestinal Function. Gaginella T S, editor. Boca Raton, FL: CRC; 1995. pp. 278–319. [Google Scholar]
- 14.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- 15.Springall D R, Riveros-Moreno V, Buttery L, Suburo A, Bishop A E, Merrett M, Moncada S, Polak J M. Histochemistry. 1992;98:259–266. doi: 10.1007/BF00271040. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigo J, Springall D R, Yttenthal O, Bentura M L, Abadia-Molina F, Riveros-Moreno V, Martínez-Murillo R, Polak J M, Moncada S. Philos Trans R Soc London B. 1994;345:175–221. doi: 10.1098/rstb.1994.0096. [DOI] [PubMed] [Google Scholar]
- 17.Guesdon J L, Ternyck T, Avrameas S. J Histochem Cytochem. 1979;27:1131–1139. doi: 10.1177/27.8.90074. [DOI] [PubMed] [Google Scholar]
- 18.Shu S, Ju G, Fan L. Neurosci Lett. 1988;85:169–171. doi: 10.1016/0304-3940(88)90346-1. [DOI] [PubMed] [Google Scholar]
- 19.Barrachina M D, Whittle B J R, Moncada S, Esplugues J V. Br J Pharmacol. 1995;114:8–12. doi: 10.1111/j.1476-5381.1995.tb14898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taché Y, Vale W, Rivier J, Brown M. Proc Natl Acad Sci USA. 1980;77:5515–5519. doi: 10.1073/pnas.77.9.5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taché Y. In: Physiology of the Gastrointestinal Tract. Johnson L R, editor. New York: Raven; 1987. pp. 911–930. [Google Scholar]
- 22.Kasting N W. Can J Physiol Pharmacol. 1988;66:22–26. doi: 10.1139/y88-004. [DOI] [PubMed] [Google Scholar]
- 23.Ivanyi T, Wiegant V M, De-Wied D. Life Sci. 1991;48:1309–1316. doi: 10.1016/0024-3205(91)90527-i. [DOI] [PubMed] [Google Scholar]
- 24.Shapiro R E, Miselis R R. J Comp Neurol. 1985;238:473–488. doi: 10.1002/cne.902380411. [DOI] [PubMed] [Google Scholar]
- 25.Laughton W B, Powley T L. Am J Physiol. 1987;252:R13–R25. doi: 10.1152/ajpregu.1987.252.1.R13. [DOI] [PubMed] [Google Scholar]
- 26.Altschuler S M, Ferenci D A, Lynn R B, Miselis R R. J Comp Neurol. 1991;304:261–274. doi: 10.1002/cne.903040209. [DOI] [PubMed] [Google Scholar]
- 27.Travagli R A, Gillis R A. Am J Physiol. 1994;266:G154–G160. doi: 10.1152/ajpgi.1994.266.1.G154. [DOI] [PubMed] [Google Scholar]
- 28.Moncada S, Palmer R M J, Higgs A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 29.Moncada S, Higgs A. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 30.Garthwaite J, Charles S L, Chess-Williams R. Nature (London) 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 31.Bredt D S, Snyder S H. Proc Natl Acad Sci USA. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bredt D S, Snyder S H. Neuron. 1992;8:3–11. doi: 10.1016/0896-6273(92)90104-l. [DOI] [PubMed] [Google Scholar]
- 33.McGuigan J E. In: Harrison’s Principles of Internal Medicine. Isselbacher K J, Braunwald E, Wilson J D, Martin J B, Fauci A S, Kasper D L, editors. New York: McGraw–Hill; 1994. pp. 1363–1382. [Google Scholar]