

Abstract
The TEL/PDGFβR fusion protein is the product of the t(5;12) translocation in patients with chronic myelomonocytic leukemia. The TEL/PDGFβR is an unusual fusion of a putative transcription factor, TEL, to a receptor tyrosine kinase. The translocation fuses the amino terminus of TEL, containing the helix-loop-helix (HLH) domain, to the transmembrane and cytoplasmic domain of the PDGFβR. We hypothesized that TEL/PDGFβR self-association, mediated by the HLH domain of TEL, would lead to constitutive activation of the PDGFβR tyrosine kinase domain and cellular transformation. Analysis of in vitro-translated TEL/PDGFβR confirmed that the protein self-associated and that self-association was abrogated by deletion of 51 aa within the TEL HLH domain. In vivo, TEL/PDGFβR was detected as a 100-kDa protein that was constitutively phosphorylated on tyrosine and transformed the murine hematopoietic cell line Ba/F3 to interleukin 3 growth factor independence. Transformation of Ba/F3 cells required the HLH domain of TEL and the kinase activity of the PDGFβR portion of the fusion protein. Immunoblotting demonstrated that TEL/PDGFβR associated with multiple signaling molecules known to associate with the activated PDGFβR, including phospholipase C γ1, SHP2, and phosphoinositol-3-kinase. TEL/PDGFβR is a novel transforming protein that self-associates and activates PDGFβR-dependent signaling pathways. Oligomerization of TEL/PDGFβR that is dependent on the TEL HLH domain provides further evidence that the HLH domain, highly conserved among ETS family members, is a self-association motif.
Chronic myelomonocytic leukemia (CMML) is classified as a myelodysplastic syndrome and is characterized clinically by dysplastic monocytosis, hypercellular bone marrow, splenomegaly, and progression to acute myelogenous leukemia. There are few effective therapies for CMML, and the etiology is poorly understood. The only known gene rearrangement in CMML is the TEL/PDGFβR (T/P) fusion that occurs as a consequence of t(5;12), a recurring cytogenetic abnormality in CMML. T/P is an unusual example of the fusion of a putative transcription factor, TEL, to the PDGFβR tyrosine kinase. The T/P gene product contains the amino-terminal 154 aa of TEL fused to the transmembrane and cytoplasmic domains of the PDGFβR. The ligand-binding domain of PDGFβR is absent in the fusion protein but an intact tyrosine kinase domain is retained, as are the binding sites for multiple src homology 2 (SH2) domain containing signaling molecules (1).
The TEL gene (GenBank data base accession no. ETV6) is rearranged in a broad spectrum of human leukemias (2–5) and encodes a member of the ETS family of transcription factors that are defined by a conserved DNA binding domain (the ets domain). A subset of ETS family members also contains a highly conserved amino-terminal domain whose function is unknown and has been variously termed the pointed domain (6), domain B, and the helix-loop-helix (HLH) domain (1, 7). Several members of the family have oncogenic potential including vEts, which was identified as part of a fusion with gag and myb in the E26 avian erythroblastosis virus, and Spi-1 and Fli-1, which are associated with murine erythroleukemia (7, 8), In these examples, the transforming ability of ETS family members requires their activity as transcription factors. In humans, examples of involvement of ETS DNA binding domains in malignancy include the EWS–Fli1 fusion associated with Ewing sarcoma and t(11;22) (9); the TLS–ERG fusion associated with acute myelogenous leukemia and t(16;21) (10); and the MN1–TEL fusion associated with myelodysplasia and t(12;22) (5). However, in contrast with EWS–fli-1, TLS–ERG, EWS–ERG, EWS–ETV1, and MN1–TEL, the T/P fusion does not incorporate the TEL DNA binding domain (Fig. 1). Instead, the TEL HLH domain is fused in frame to the transmembrane and cytoplasmic domain of PDGFβR, suggesting a different functional role for TEL in the T/P fusion than other fusions involving ETS proteins. The reciprocal PDGFβR/TEL fusion, which would contain the TEL DNA binding domain, is not expressed in patients with t(5;12) CMML (11).
Figure 1.
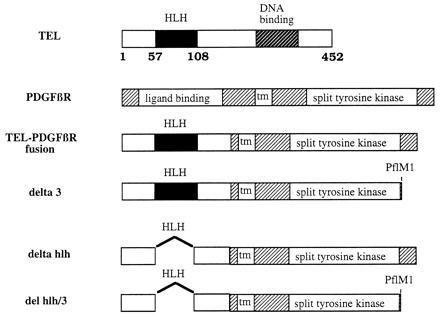
Schematic representation of TEL, PDGFβR, T/P, and mutations of T/P.
PDGFβR is a well-characterized plasma membrane receptor with endogeneous tyrosine kinase activity, which is autophosphorylated in response to binding of dimeric platelet-derived growth factor (PDGF) ligand (1). PDGF binding induces diverse physiologic effects, depending on the cell type in which PDGFβR is expressed and what biological responses are measured. Biological responses that have been ascribed to signal transduction through the PDGFβR include turnover of phosphoinositol, Ca2+ flux, cellular migration, angiogenesis, mitogenesis, membrane ruffling, and cytoskeletal rearrangement. Depending on the cell type in which PDGFβR is expressed, autophosphorylation of tyrosine residues is accompanied by binding of a variety of proteins that mediate downstream signal transduction events. Autophosphorylation of PDGFβR can lead to association with and activation of phospholipase Cγ1 (PLCγ), phosphoinositol-3-kinase (PI3-K), Src, SH-PTP2 (now designated SHP2), RAS, NCK, GRB2, and SHC, as well as activation of STAT 1 and 3 (1, 12–15). Aberrant activation of the PDGFβR leading to cell transformation has been described in several circumstances. v-Sis, the transforming protein of simian sarcoma virus, has near identity to the PDGF-B isoform, the preferred ligand for PDGFβR, and transforms cells through activation of the PDGFβR (16). PDGF-B itself induces a myeloproliferative disease when constitutively expressed in mouse bone marrow transplantation models (17). In addition, the bovine papillomavirus E5 protein transforms cells by mediating dimerization and constitutive activation of PDGFβR (18). It is therefore plausible that mutations of the PDGFβR itself could confer transforming activity, but before the cloning of the T/P fusion, no such mutations or rearrangements had been identified.
Because T/P is exclusively associated with hematopoietic malignancy, we chose to study the transforming properties of T/P in the murine hematopoietic cell line Ba/F3. Demonstration of transforming activity of putative oncoproteins is a critical step in the analysis of human cancer genetics. Transformation of cells by an oncoprotein documents relevance to disease pathogenesis and establishes a model for studying disease mechanisms. Here we report that the consequence of the t(5;12) is generation of a mutant PDGFβR fusion protein that transforms hematopoietic cells and requires functional motifs contributed by both the TEL and PDGFβR moieties. This establishes the transforming activity of T/P and provides a model system for studying the mechanisms of transformation by this novel fusion protein.
MATERIALS AND METHODS
Cell Lines and Transfections.
Ba/F3 cells were a kind gift of Alan D’Andrea (Dana–Farber Cancer Institute). Cells were maintained in interleukin 3 (IL-3) medium [RPMI 1640 medium with 10% fetal bovine serum (FBS) and 0.5–1 ng of recombinant IL-3 (R & D Systems)] in a 5% CO2 incubator at 37°. NIH 3T3 cells were maintained in DMEM with 10% FBS. Ba/F3 cells were electroporated as follows: 1 × 107 cells were washed in phosphate-buffered saline (PBS) and incubated for 10 min at room temperature with 20 μg of plasmid DNA in PBS. Cells were electroporated at 350 mV/960 μF in a Bio-Rad apparatus. Following a 10-min incubation at room temperature cells were plated in 10 ml of IL-3 medium for 48 hr and then selected in IL-3 medium plus 1 mg of G418 per ml. Neo-resistant cells were subcloned by limiting dilution. NIH 3T3 cells were transfected by calcium phosphate precipitation and selected in G418. For soft colony agar assays, 5 × 104 NIH 3T3 cells stably transfected with pSRα alone, pSRα-BCR-ABL, or pSRα-T/P were plated in soft agar as previously described (19). For growth curves, 2 × 104 Neo-resistant cells (pcDNA3, Y635K, ΔHLH) or IL-3-independent cells (T/P) were plated on day 0, and viable cells were counted on each day by trypan blue exclusion.
DNA Constructs.
The retroviral expression vector pSRa/MSV/TKneo (pSRa) has been described (28) and was provided by O. Witte (University of California, Los Angeles). pcDNA3 was purchased from Invitrogen. A full-length T/P was reconstructed in pSRa by splicing together a wild-type TEL cDNA, a wild-type PDGFβR cDNA (a kind gift of J. Cooper), and a reverse transcription-PCR-generated fragment spanning the T/P junction. The ΔHLH mutant was generated by cutting within the TEL cDNA at BspMI and EcoNI sites, blunting with mung bean nuclease, and religating, leading to deletion of nucleotides 195–348 of the TEL gene. Deletions constructs were confirmed by sequencing. pLXSN PDGFβR Y635K was a kind gift of J. Cooper (University of Washington, Seattle) and Adam Kashishian (Beth Israel Hospital, Boston, MA). A KspI/HindIII fragment from this construct was cloned into the KspI/HindIII sites of pSRα T/P.
In Vitro Transcription/Translation.
All constructs were cloned into pcDNA3 (Invitrogen). T/P Δ3 and T/P ΔHLH Δ3 constructs were generated by digesting pcDNA3 T/P and pcDNA3 T/P ΔHLH constructs, respectively, with PflMI [which digests the PDGFβR portion of the fusion protein 3′ of the kinase domain (nucleotide 1818 of the human PDGFβR)] and blunting with T7 polymerase. The plasmid was then digested with EcoRI, and the EcoRI/PflMI fragment was cloned into the EcoRI/EcoNI sites of pcDNA3. In vitro transcription/translation was performed using a rabbit reticulocyte lysate kit (TNT system; Promega) according to the manufacturer’s specifications. Proteins were labeled with [35S]methionine incorporation. One-half of the reaction was removed, diluted to 250 μl in lysis buffer (150 mM NaCl/50 mM Tris·HCl/1% Triton X-100 plus protease and phosphatase inhibitors) and immunoprecipitated with an antibody to the carboxyl-terminal portion of the PDGFβR molecule (Upstate Biotechnology). Total reaction mixtures and immunoprecipitates were resolved by SDS/PAGE, and the separated proteins were visualized by fluorography using Amplify (Amersham) according to the manufacturer’s instructions.
Immunoprecipitaton and Western Blotting.
Cells were washed once in PBS and lysed in lysis buffer (150 mM NaCl/50 mM Tris·HCl/1% Triton X-100 plus protease and phosphatase inhibitors). For immunoprecipitates, 1000 μg of total cell lysate was incubated with appropriate antibody for 2 hr on ice, and then 100 μl of formaldehyde-fixed Staphylococcus aureus (Pansorbin; Calbiochem) was added for 20 min at 4°. Immunoprecipitates were washed three times in lysis buffer and then boiled in loading buffer for 5 min. Antibodies were as follows: for Western blotting, PDGFβR and PLCγ (Upstate Biotechnology), PI3 kinase and SHP2 (Transduction Laboratories, Lexington, KY), and anti-phosphotyrosine–4G10, a kind gift of Tom Roberts (Dana–Farber Cancer Institute); and for immunoprecipitates, PDGFβR (PharMingen; anti-tail antibody). For Western blots, blots were blocked in either 5% dry milk in TBST (0.1% Tween-20/0.01 M Tris·HCl, pH 7.6/150 mM NaCl) or 5% bovine serum albumin in TBST (for 4G10 Western blots), rinsed in TBST for 5 min and incubated for 2 hr at room temperature in primary antibody. Blots were washed and incubated with horseradish peroxidase-conjugated secondary antibody and visualized using enhanced chemiluminescence (ECL; Amersham).
RESULTS
T/P Transforms Hematopoietic Cells to Factor Independence.
Because T/P is associated with the hematologic malignancy CMML, we characterized transforming activity of T/P in the murine hematopoietic cell line Ba/F3. Ba/F3 cells are dependent on IL-3 for growth and have an extremely low rate of spontaneous reversion to factor independence. In addition, Ba/F3 cells express no detectable endogeneous PDGFβR (M.C., unpublished observations). Ba/F3 cells were electroporated with the control vector pcDNA3 or pcDNA3–T/P, which contains the full-length T/P fusion cDNA. Cells were allowed to recover for 48 hr in growth medium plus IL-3 and were then selected for G418 resistance for ≈10 days. G418-resistant cells were washed free of IL-3 and replated in growth medium alone. In five separate experiments, IL-3-independent cells were obtained after 10–21 days in cells transfected with T/P. No IL-3-independent cells were obtained when Ba/F3 cells were transfected with vector alone. Ba/F3–T/P cells grow in the absence of IL-3 at a rate comparable to control cells in the presence of IL-3 (Fig. 2). Western blotting confirmed that the T/P protein was expressed in all factor-independent cells studied (n = 3; Fig. 2b, lanes 2 and 3). Two distinct bands are detected, which may be explained in part by alternate start sites for translation for TEL (20). T/P has a calculated molecular mass of 76 kDa but migrates with an apparent molecular weight of ≈100 kDa after isolation from transfected Ba/F3 cells, in vitro translation mixtures, or primary patient material (data not shown).
Figure 2.
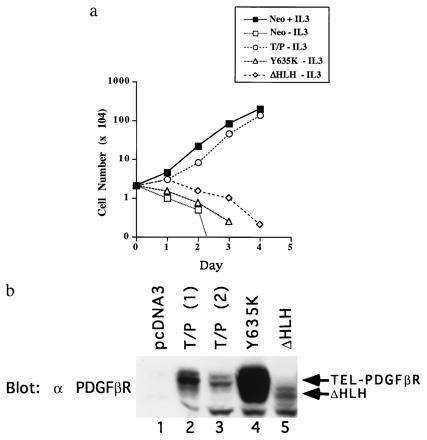
T/P transforms Ba/F3 cells to IL-3 factor independence. (a) Ba/F3 cells were transfected with pcDNA3, pcDNA3 T/P, the kinase-inactive mutant, Y635K, or the ΔHLH mutant and selected for G418 resistance. G418-resistant cells were selected for growth in the absence of IL-3. Neo-resistant (2 × 104 cells; pcDNA3, Y635K, and ΔHLH) or IL-3-independent cells (T/P) were washed free of IL-3 and plated on day 0 in RPMI 1640/10% FCS. Viable cells were counted on each day. (b) Expression of T/P and mutants in Ba/F3 cells. Transfected Ba/F3 cells were lysed in 1% Triton X-100/150 mM NaCl/50 mM Tris, pH 8.0, plus protease inhibitors. Lysates were separated by SDS/PAGE, transferred to nitrocellulose, and blotted with anti-PDGFβR antisera. Lanes 2 and 3 show two separate transfections with wild-type T/P.
To address the possibility of an autocrine mechanism of transformation by T/P (e.g., induction of endogenous IL-3 expression), Ba/F3–T/P cells were grown to confluence and the T/P-conditioned medium was harvested by centrifugation and filtration to remove residual cells. Normal Ba/F3 cells showed no growth or inhibition of apoptosis in 10% T/P-conditioned medium (data not shown), suggesting that IL-3-dependent growth was not the consequence of autocrine production of IL-3 or other growth factors.
T/P Does not Transform NIH 3T3 Cells.
NIH 3T3 cells were transfected with pSRa or pSRa-T/P constructs and stable transfectants selected in G418. Expression of the T/P protein was confirmed by Western blot analysis (data not shown). Cells (1 × 104) were plated in soft agar and macroscopic colonies were scored after 21 days as described (21). NIH 3T3 cells transformed by the fusion protein p185bcr/abl were used as a positive control. In two separate experiments, no colonies were detected in 3T3-T/P cells, whereas >150 colonies were present on plates seeded with p185bcr/abl-expressing cells (Table 1). In addition, the NIH 3T3 cells transfected with T/P were maintained in continuous culture for several months. These cells did not form foci and showed no morphologic changes.
Table 1.
T/P does not transform NIH 3T3 cells
| Transfected DNA | No. of colonies* |
|---|---|
| pSRα | 0 |
| T/P | 0 |
| BCR–ABL | 177 |
NIH 3T3 cells were transfected with the indicated constructs and stable transfectants selected in G418. Cells (1 × 104) were plated in soft agar and incubated at 37° in a 5% CO2 incubator. Macroscopic colonies were counted at day 21.
Average of two experiments.
T/P Is Constitutively Tyrosine-Phosphorylated.
The wild-type PDGFβR protein is inducibly tyrosine-phosphorylated after ligand binding, so we examined T/P protein expressed in Ba/F3 cells for tyrosine phosphorylation. Ba/F3–T/P cells grown in medium lacking IL-3 (Fig. 3, lane 2), and vector-transfected cells (Fig. 3, lane 1) were lysed and proteins were immunoprecipitated using an anti-PDGFβR antisera. Immunoprecipitates were separated by SDS/PAGE and transferred to nitrocellulose. Blots were probed using an antiphosphotyrosine antibody. A prominent 100-kDa tyrosine-phosphorylated protein was detected in Ba/F3–T/P cells but was not detected in vector-transfected control cells (Fig. 3 Lower). Stripping and reprobing with an anti-PDGFβR antisera confirmed that the 100-kDa protein was T/P (Fig. 3 Upper).
Figure 3.
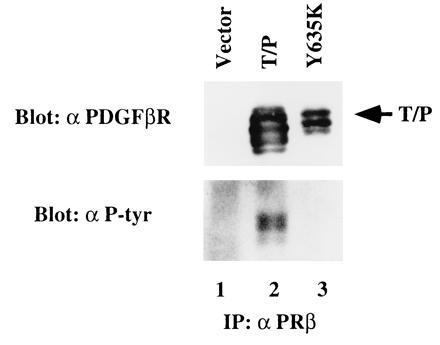
T/P is constitutively tyrosine-phosphorylated in Ba/F3 cells. Ba/F3 cells were transfected with pcDNA3 alone (lane 1), T/P (lane 2), or a kinase-inactive mutant with a tyrosine-to-lysine mutation at the site corresponding to Y635 in the human PDGFβR (Y635K; lane 3). Cells were selected in 1 mg/ml of G418, and T/P cells were subsequently selected for growth in the absence of IL-3. Cells were lysed and immunoprecipitated with an antibody to the human PDGFβR (Upstate Biotechnology). Immunoprecipitates were separated by SDS/PAGE, transferred to nitrocellulose, and Western blotted using the indicated antisera and enhanced chemiluminescence (ECL) detection methods. All constructs direct the synthesis of a doublet due to the use of an alternative start site for translation in the TEL portion of the fusion cDNA.
Transformation by T/P Requires the Kinase Activity of the PDGFβR Cytoplasmic Domain.
To test the hypothesis that T/P transforms cells through constitutive activation of the PDGFβR kinase, we constructed a kinase-deficient T/P mutant. A point mutation corresponding to a kinase inactivating mutation in the PDGFβR (Y635K) was introduced into the T/P cDNA. The kinase-inactive construct, designated T/P Y635K, was transfected into Ba/F3 cells, and cells were selected for G418 resistance as above. Western blot analysis confirmed expression of T/P Y635K and demonstrated that, unlike the wild-type T/P, T/P Y635K is not tyrosine-phosphorylated (Fig. 3 Upper, lane 3). Stable transfectants expressing T/P Y635K were unable to grow in medium lacking IL-3 (Fig. 2) providing convincing evidence that the tyrosine kinase activity of PDGFβR is necessary for T/P transformation of Ba/F3 cells.
T/P Oligomerizes in Vitro.
Dimerization is known to be a prerequisite for activation of the wild-type PDGFβR. We therefore tested for in vitro oligomerization of T/P by in vitro transcription and translation of T/P, and T/P Δ3 (Fig. 1), which contains a carboxyl-terminal truncation of the PDGFβR. As expected, anti-PDGFβR antisera, which was specific for the carboxyl terminus of PDGFβR-immunoprecipitated T/P (Fig. 4, lanes 1 and 7), but did not recognize the T/P Δ3 truncation mutant (Fig. 4, lanes 2 and 8). However, when T/P and T/P Δ3 were cotranslated, both were immunoprecipitated by antibody directed against the carboxyl terminus of PDGFβR (Fig. 4, lanes 3 and 9). These data provide evidence for an interaction between the T/P and T/P Δ3 proteins and demonstrate that T/P self-associates into dimeric or higher order complexes in vitro.
Figure 4.
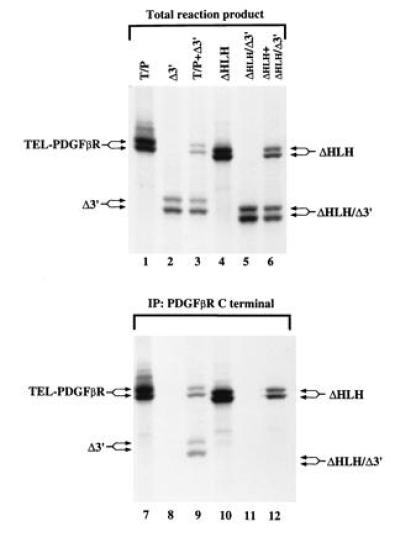
T/P dimerizes in vitro and dimerization requires the HLH domain. cDNA constructs for the indicated mutations of T/P were cloned into the pcDNA3 expression vector, and in vitro transcription/translation was performed according to the manufacturer’s instruction (Promega TNT kit) using radiolabeled [35S]methionine. Quantity of DNA added was adjusted to give approximately equal amounts of each translated protein. For lanes 7–12, one-half of the reaction mixture was removed and immunoprecipitated with an antisera to the carboxyl terminus of human PDGFβR. Total reaction products or total immunoprecipitates were separated by SDS/PAGE. The gel was fixed, treated with Enhance (Amersham), dried, and exposed to film at −70° for 30 min.
T/P Oligomerization and Transformation of Ba/F3 Cells Requires the HLH Domain of TEL.
The TEL HLH domain is conserved among ETS family members and has been postulated to serve as a self-association motif (11, 20). To test the hypothesis that the TEL HLH domain mediates T/P oligomerization, T/P containing a 51-aa in-frame deletion of the HLH domain was constructed (T/P ΔHLH, Fig. 1). A corresponding mutant was prepared containing a carboxyl-terminal truncation of the PDGFβR moiety (T/P ΔHLH Δ3, Fig. 1), and tested in in vitro transcription/translation assays as described above. As expected, antisera directed against the carboxyl terminus of PDGFβR immunoprecipitated the T/P ΔHLH mutant (Fig. 4, lanes 4 and 10) but did not recognize the carboxyl terminal truncation mutant T/P ΔHLH Δ3 (Fig. 4, lanes 5 and 11). When the two HLH deletion mutants were cotranslated, only the T/P ΔHLH mutant was immunoprecipitated with antisera to the PDGFβR carboxyl terminus (Fig. 4, lanes 6 and 12). These data provide evidence that deletion of the TEL HLH domain abrogates the ability of the T/P fusion to self-associate and are consistent with the hypothesis that the TEL HLH domain is a self-association motif.
To determine whether the TEL HLH domain is required for transformation of Ba/F3 cells, T/P ΔHLH was transfected into Ba/F3 cells and cells were selected in G418. Expression of T/P ΔHLH was confirmed by Western blot analysis (Fig. 2b, lane 5). Ba/F3 cells, which were stably transfected with T/P ΔHLH, were incapable of growth in the absence of IL-3 (Fig. 2). Immunoprecipitation of T/P ΔHLH from these cells and Western blotting with an anti-phosphotyrosine antibody demonstrate that T/P ΔHLH is not tyrosine-phosphorylated (data not shown). These data show that the TEL HLH domain is required for transforming activity as well as for oligomerization.
T/P Constitutively Associates with Signaling Molecules That Are Activated by Wild-Type PDGFβR.
We hypothesized that T/P might cause proliferation of myeloid cells through constitutive activation of mitogenic signaling pathways used by the wild-type PDGFβR. To explore this possibility, the T/P protein was immunoprecipitated from Ba/F3–T/P cells growing in the absence of IL-3 and analyzed by Western blotting for interaction with PLCγ, SHP2, PI3 kinase (p85), and RAS-GAP. As a positive control, Ba/F3 cells transfected with the wild-type PDGFβR were also analyzed. T/P and PDGFβR associate with signaling molecules, including PLCγ, SHP2, PI3 kinase (Fig. 5), and RAS-GAP (not shown). Collectively, these data are consistent with the hypothesis that self-association of T/P leads to constitutive tyrosine kinase activation, autophosphorylation, and interaction with signaling molecules known to be relevant to mitogenic pathways for the wild-type PDGFβR.
Figure 5.
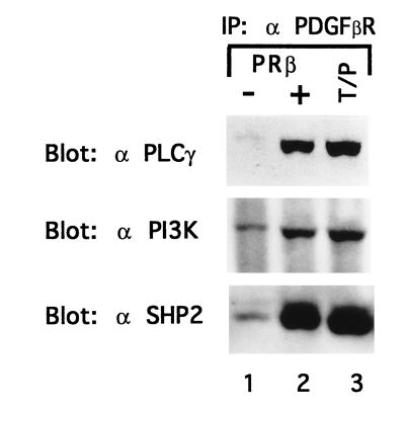
T/P associates with signaling molecules. Ba/F3–PDGFβR and Ba/F3 T/P cells were lysed and immunoprecipitated with anti-PDGFβR antisera. Immunoprecipitates were separated by SDS/PAGE, transferred to nitrocellulose, and blotted with the indicated antibodies. Ba/F3–PDGFβR were either deprived of IL-3 for 4 hr (−) or stimulated with 100 ng of recombinant human PDGF B for 10 min at 37° (+).
DISCUSSION
The t(5;12) is a recurring translocation that is associated with the clinical phenotype of CMML. The consequence of t(5;12) in all patients examined thus far is generation of a T/P fusion transcript, which contains the TEL HLH and the PDGFβR tyrosine kinase functional domains. The reciprocal PDGFβR/TEL transcript, which could be generated by the balanced reciprocal translocation, is not expressed. These data suggest an important role for the T/P fusion protein in pathogenesis of CMML and for the TEL HLH domain and PDGFβR tyrosine kinase activity.
Here we report that T/P is an oncoprotein that transforms the hematopoietic cell line Ba/F3 to growth factor independence. Mutational analysis shows that transformation is dependent upon both the TEL HLH domain and PDGFβR tyrosine kinase activity. T/P does not transform the nonhematopoeitic cell line NIH 3T3, although NIH 3T3 cells contain functional PDGFβR that is capable of signaling mitogenesis in response to the PDGF BB ligand. The finding that T/P transforms hematopoietic cells, but not fibroblasts, may be relevant to the observed hematopoietic tissue specificity of the T/P fusion protein, although other explanations are possible. For example, it is possible that the wild-type PDGFβR in NIH 3T3 cells interferes with transformation by T/P.
PDGFβR dimerization mediated by the dimeric PDGF ligand is necessary for tyrosine kinase activation and subsequent activation of PDGF dependent signaling pathways. Because the PDGF ligand-binding domain of PDGFβR is replaced by a portion of the TEL protein in the T/P fusion, we hypothesized that the TEL portion of the fusion contributed a dimerization or oligomerization motif that would constitutively activate the PDGFβR kinase. In support of this hypothesis, we have shown that the TEL HLH domain, conserved among ETS family members, mediates oligomerization of T/P and is required for T/P transformation of Ba/F3 cells. Other examples of constitutive activation of receptors that lead to transformation include the v-sis oncogene, which transforms cells via activation of PDGFβR (16), mutations in the erythropoietin receptor, which induce constitutive dimerization (22), and dimerization and activation of PDGFβR by bovine papillomavirus E5 (18). Of note, Ba/F3 cells transfected with the wild-type PDGFβR are not factor-independent, although they respond to PDGF, as shown in Fig. 5. We have not as yet compared the growth rate of cells transformed with T/P to those transfected with PDGFβR and stimulated with PDGF. It will be important to do this to understand if T/P is simply constitutively activating PDGFβR-dependent signaling pathways or is transforming cells through other mechanisms.
The finding that the TEL HLH domain is necessary for T/P oligomerization suggests that the domain is a self-association motif. Additional support for this observation includes TEL HLH-mediated oligomerization of the TEL–ABL (20) and TEL–AML1 fusions (23), and that wild-type TEL can self-associate (T.R.G., unpublished observations). The HLH domain has weak homology with the HLH domain of basic HLH transcription factors, such as MYC and MYO-D (24), and has been variously termed the 5′ Ets domain, the pointed domain, and domain B. However, the structure and function of this conserved region in ETS proteins are not known. Analysis of structure–function relationships in leukemogenic TEL fusion proteins suggests that the HLH domain in TEL may serve as self-association or protein–protein interaction motif, although it is unknown whether the HLH domain serves a similar function in other ETS family members. Given the interaction through this domain, it is possible that T/P could interact with the wild-type TEL protein and that this may have a role in transformation by T/P. Future experiments will address this question.
We have further demonstrated that T/P is constitutively tyrosine-phosphorylated and that PDGFβR tyrosine kinase activity is necessary for transformation. Western blotting showed that T/P associates with signaling molecules, which have been reported to be activated by the wild-type PDGFβR after ligand binding including PLCγ, SHP2, and PI3 kinase. PDGFβR association with all of these molecules is not necessary for stimulation of DNA synthesis or mitogenesis in response to ligand binding. Valius and Kazlauskas (25) have reported that in Hep G2 cells, PI3 kinase, and PLCγ are the critical mediators of the PDGFβR mitogenic activity. However, analysis of PDGFβR-induced mitogenesis in CHO and Ba/F3 cells has demonstrated that the critical mediators of mitogenesis vary in different cell types. Bovine papillomavirus E5 mediates constitutive dimerization of PDGFβR and transformation of Ba/F3 cells (17). However, in contrast with the wild-type receptor, transformation in the bovine papillomavirus E5/PDGFβR system is not affected by point mutations that abrogate PDGFβR interactions with PLCγ, NCK, Grb2, PI3K, Ras-GAP, or SHP-2. It will be necessary to prepare point mutations of specific tyrosine residues in the PDGFβR portion of the T/P fusion to determine which signaling pathways are critical for transformation by T/P.
In summary, our data is consistent with a model of transformation in which T/P oligomerizes via the HLH domain of TEL, leading to constitutive activation of the PDGFβR tyrosine kinase and stimulation of PDGFβR-dependent signaling pathways. The model suggests several potential strategies for reversion of transformation by T/P. Since transformation by T/P appears to rely on TEL HLH-mediated oligomerization, it may be possible to interfere with the ability of T/P to oligomerize and transform cells by overexpression of wild-type TEL, a TEL HLH peptide, or a kinase inactive T/P mutant. Alternatively, since kinase inactive mutants of T/P are nontransforming, specific inhibitors of the PDGFβR kinase (26, 27) may also inhibit transformation mediated by T/P. It should be possible to develop in vitro and in vivo models of transformation to explore these possibilities.
Acknowledgments
This work would not have been possible without the support and advice of Cheryl Miller, Dwayne Barber, and Terri Laufer. We thank Owen Witte for the pSRa expression vector and Alan D’Andrea for Ba/F3 cells. This work was supported in part by National Institutes of Health Grants PO1DK50654-01 and P01CA66996-01 and the Lawrence Family Foundation (D.G.G.), American Cancer Society Grant PRTA20 (M.C.). T.R.G. is a recipient of a Burroughs–Wellcome Fund Career Award in the Biomedical Sciences. D.G.G. is an Assistant Investigator of the Howard Hughes Medical Institute.
Footnotes
Abbreviations: PDGF, platelet-derived growth factor; PDGFβR, PDGF β receptor; CMML, chronic myelomonocytic leukemia; HLH, helix-loop-helix; PLCγ, phospholipase Cγ1; PI3-K, phosphoinositol-3-kinase; T/P, TEL/PDGFβR; IL-3, interleukin 3.
References
- 1.Claesson-Welsh L. J Biol Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- 2.Golub T R, Barker G F, Bohlander S K, Hiebert S W, Ward D C, Bray-Ward P, Morgan E, Raimondi S C, Rowley J D, Gilliland D G G. Proc Natl Acad Sci USA. 1995;92:4917–4921. doi: 10.1073/pnas.92.11.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shurtleff S, Buijs A, Behm F, Rubnitz J, Raimondi S, Hancock M, Chan G, Pui C, Grosveld G, Downing J. Leukemia. 1995;9:1985–1989. [PubMed] [Google Scholar]
- 4.Papadopoulos P, Ridge S A, Boucher C A, Stocking C, Wiedemann L M. Cancer Res. 1995;55:34–38. [PubMed] [Google Scholar]
- 5.Buijs A, Sherr S, van Baal S, van Bezouw S, van der Plas D, van Kessel A D, Riegman P, Deprez R L, Zwarthoff E, Hagemeijer A, Grosveld G. Oncogene. 1995;10:1511–1519. [PubMed] [Google Scholar]
- 6.Schneikert J, Lutz Y, Waslyk B. Oncogene. 1992;7:249–256. [PubMed] [Google Scholar]
- 7.Crepieux P, Coll J, Stehelin D. Crit Rev Oncog. 1994;5:615–638. [PubMed] [Google Scholar]
- 8.Ben-David Y, Giddens E B, Letwin K, Bernstein A. Genes Dev. 1991;5:908–918. doi: 10.1101/gad.5.6.908. [DOI] [PubMed] [Google Scholar]
- 9.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, Aurias A, Thomas G. Nature (London) 1992;359:162–165. doi: 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- 10.Ichikawa H, Shimizu K, Hayashi Y, Ohki M. Cancer Res. 1994;54:2865–2868. [PubMed] [Google Scholar]
- 11.Cochran B W, Reffel A C, Stiles C D. Cell. 1983;33:939–947. doi: 10.1016/0092-8674(83)90037-5. [DOI] [PubMed] [Google Scholar]
- 12.Lechleider R J, Sugimoto S, Bennett A M, Kashishian A S, Cooper J A, Shoelson S E, Walsh C T, Neel B G. J Biol Chem. 1993;268:21478–21481. [PubMed] [Google Scholar]
- 13.Li W, Nishimura R, Kashishian A, Batzer A G, Kim W J, Cooper J A, Schlessinger J. Mol Cell Biol. 1994;14:509–517. doi: 10.1128/mcb.14.1.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arvidsson A K, Rupp E, Nanberg E, Downward J, Ronnstrand L, Wennstrom S, Sclessinger J, Heldin C H, Claesson-Welsh L. Mol Cell Biol. 1994;14:6715–6726. doi: 10.1128/mcb.14.10.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waterfield M D, Scrace G T, Whittle N, Stroobant P, Johnsson A, Wasteson A, Westermark B, Heldin C H, Huang J S, Dueel T F. Nature (London) 1983;304:35–39. doi: 10.1038/304035a0. [DOI] [PubMed] [Google Scholar]
- 16.Yan X-Q, Brady G, Iscove N N. Oncogene. 1994;9:163–173. [PubMed] [Google Scholar]
- 17.Drummond-Barbosa D, Vaillancourt R R, Kazlauskas A, DiMaio D. Mol Cell Biol. 1995;15:2570–2581. doi: 10.1128/mcb.15.5.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lugo T G, Witte O N. Mol Cell Biol. 1989;9:1263–1270. doi: 10.1128/mcb.9.3.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golub T R, Goga A, Barker G, Afar D, McLaughlin J, Bohlander S, Rowley J, Witte O, Gilliland D G. Mol Cell Biol. 1996;16:4107–4116. doi: 10.1128/mcb.16.8.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lugo T G, Pendergast A, Muller A J, Witte O N. Science. 1990;247:1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 21.Golub T R, Barker G F, Lovett M, Gilliland D G. Cell. 1994;77:307–316. doi: 10.1016/0092-8674(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura A, Longmore G, Lodish H F. Nature (London) 1990;348:647–649. doi: 10.1038/348647a0. [DOI] [PubMed] [Google Scholar]
- 23.McLean, T. W., Ringold, S., Stegmaier, K., Neuberg, D., Tantravahi, R., Ritz, J., Koeffler, H. P., Takeuchi, S., Janssen, J. W. G., Seriu, T., Bartram, C. R., Sallan, S. E., Gilliland, D. G. & Golub, T. R. (1996) Blood, in press. [PubMed]
- 24.Seth A, Papas T S. Oncogene. 1990;5:1761–1767. [PubMed] [Google Scholar]
- 25.Valius M, Kazlauskas A. Cell. 1993;73:321–334. doi: 10.1016/0092-8674(93)90232-f. [DOI] [PubMed] [Google Scholar]
- 26.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Regenass U, Lydon N B. Proc Natl Acad Sci USA. 1995;92:2558–2562. doi: 10.1073/pnas.92.7.2558. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker B J, Lydon N B. Cancer Res. 1995;56:100–104. [PubMed] [Google Scholar]
- 28.Muller A J, Young J C, Pendergast A M, Pondel M, Landau N R, Littman D R, Witte O N. Mol Cell Biol. 1991;11:1785–1792. doi: 10.1128/mcb.11.4.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]