

Abstract
The vulnerability of the nervous system to advancing age is all too often manifest in neurodegenerative disorders such as Alzheimer's and Parkinson's diseases. In this review article we describe evidence suggesting that two dietary interventions, caloric restriction (CR) and intermittent fasting (IF), can prolong the health-span of the nervous system by impinging upon fundamental metabolic and cellular signaling pathways that regulate life-span. CR and IF affect energy and oxygen radical metabolism, and cellular stress response systems, in ways that protect neurons against genetic and environmental factors to which they would otherwise succumb during aging. There are multiple interactive pathways and molecular mechanisms by which CR and IF benefit neurons including those involving insulin-like signaling, FoxO transcription factors, sirtuins and peroxisome proliferator-activated receptors. These pathways stimulate the production of protein chaperones, neurotrophic factors and antioxidant enzymes, all of which help cells cope with stress and resist disease. A better understanding of the impact of CR and IF on the aging nervous system will likely lead to novel approaches for preventing and treating neurodegenerative disorders.
Keywords: Caloric restriction, Intermittent fasting, Aging
1. Introduction
Brain disorders of aging have recently become leading causes of disability and death, due to numerous advances in the prevention and treatment of cardiovascular disease and cancers. Several prominent risk factors for major age-related diseases, such as cardiovascular disease, type 2 diabetes and cancers, are also risk factors for many neurodegenerative diseases. These risk factors include a high calorie diet, vitamin insufficiencies (e.g. folic acid and antioxidants) and a sedentary life-style. Research efforts on neurodegenerative disorders have rapidly expanded in the past decade and those efforts have led to many promising therapeutic interventions to increase both health-span and lifespan. Many people live for eight or more decades and enjoy a well-functioning brain throughout their life-span. We therefore know that the human brain is capable of aging successfully. We are now at a stage where our knowledge of both the genetic and environmental factors which have been linked to unsuccessful brain aging, and their cellular and molecular consequences, can be utilized to provide the general population with advice on aging successfully. In this review, we will discuss two dietary strategies, caloric restriction and intermittent fasting, which could potentially be used to mediate successful aging and forestall the onset of certain neurodegenerative disorders (Fig. 1).
Fig. 1.
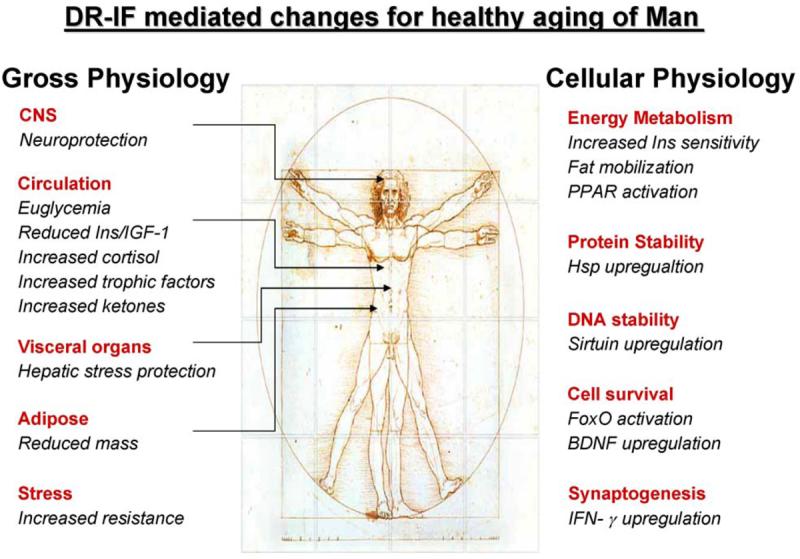
Dietary restriction and the healthy aging of man. Taking Da Vinci's Man as a paragon of humanity we have described how he may live beyond the years normally ascribed to renaissance homo sapiens through alterations in caloric intake. Both gross and cellular physiology is profoundly affected by caloric restriction (CR) or intermittent fasting (IF) regimes. With respect to gross physiology there is of course a significant reduction of body fat and mass, which supports a healthy cardiovascular system and reduces incidents of myocardial infarction. In addition to cardioprotection a greater tolerance to stress is induced in the liver, the nutrient core of homo sapiens. The presence of alternative energy stores such as ketone bodies (e.g. β-hydroxybutyrate) enable homo sapiens to survive additional stresses of life. Excessive and deleterious blood glucose is curtailed by an enhanced sensitivity to insulin (Ins) and glucose and its utilization as an energy source. The elevation of neurotrophic factors also supports the maintenance of complex neuronal circuits required for memory retention and cognition. At the molecular level many of the beneficial effects of CR/IF are recapitulated. Proteins and nucleic acids are protected from damaging post-transaltional modifications via upregulations of sirtuin histone deacetylases and heat shock proteins (Hsp). To maintain Man during the beneficial periods of fasting, peroxisome proliferator-activated receptors (PPAR) are activated to mobilize fat stores for energy usage. During these times of energy deficit, cell survival is supported by the activation of forkhead box-other (FoxO) transcription factors and through the generation of neurotrophic agents such as brain-derived neurotrophic factor (BDNF). Inflammatory cytokines, upregulated by CR/IF can even serve to allow enhanced synaptic strength during the times of energy deficit.
2. Molecular actions involved in aging and degeneration
An increasing number of genetic and environmental factors are being identified that can render neurons vulnerable to the aging process. An understanding of how such causal or predisposing risk factors promote neuronal dysfunction and/or death is critical for developing approaches to preserve functional neuronal circuits. Similarly to other organ systems, cells in the brain encounter a cumulative burden of oxidative and metabolic stress that may be a universal feature of the aging process. Increased oxidative stress during brain aging can be found in each of the major classes of cellular molecules, including proteins, lipids and nucleic acids. Some oxidative modifications of proteins that have been observed in neurons during aging include carbonyl formation (Butterfield et al., 1997; Cakatay et al., 2001; Dubey et al., 1996), covalent modifications of cysteine, lysine and histidine residues by the lipid peroxidation product 4-hydroxynonenal (Papaioannou et al., 2001), nitration of proteins on tyrosine residues (Sloane et al., 1999), and glycation (Munch et al., 2000). A common oxidative modification of DNA, observed during brain aging is the formation of 8-hydroxydeoxyguanosine (Sohal et al., 1994). Each of these modifications of proteins, lipids and nucleic acids are also exacerbated in numerous degenerative disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD). AD can be caused by mutations in the genes encoding the amyloid precursor protein (APP) and/or presenilin-1 (PS-1) or -2 (PS-2). Each of these mutations results in an increased production of amyloid-β peptide which itself can increase the oxidative burden on neurons. AD leads to a progressive deterioration of cognitive function with a loss of memory. Neuronal injury is thus present in regions of the brain that involve the hippocampus and the cortex. AD is characterized by two main pathological hallmarks that consist of extracellular plaques of amyloid-β peptide aggregates, and intracellular neurofibrillary tangles composed of the hyperphosphorylated microtubule-associated protein tau. The β-amyloid deposition that constitutes the plaques is composed of a 39–42 amino acid peptide (Aβ), which is the proteolytic product of the APP protein. Interestingly, the APP and presenilin mutations have also been shown to decrease levels of a secreted form of APP that has been shown to promote synaptic plasticity (learning and memory) and survival of neurons (Furukawa et al., 1996; Ishida et al., 1997). PD is also a relatively common progressive neurodegenerative disorder affecting approximately 1% of the population older than the age of 65 years and approximately 4–5% of the population older than the age of 85. It is caused by a selective degeneration of the dopamine neurons in the substantia nigra. PD is characterized by tremor, rigidity and slowness of movements. Non-motor features, such as dementia and dysautonomia, occur frequently, especially in the advanced stages of the disease.
3. Life-span and health-span extension by caloric restriction and intermittent fasting
Throughout history, numerous societies have recognized the beneficial effects on health and general wellbeing of limiting food intake for certain periods of time, either for religious reasons or when food was scarce. The first widely recognized scientific study of restricted diets and their ability to extend life-span was published by McCay et al. (1935). McCay showed that feeding rats with a diet containing indigestible cellulose dramatically extended both mean and maximum life-span in these animals (McCay et al., 1935). Many studies have confirmed this result and extended it to mice (Weindruch and Walford, 1988; Sprott, 1997) and other species including fruitflies (Chapman and Partridge, 1996), nematodes (Houthoofd et al., 2002), water fleas, spiders and fish (Weindruch and Walford, 1988).
In this review we will attempt to indicate how, with dietary alteration, not only can life-span be extended but also potentially, health-span, i.e. the time of our lives in which we have a disease/pathology free disposition. We shall also investigate through which molecular mechanisms the benefits, on the whole organism, of dietary intake modification are derived. Variations of this basic dietary regime, now known as caloric restriction (CR), are the most effective way of extending the life-span of mammals without genetically altering them. More recently, another variation of CR, intermittent fasting (IF) or every other day feeding (EODF), has also been shown to extend life-span and have beneficial health effects. (Sohal and Weindruch, 1996; Goodrick et al., 1982; Ingram and Reynolds, 1987). Rodents maintained on calorie-restricted diets are generally smaller and leaner and have less body fat and smaller major organs than ad libitum fed animals (Weindruch and Sohal, 1997). They are generally more active, which may relate to the need to search for food (Hart and Turturro, 1998; Martin et al., unpublished data), and the normal age-related decrease in physical activity is markedly reduced in calorie-restricted animals (Means et al., 1993). However, these animals are more vulnerable to cold temperatures (Johnson et al., 1982), which is a major source of mortality for small mammals (Berry and Bronson, 1992). The amount by which life-span is extended has been shown to increase progressively as caloric intake is reduced, until the point of starvation. The time of onset of the dietary restriction (e.g. pre- or post-pubertal) and the duration of the CR regime also determine the amount by which life-span is extended. Crucially both CR and IF can diminish the severity of risk factors for diseases such as diabetes and cardiovascular disease in rodents (Anson et al., 2003; Wan et al., 2003). In many studies, implementation of the IF dietary regime results in an approximately 20–30% reduction in caloric intake over time. Maintenance of rats on this alternate day CR feeding regimen for 2–4 months results in resistance of hippocampal neurons to chemically induced degeneration (Bruce-Keller et al., 1999a,b). This reduced damage to hippocampal neurons is also correlated with a striking preservation of learning and memory in a water maze spatial learning task. Thus these dietary regimes could have a significant benefit for debilitating and prevalent neurodegenerative disorders such as Alzheimer's, Huntington's and Parkinson's diseases.
4. Molecular mechanisms of neuroprotection by CR and IF
Data from the animal studies described in this review show that neurons in the brains of rats and mice maintained on CR or IF regimens exhibit increased resistance to oxidative, metabolic and excitotoxic insults. The critical question to ask with respect to these studies is, what are the underlying molecular mechanisms that account for the protection against this myriad of potent cellular insults? Investigators have addressed this important question by measuring numerous proteins and lipids that are known to play a role in protecting neurons against many different insults. We shall discuss and demonstrate what a complex physiological response to CR/IF occurs in the organism and how this may eventually translate to healthy aging.
4.1. Stress responses
From nature we know that the acquisition of available food forms one of the most profound behavior sets and thus removal of adequate food sources acts as a great driving force for engrained behavior and causes a certain degree of psychological and physiological stress in the organism. This paradigm, as with so many aspects of biology, extends even to the fundamental physiological and cellular processes within the organism. To exemplify this, several different stress proteins have been measured in the brains from rats maintained on either ad libitum or CR diets for 3 months. Examples of such stress proteins include heat-shock proteins and glucose-regulated proteins. These molecular chaperone proteins interact with many different proteins in cells and function to ensure their proper folding, on the one hand, and degradation of damaged proteins, on the other hand (Frydman, 2001; Gething, 1999). They may also interact with, and modify the function of, apoptotic proteins including caspases (Beere et al., 2000; Ravagnan et al., 2001). Levels of some of these chaperone proteins may be increased during the aging process as a protective response (Lee et al., 1999, 2000a,b). Cell culture and in vivo studies have shown that heat-shock protein-70 (HSP-70) and glucose-regulated protein 78 (GRP-78) can protect neurons against injury and death in experimental models of neurodegenerative disorders (Lowenstein et al., 1991; Yu and Mattson, 1999). Levels of HSP-70 and GRP-78 were found to be increased in the cortical, hippocampal and striatal neurons of the CR rats compared to the age-matched ad libitum fed animals (Lee et al., 1999; Mattson, 1998). Previous studies in this and other laboratories have provided evidence that HSP-70 and GRP-78 can protect neurons against excitotoxic and oxidative injury (Warrick et al., 1999; Yu et al., 1999), which suggests that they contribute to the neuroprotective effect of CR. These data may demonstrate that CR can induce a mild stress response in neurons, presumably due to a reduced energy, primarily glucose, availability. In addition to these subcellular stress responses it has been reported that IF results in increased levels of circulating corticosterone (Wan et al., 2003), which is usually associated positively with the stress-state of the organism. In contrast to detrimental stressors, such as chronic uncontrollable stress, which endanger neurons through glucocorticoid receptor activation, IF downregulates glucocorticoid receptors with maintenance of mineralocorticoid receptors in neurons which can act to prevent neuronal damage and death (Lee et al., 2000a,b). It may be that alternating periods of anabolism and catabolism, occurring during IF, may play a mechanistic role in triggering increases in cellular stress resistance and the repair of damaged proteins and cells.
Excessive neurological stress often takes the form of elevated levels of glutamatergic neurotransmission, e.g. in post ischemic events or epileptic seizures there can be an overload of cells with calcium, induced by the overt glutamate release that results in eventual cell death. This form of excitoxic cell death can be mimicked by the injection of kainic acid (KA) into the cerebral ventricles/brain regions of experimental animals. When the excitotoxic KA is injected into the dorsal hippocampus of mice it induces seizures and damage to pyramidal neurons in regions CA3 and CA1 (Duan et al., 2001). A significant increase in the survival of CA3 and CA1 neurons in the IF mice compared with mice fed ad libitum, after the kainic insult has been demonstrated (Anson et al., 2003).
4.2. Neurotrophic factors
As both IF and CR induce a mild stress response in brain cells this can result in the activation of compensating mechanisms, e.g. the upregulation of neurotrophic factors such as BDNF and glial cell line-derived neurotrophic factor (GDNF) as well as the aforementioned heat shock proteins (Bruce-Keller et al., 1999a,b; Duan and Mattson, 1999; Duan et al., 2003; Maswood et al., 2004). IF regimens have been demonstrated to ameliorate and attenuate neuronal damage and improve the functional outcome in animal models of neurological trauma such as stroke (Yu and Mattson, 1999) and also neurodegenerative disorders such as Parkinson's disease (Duan and Mattson, 1999), and Huntington's disease (Duan et al., 2003). The neuroprotective mechanism of IF is not known, but it has been reported that IF induces the production of brain-derived neurotrophic factor (BDNF) which was associated with increased hippocampal neurogenesis in rats and mice (Lee et al., 2002). One of the primary neuroprotective mechanisms attributed to BDNF appears to be the ability of BDNF-mediated activation of its cognate TrkB receptor which then entrains stimulation of multiple signaling pathways. Prominent amongst these TrkB signaling pathways is the phosphatidyl inositol 3-kinase (PI3K)/protein kinase B (Akt) pathway that has been implicated in several of the CR/IF protective mechanisms that will be discussed at greater length in this review.
4.3. Ketone bodies
Dietary fasting is known to result in an increased production of ketone bodies, e.g. β-hydroxybutyrate, which can be used by the organism as an energy source in the face of limited glucose availability (Mitchell et al., 1995; Vazquez et al., 1985). With respect to ketogenesis it appears that IF regimens seem to be more amenable to this energy production pathway than more strict CR protocols. Mice on IF regimens have been shown to weigh on average more than mice on CR regimens. They also have larger adipose reserves and a greater ketogenic response than CR mice. IF dietary regimes can develop a two-fold increase in the fasting serum concentration of β-hydroxybutyrate compared with mice fed ad libitum (Anson et al., 2003). This shift to ketogenesis may play a direct role in the cytoprotective effects of IF, because it has been reported that rats fed a ketogenic diet exhibit increased resistance to seizures (Bough et al., 1999), and that β-hydroxybutyrate itself can protect neurons in rodent models of Alzheimer's and Parkinson's diseases (Kashiwaya et al., 2000). Ketogenic diets, which promote a metabolic shift from glucose utilization to ketogenesis, are also prescribed for some patients with epilepsy (Gilbert et al., 2000) as this is prophylactic against the progressive excitotoxic neuronal damage and degradation that can occur if the condition is untreated.
4.4. Glucose/insulin signaling
During fasting or dietary restriction the primary alteration to the organism is the availability of glucose for oxidative respiration. The mechanisms by which energy is derived from alternate sources or how the remaining glucose is handled are germane to the extrapolation of the health benefits of CR/IF regimens. The importance of glucose handling efficiency for healthy aging can be demonstrated by the fact that glucose levels in the blood, integrated over time, have been postulated to lead to high levels of non-enzymatic glycation, a form of protein damage. CR has been shown to specifically attenuate oxyradical production and damage (Weindruch and Sohal, 1997) and non-enzymatic glycation (Cefalu et al., 1995).
Both IF and CR regimens have similar effects on insulin and glucose levels, i.e. reduction, yet interestingly they have different effects on serum IGF-1 levels and serum β-hydroxybutyrate levels, i.e. both these parameters are increased with IF compared to CR (Dunn et al., 1997; Anson et al., 2003). A longitudinal study on male rats (Masoro et al., 1992) demonstrated that CR decreased the mean 24-h plasma glucose concentration by about 15 mg/dl and the insulin concentration by about 50%. CR regime animals utilized glucose at the same rate as did the rats fed ad libitum, despite the lower plasma glucose and markedly lower plasma insulin levels. Therefore, it is proposed that CR either increases glucose effectiveness or insulin responsiveness or both, and that the maintenance of low levels of glucose and insulin control the beneficial and life-extending actions of CR. CR has also been found to reduce plasma glucose and insulin concentrations in fasting rhesus monkeys (Kemnitz et al., 1994). In addition, CR can increase insulin sensitivity in rhesus and cynomolgus monkeys (Lane et al., 1995 and Cefalu et al., 1997). A major reason for this emphasis being placed on the insulin–glucose control system in aging is the finding that loss-of-function mutations of the insulin signaling system result in life extension in three species: C. elegans (Kenyon et al., 1993; Wolkow et al., 2000), D. melanogaster (Clancy et al., 2001), and mice (Bluher et al., 2003). Overall, from many experimental studies, CR and IF seem to chronically reduce the circulating levels of insulin resulting in an eventual enhanced glucose mobilization and an enhanced insulin sensitivity, both of which serve to maintain a supply of glucose for the vital organs, central nervous system and gonads to support these critical organs in time of limited energy intake. The actual reduction of insulin receptor signaling mediated by reduced plasma insulin levels has an impact also on several other factors that profoundly impact upon the cellular response to CR/IF; this will be discussed in later sections.
4.5. Cytokines
There is mounting evidence to suggest that inflammatory processes could be critically involved in the development of age-related pathologies such as those observed in Alzheimer's disease. The activation of microglia in response to injury or during aging causes the induction of an inflammatory-like response. This response is typified and initiated by an enhanced expression of interleukin-1 in the stimulated microglia (for review see, Griffin, 2006). With this in mind it is therefore unsurprising that inflammatory cytokines may also be implicated in the CR/IF-mediated processes that ameliorate this neurodegeneration. Recent findings suggest that IFN-γ is an important mediator of neuronal plasticity, e.g. IFN-γ may enhance synaptogenesis, regulate synaptic plasticity and control neurogenesis (Brask et al., 2004; Vikman et al., 2001; Improta et al., 1988; Wong et al., 2004). It was recently reported that levels of IFN-γ are increased in circulating leukocytes of monkeys that had been maintained on a CR diet (Mascarucci et al., 2002). It has also been demonstrated that CR elevates the expression of IFN-γ in the hippocampus where it exerts an excitoprotective action of IFN-γ (Lee et al., 2006). Cytokines can also be produced by visceral organs outside the immune system and the central nervous system. Adipose tissue, which accumulates during aging and is specifically reduced upon CR or IF regimens, can act as an endocrine organ, which produces trophic hormones that are active throughout the body (Bordone and Guarente, 2005), e.g. tumour necrosis factor-α (TNFα). TNFα has also been shown to trigger insulin resistance in animals (Feinstein et al., 1993). In vitro cell-culture studies have shown that TNFα renders cells insulin resistant through a downregulation of glucose transporter synthesis as well as through interference with insulin receptor signaling pathways (Stephens et al., 1997) which we have seen are critically involved in healthy aging. In vivo, the absence of the TNFα receptor significantly improves insulin sensitivity which mimics the insulin-related effects seen in CR/IF animals. Interestingly, it has been shown that CR attenuates the age-related upregulation of nuclear factor (NF)-κB (Kim et al., 2000), which is a transcription factor that induces the expression of TNFα (Bordone and Guarente, 2005) in adipose tissue and the production of inflammatory cytokines in immune cells. Thus attenuation of TNF-α-induced insulin resistance may enhance the glucose utilization capacity of the organism, thus fending off the detrimental effects of excessive blood glucose that may occur in times of poor health and with advancing age.
4.6. Satiety and adipose-generated hormones
Leptin and adiponectin are two hormones that are typically associated with the feedback control of appetite and satiety. Both of these factors are produced by adipose tissue (Meier and Gressner, 2004) which is of course profoundly affected by CR/IF regimes. In addition to its role in satiety, leptin, released into the circulation, reduces the level of stress hormones (Barzilai and Gupta, 1999) and increases thyroid activity and thyroid-hormone levels which both result in increased energy expenditure (Legradi et al., 1997). As we have seen, CR regimens tend to upregulate stress hormones in a tolerable manner and in addition they can downregulate thyroid hormones, potentially through this attenuation of circulating leptin levels (Barzilai and Gupta, 1999). However leptin's role in mediating the beneficial effects of CR may be secondary to its satiety role as it has been demonstrated that mice that lack leptin unfortunately demonstrate a reduced life-span, compared to ad libitum animals, and are obese (Allison et al., 2001). Adiponectin has been shown to trigger increased insulin sensitivity (Meier and Gressner, 2004; Pajvani and Scherer, 2003) via upregulation of AMP-activated protein kinase (AMPK: Wu et al., 2003). This kinase regulates glucose and fat metabolism in muscle in response to energy limitation (Musi et al., 2001), and has been shown to protect neurons against metabolic stress (Culmsee et al., 2001). Importantly, adiponectin levels rise during CR, which suggests that this adipose-derived hormone might also have an important contributory role in the physiological shift to an enhanced insulin sensitivity in these animals (Combs et al., 2003). Recent findings show that mice that have been genetically engineered to be lean live longer. Indeed, tissue-specific knockout of the insulin receptor in adipose cells prevents the tissue from storing fat, which gives rise to lean animals that live significantly longer than wild-type mice (Bluher et al., 2003). These data suggest that visceral adipose might be especially important in driving insulin resistance and pathogenesis (Bjorntorp, 1991).
4.7. Sirtuins
As lower organisms, e.g. yeast and nematode worms, possess a considerably shorter life-span than mammals they have proved useful for the discovery of the molecular determinants of healthy longevity. It has become apparent that amongst the multiple factors that have been identified that control life-span in these lower organisms, many of these also link the alteration of caloric intake to the increase in health-span so desired by dietary interventions of disease processes.
One of the primary genetic determinants of replicative life-span to emerge from genetic studies in yeast is the silent information regulator 2 (SIR2). The SIR2 gene was denoted because it mediates a specific gene silencing action (Rine and Herskowitz, 1987). Inhibitory mutations of SIR2 can shorten life-span, and increased gene dosage of SIR2 extended life-span (Kaeberlein et al., 1999). The SIR2 ortholog in C. elegans was similarly shown to be a key determinant of the life-span in that organism (Tissenbaum and Guarente, 2001). As yeast and C. elegans diverged from a common ancestor about one billion years ago this may suggest that descendants of that ancestor, including mammals, will possess SIR2-related genes involved in regulating their life-span. As dietary regulation has also shown to be a powerful modulator of life-span it is reasonable to speculate that CR/IF and SIR2 genes may converge to play an important role in these multiple and complex physiological pathways. Mammalian homologues of the yeast SIR2 gene have subsequently been found and interestingly the SIR2 ortholog, SIRT1, may in part mediate a broad array of physiological effects that occur in animals on a modified diet, beit CR or IF. The family of proteins discovered that are encoded for by the mammalian SIR2 homologues are collectively termed sirtuins. Several recent reports have shown increases in SIRT1 protein levels in response to food deprivation (Nemoto et al., 2004; Cohen et al., 2004). In addition SIR upregulation has been shown in response to cell stressors, such as high osmolarity (Lin et al., 2002), thus the sirtuin family of proteins could be actively regulated by the mild, controllable stress induced by CR/IF. Sirtuins possess a relatively rare enzymatic capacity as they are NAD-dependent histone deacetylases (Imai et al., 2000; Landry et al., 2000). The mammalian SIRT1 gene product enzyme can, in addition to histones, deacetylate many other substrates. In this regard, SIRT1 was recently shown to deacetylate and downregulate NF-κB (Yeung et al., 2004). It is intriguing to speculate that the upregulation of SIRT1 by CR contributes to the observed increase in insulin sensitivity and reduction in inflammation, potentially through the control of the NF-κ/TNFα pathways.
Lin et al. (2002, 2004) have proposed a molecular pathway for SIR2 activation that potentially connects alterations in caloric intake to life-span extension. Upon CR/IF there is an initial increase in oxygen consumption and respiration, at the expense of fermentative processes. Fermentation is a typical mechanism by which cells can generate ATP and also store excess energy in the form of ethanol when glucose is abundant. This metabolic shift triggers a concomitant reduction in NADH levels. NADH acts as a competitive inhibitor of SIR2, so its reduction during CR/IF periods would be expected to upregulate the enzyme and thereby extend the organism's life-span in line with yeast and C. elegans studies. Consistent with this, ablation of mitochondrial electron transport blocked the effect of CR on life-span, and overexpressing NADH dehydrogenase, the enzyme that shunts electrons from NADH to the electron transport chain, increased the animal's life-span. Thus it appears then that CR/IF induces a more efficient use of glucose via an increase in respiration. In addition to this there is a transition in muscle cells from using glucose, which is to some extent, metabolized in ad libitum animals fermentatively (producing lactate), toward the use of fatty acids, which are oxidatively metabolized. This shift spares glucose for the brain, preventing neurodegeneration, and correlates with the characteristic enhancement of insulin sensitivity in muscle and liver seen in CR. Although the actions of sirtuins in the nervous system are only beginning to be explored, it has been reported that SIR2 (SIRT1 in mammals) activation through increased gene dosage or treatment with the sirtuin activator resveratrol can protect neurons against the pathogenic effects of polyglutamine-expanded huntingtin proteins in worm and mouse models of Huntington's disease (Parker et al., 2005).
Sirtuins also seem to play a role in mediating the effective role of adipose tissue in the physiological transference of the benefits of CR/IF regimes to the organism. One of the most important regulators of adipose tissue function is the peroxisome proliferator-activated transcription factor receptor gamma (PPARγ: Tontonoz et al., 1994). This receptor acts as a nuclear transcription factor that controls multiple genes connected to cell survival and responses to metabolic alterations. One PPARγ gene target, the aP2 gene, encodes a protein that assists fat storage. SIRT1 can act as a repressor of PPARγ, thereby downregulating genes such as the mouse aP2 gene (Picard et al., 2004). During fasting SIRT1 activation is followed by an enhanced binding to the aP2 promoter in adipose tissue. This causes a repression of aP2 gene expression causing an eventual promotion of fat mobilization into the blood to aid the organism's energy balance. Therefore, according to Bordone and Guarente (2005), upon manipulation of caloric intake there is a reactionary activation of SIRT1 in adipose tissue, which acts to reduce fat stores and probably resets hormonal levels to change the pace of aging. This strategy also makes evolutionary sense when it is considered that successful reproduction is also regulated by body fat and is shut off during CR, only to resume when available energy supplies become more abundant.
4.8. Peroxisome proliferator-activated receptor (PPAR) and co-factors
PPARs, as we have seen, are members of the nuclear hormone receptor subfamily of transcription factors. PPARs form functional heterodimers with retinoid X receptors (RXRs) and these heterodimers regulate transcription of various genes. There are three known subtypes of PPARs, α, δ and γ. These nuclear receptor transcription factors regulate genes involved in nutrient transport and metabolism as well as resistance to stress. PPARs themselves also recruit other proteins in addition to the RXR to mediate their complete function. One such protein is the peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1 (PGC-1). This coactivator has been shown to be closely regulated by dietary alteration in lower organisms and higher mammals. PGC-1 exists in two isoforms, α and β, and these isoforms have emerged as prominent regulators of the adaptive responses to caloric deprivation. PGC-1 regulates the ligand-dependent and -independent activation of a large number of nuclear receptors including the PPARs. There has been reported an age-dependent reduction in PGC-1α (Ling et al., 2004) which may exacerbate the aging process. However in mice and primates CR has been shown to reverse this age-dependent decrease in PGC-1α, PPAR and regulated genes (Weindruch et al., 2002; Kayo et al., 2001).
PGC-1α, the first PGC family member identified was characterized as a protein that interacts with the PPARγ to regulate brown fat differentiation during adaptation to cold stress (Puigserver et al., 1998). This cold stress may be regarded as analogous to the physiological and psychological stress induced by caloric restriction. During CR/IF periods, when insulin levels are low, PGC-1α and PGC-1β gene expression is enhanced in rodents (Puigserver and Spiegelman, 2003; Herzig et al., 2001). PGC-1α was also induced in the livers of mice (Corton et al., 2004) and rats (Zhu et al., 2004) after longer term CR. PGC-1α and β can coordinately regulate genes involved in gluconeogenesis and fatty acid β-oxidation in a number of organs during fasting (Lin et al., 2002a, 2004a; Kamei et al., 2003; Kressler et al., 2002). Both these processes are beneficial to the maintenance of a healthy energy balance in times of limited food. Hence through PPAR activation extra supplies of glucose can be mobilized and alternate energy sources can be exploited. As well as PGC regulation during fasting, PPARα is also upregulated by fasting in liver, small and large intestine, thymus (Escher et al., 2001), and pancreas (Gremlich et al., 2005). A large number of genes involved in fatty acid β-oxidation, known to be regulated by PPARα are also increased in expression in response to fasting. During periods of fasting PPARα knock-out mice exhibit an inability to regulate genes involved in fatty acid β- and ω-oxidation and ketogenesis in the liver, kidney and heart along with lack of control of blood levels of glucose or ketone bodies bodies (Kroetz et al., 1998; Sugden et al., 2001; Leone et al., 1999).
Not only is the liver the energy-regulating core of mammals but it also represents one of the most significant stores of glycogen, nutrients and vitamins. One would therefore expect that there would be a critical link between alterations of caloric intake and resultant hepatic function. Thus it has been shown that CR protects the liver from a wide range of environmental stressors, many of which induce damage through circulating inflammatory mediators (Kim et al., 2002; Bokov et al., 2004). PPARα has been shown to regulate hepatic responses to diverse forms of stress. Mice pre-exposed to PPARα agonists exhibit decreased cellular damage, increased tissue repair, and decreased mortality after exposure to a number of physical and chemical hepatic stressors (Anderson et al., 2002; Wheeler et al., 2003). It appears that functional PPARαs are crucial for the CR-mediated protection of the liver from damage induced by hepatotoxicants like thioacetamide. Specifically, it was demonstrated by Corton et al. (2004) that PPARα knock-out mice, in contrast to wild-type mice, were not protected from thioacetamide by CR regimes. Lipid peroxide levels, associated with oxidative cell stress in the periphery and the central nervous system, are also significantly increased in aging. PPARα knock-out mice show a marked elevation in lipid peroxidation products compared to wild-type mice (Poynter and Daynes, 1998). Thus PPARα may influence aging through the regulation of multiple damage and repair processes after exposure to a plethora of endogenous or environmental stressors.
PGC-1 isoforms are transcriptionally or posttranslationally regulated in mammals by several signaling pathways implicated in the connection between CR/IF and life-span extension. These include forkhead box “other” (FoxO) transcription factors (through an insulin/insulin-like growth factor-I -dependent pathway), glucagon-stimulated cellular AMP (cAMP) response element binding protein (CREB), stress-activated protein kinases (p38 and c-jun N-terminal kinase) and unsurprisingly SIRT1. We shall discuss next how these factors interact to control the molecular mechanisms of CR/IF that impact upon translation to healthy aging.
4.9. FoxO transcription factors
In mammals, insulin and IGF-I bind to either insulin or IGF-1 receptors activating multiple signaling pathways. With respect to the aging process and the amelioration of degenerative disorders it seems that the most important pathway entrained by insulin/IGF-1 is the canonical phosphatidylinositol 3-kinase (PI3K) and serine–threonine protein kinases (Akt-1/Akt-2/protein kinase B [PKB]) signaling cascade. In C. elegans, this pathway determines responses to longevity and environmental stress (Guarente and Kenyon, 2000). Mutations in C. elegans which inactivate the insulin/IGF-I pathway, including Daf-2, the receptor for insulin/IGF-I or the PI3K ortholog Age-1, increase life-span as well as temperature and oxidative stresses. These effects require reversal of negative regulation of the stress resistance factor, Daf-16 (Libina et al., 2003). Daf-16 encodes a transcription factor containing a “forkhead” DNA binding domain. Overexpression of Daf-16 in worms (Henderson and Johnson, 2001) or an ortholog in flies (Giannakou et al., 2004) significantly extends their life-span. Daf-16 regulates the expression of an array of genes involved in xenobiotic metabolism and stress resistance (Murphy et al., 2003). Mammalian homologs of Daf-16 fall into the family of FoxO factors. There are four main groups of mammalian FoxOs, FoxO1, FoxO3, FoxO4 and FoxO6. FoxO transcription factors belong to the larger Forkhead family of proteins, a family of transcriptional regulators characterized by the conserved ‘forkhead box’ DNA-binding domain (Kaestner et al., 2000). These FoxO proteins control a wide array of genes that all are linked by a common mechanism in that they serve to control energy metabolim in the organism in response to environmental changes, e.g. restriction of available food. For example FoxOs control genes involved in glucose metabolism (glucose 6-phosphatase and phosphoenolpyruvate carboxylase: Nakae et al., 2001; Yeagley et al., 2001); cell death (Fas-ligand), reactive oxygen species detoxification (catalse and manganese superoxide dismutase, Kops et al., 2002) and DNA repair (growth arrest and DNA damage-inducible protein 45 and damage-specific DNA-binding protein 1, Tran et al., 2002).
Insulin receptor stimulation, during caloric intake, leads to activation of the PI3K/Akt pathway and resultant phosphorylation of FoxOs in mammals. Phosphorylated FoxO factors are recognized by 14-3-3 proteins which facilitate their transport out of the nucleus, reducing their transcriptional activity. Thus upon CR/IF there is a complex interplay between activation and inactivation of these FoxO factors. There are potentially beneficial effects of FoxO activation and inactivation depending upon the prevailing cellular conditions. Mammalian FoxO family members carry out functions that determine cell survival during times of stress including regulation of apoptosis, cell-cycle checkpoint control, and oxidative stress resistance (Coffer, 2003; Furukawa-Hibi et al., 2002). Activation of FoxO3 or FoxO4 leads to increases in cell-cycle G1 arrest (van der Horst et al., 2004) and increases in apoptosis (Motta et al., 2004) presumably as a way to eliminate cells damaged by oxidative stress. Thus alterations in the capacity to activate the PI3K/Akt pathways can have dramatic effects upon cell survivability and this process may be critical in transferring the positive effects of CR/IF to the organism. CR uncouples insulin/IGF-I signaling to FoxO factors by markedly reducing plasma IGF-I and insulin levels in rats (Sonntag et al., 1999). These decreases in circulating insulin/IGF-I levels result in decreased Akt phosphorylation in liver (Al-Regaiey et al., 2005) and decreased PI3K expression in muscle (Argentino et al., 2005). In addition there is a compensatory increase in the expression of FoxO family members by fasting (Imae et al., 2003; Furuyama et al., 2003) or CR (Al-Regaiey et al., 2005; Tsuchiya et al., 2004). Therefore, when insulin signaling is decreased, e.g. during CR/IF there are not only increases in nuclear/cytoplasmic FoxO ratios but FoxO factor expression as well (Imae et al., 2003; Furuyama et al., 2003; Al-Regaiey et al., 2005; Tsuchiya et al., 2004). Overall, multiple studies have revealed that downregulation of insulin/IGF-I signaling results in increases in the activity of FoxO factors, that critically regulate cell survival mechanisms, and that these alterations are found consistently in many diverse models of longevity among different species.
Many of the genes regulated by FoxOs are similarly regulated by the tumor suppressor p53, which has led to the speculation that these two genes may work in concert to prevent both deleterious aging and tumor growth. Consistent with this possibility, p53 and FoxO are both phosphorylated and acetylated in response to oxidative stress stimuli and UV radiations (Vousden and Lu, 2002; Brunet et al., 2004). In addition, both p53 and FoxOs bind to SIRT1 deacetylase (Luo et al., 2001; Vaziri et al., 2001). FoxO and p53 seem to be functionally linked as p53 can inhibit FoxO function by inducing serum and glucocorticoid induced kinase (SGK) -mediated phosphorylation of FoxO3 resulting in its relocation from the nucleus to the cytoplasm (You et al., 2004). FoxO3 has been found to prevent p53 from repressing SIRT1 gene expression. FoxO-induced repression of p53 appears to be mediated by the direct interaction between FoxO3 and p53 (Nemoto et al., 2004). That FoxO factors induce SIRT1 expression is consistent with the observation that SIRT1 expression is increased in rodent tissues when insulin and IGF-1 are lowered by CR (Cohen et al., 2004). In turn, SIRT1 itself can bind to and deacetylate p53 and FoxO transcription factors, controlling their activity. Mice harboring a mutation, which results in the activation of p53, display a significant reduction of life-span and exhibit signs of premature aging (Tyner et al., 2002). Interestingly, while activation of p53 in these mouse models reduces life-span, p53 activation still allows an increased resistance to cancer (Tyner et al., 2002), demonstrating that p53 causes tumor suppression at the expense of longevity.
One of the most important recent fields of caloric restriction study is the demonstration that CR may be able to prevent the generation of multiple forms of cancer itself. For example, in mice with genetically attenuated p53 levels CR increased the latency of spontaneous tumor development (mostly lymphomas) by approximately 75% (Hursting et al., 2001). It is therefore clear that there is a subtle and complicated relationship between these related factors that are linked together by changes in dietary energy intake.
In addition to negative regulation by insulin/IGF-1 signaling and p53, FoxO factors are regulated by the CREB binding protein (CBP) and a related protein, p300. Interestingly, cellular overexpression of CBP (Daitoku et al., 2004) or p300 (Fukuoka et al., 2003) enhances the ability of FoxO factors to activate functional gene expression. SIRT1 again seems to play a central role in adaptive changes to energy regulation as it can reverse the negative regulation of FoxO family members by CBP. Like PGC-1, SIRT1 levels are increased during CR in rat liver and are negatively regulated by insulin and IGF-I (Cohen et al., 2004). Additionally, the related family member SIRT3, a mitochondrial protein, exhibits increased expression in white and brown fat upon CR (Shi et al., 2005).
FoxOs seem to exist at a nexus between mechanisms that connect cellular stress responses to eventual survival mechanisms. For instance the stress-related protein kinase cJun N-terminal kinase 1 (JNK-1), which serves as a molecular sensor for various stressors actively can control FoxO transcriptional action. In C. elegans, JNK-1 directly interacts with and phosphorylates the FoxO homologue Daf-16, and in response to heat stress, JNK-1 promotes the translocation of Daf-16 into the nucleus. Overexpression of JNK-1 in C. elegans leads to increases in life-span and increased survival after heat stress (Oh et al., 2005). In D. melanogaster as well, mild activation of JNK leads to increased stress tolerance and longevity (Wang et al., 2003) dependent on an intact FoxO (Wang et al., 2005).
In conclusion it seems that FoxO transcription factors are promising candidates to serve as molecular links between dietary modifications and longevity. In conditions such as CR/IF where the circulating levels of insulin/IGF-1 are attenuated to improve euglycemia, FoxO nuclear translocation results in the upregulation of a series of target genes that promote cell cycle arrest, stress resistance, and apoptosis. External stressful stimuli also trigger the relocalization of FoxO factors into the nucleus, thus allowing an adaptive response to stress stimuli. Consistent with the notion that stress resistance is highly coupled with life-span extension, activation of FoxO transcription factors in worms and flies increases longevity. FoxO proteins translate environmental stimuli, including the stress induced by caloric restriction into changes in gene expression programs that may coordinate organismal healthy aging and eventual longevity.
5. Caloric restriction in humans?
We are approaching a comprehensive understanding of the various molecular mechanisms by which changes in caloric intake can be transferred to an enhanced survival of cells during the aging process. However the question remains whether CR and IF will have beneficial effects on humans. To date, there have been no well-controlled scientific studies to determine the effects of long-term CR on humans. Currently there are studies ongoing involving 30% CR in non-human primates (rhesus monkeys) and data so far from these studies look promising, in that they have supported the life- and health-extending properties of this dietary regime (Lane et al., 1995, 1996).
However, the excessive loss of body fat and the concomitant decline in sex steroids can lead to menstrual irregularities, amenorrhea, bone thinning and the development of osteoporosis in females. Perhaps a variation of the CR/IF protocols in which there is a milder caloric restriction combined with a change in feeding frequency may have a greater likelihood of compliance amongst human subjects. Hopefully this more gentle alteration of dietary food intake will still retain the benefits of the experimental regimes employed so far. It is worth noting that to date most studies using CR have compared the beneficial effects of CR to overweight (or even obese) age-matched control animals. It is unclear whether animals with a healthy bodyweight, that are able to partake in regular exercise and have some form of mental stimulation (as they would do in the wild), would benefit from a CR regime. Recent studies carried out with human subjects, subject to 25% CR, are however attempting to address this as they are employing control subjects with normal body-mass indices.
The development of a chemical CR mimetic may be a promising therapeutic avenue for the treatment of neurodegenerative diseases and to delay the aging process, as it would provide similar health benefits to CR (such as extending health- and life-span), while circumventing the long-term need to reduce food intake. However, it remains to be seen whether a CR mimetic would be a feasible drug to produce, especially since the appreciation of the processes whereby CR exerts its protective effects are still somewhat incomplete and the underlying mechanisms are proving to be very complex. One must also not discount the psychological effects of food intake in higher, more introspective, organisms such as humans. We possess an almost unique emotional connection with a huge variety of foodstuffs. Therefore removal of this psychological succour, during a CR/IF-like regime may partially counteract the physiological benefits of these paradigms.
The main factor that may negate the widespread implementation of CR/IF as an effective geronto-therapeutic is potentially the modern Western lifestyle of near constant work and persistently high stress levels. Hence, to build the society and technological advances that we are used to, we have left behind the feeding patterns of our ancient ancestors in favor of constant mental activity and limited physical exercise. Due to increases in our day to day activity we have an increased energy (mainly glucose) requirement while our physiology is largely still geared to a feast and famine pattern of energy intake characteristic of our hunter-gatherer homo sapiens ancestors. This dilemma between our modern society/behavior and our ancient physiology will represent a recurring problem for gerontology for years to come. Hopefully, with our rapidly advancing appreciation of our aging process we will not need to wait for our physiological evolution to catch up with our lifestyle.
Acknowledgement
This work was supported by the National Institute on Aging Intramural Research Program.
References
- Allison DB, Miller RA, Austad SN, Bouchard C, Leibel R, Klebanov S, Johnson T, Harrison DE. Genetic variability in responses to caloric restriction in animals and in regulation of metabolism and obesity in humans. J. Gerontol. A Biol. Sci. Med. Sci. 2001;56:55–65. doi: 10.1093/gerona/56.suppl_1.55. [DOI] [PubMed] [Google Scholar]
- Al-Regaiey KA, Masternak MM, Bonkowski M, Sun L, Bartke A. Long-lived growth hormone receptor knockout mice: interaction of reduced IGF-1/insulin signaling and caloric restriction. Endocrinology. 2005;146:851–860. doi: 10.1210/en.2004-1120. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Yoon L, Richard EB, Dunn CS, Cattley RC, Corton JC. Delayed liver regeneration in peroxisome proliferator-activated receptor-alpha-null mice. Hepatology. 2002;36:544–554. doi: 10.1053/jhep.2002.35276. [DOI] [PubMed] [Google Scholar]
- Anson RM, Guo Z, de Cabo R, Iyun T, Rios M, Hagepanos A, et al. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6216–6220. doi: 10.1073/pnas.1035720100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argentino DP, Dominici FP, Munoz MC, Al-Regaiey K, Bartke A, Turyn D. Effects of long-term caloric restriction on glucose homeostasis and on the first steps of the insulin signaling system in skeletal muscle of normal and Ames dwarf (Prop1(df)/Prop1(df)) mice. Exp. Gerontol. 2005;40:27–35. doi: 10.1016/j.exger.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Barzilai N, Gupta G. Revisiting the role of fat mass in the life extension induced by caloric restriction. J. Gerontol. A Biol. Sci. Med. Sci. 1999;54:B89–B96. doi: 10.1093/gerona/54.3.b89. [DOI] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell. Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Berry RJ, Bronson FH. Life-history and bioeconomy of the house mouse. Biol. Rev. Cambridge Philos. Soc. 1992;67:519–550. doi: 10.1111/j.1469-185x.1992.tb01192.x. [DOI] [PubMed] [Google Scholar]
- Bjorntorp P. Metabolic implications of body fat distribution. Diab. Care. 1991;14:1132–1143. doi: 10.2337/diacare.14.12.1132. [DOI] [PubMed] [Google Scholar]
- Bough KJ, Valiyil R, Han FT, Eagles DA. Seizure resistance is dependent upon age and calorie restriction in rats fed a ketogenic diet. Epilepsy Res. 1999;35:21–28. doi: 10.1016/s0920-1211(98)00125-9. [DOI] [PubMed] [Google Scholar]
- Brask J, Kristensson K, Hill RH. Exposure to interferon-gamma during synaptogenesis increases inhibitory activity after a latent period in cultured rat hippocampal neurons. Eur. J. Neurosci. 2004;19:3193–3201. doi: 10.1111/j.0953-816X.2004.03445.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann. Neurol. 1999a;45:8–15. [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299:572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- Bokov A, Chaudhuri A, Richardson A. The role of oxidative damage and stress in aging. Mech. Ageing Dev. 2004;125:811–826. doi: 10.1016/j.mad.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat. Rev. Mol. Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Umberger G, McFall R, Mattson MP. Food restriction reduces brain damage and improves behavioral outcome following excitotoxic and metabolic insults. Ann. Neurol. 1999b;45:8–15. [PubMed] [Google Scholar]
- Butterfield DA, Howard BJ, Yatin S, Allen KL, Carney JM. Free radical oxidation of brain proteins in accelerated senescence and its modulation by N-tert-butyl-alpha-phenylnitrone. Proc. Natl. Acad. Sci. U.S.A. 1997;94:674–678. doi: 10.1073/pnas.94.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakatay U, Telci A, Kayali R, Tekeli F, Akcay T, Sivas A. Relation of oxidative protein damage and nitrotyrosine levels in the aging rat brain. Exp. Gerontol. 2001;36:221–229. doi: 10.1016/s0531-5565(00)00197-2. [DOI] [PubMed] [Google Scholar]
- Cefalu WT, Bell-Farrow AD, Wang ZQ, Sonntag WE, Fu MX, Baynes JW, Thorpe SR. Caloric restriction decreases age-dependent accumulation of the glycoxidation products, N epsilon-(carboxymethyl)-lysine and pentosidine, in rat skin collagen. J. Gerontol. A Biol. Sci. Med. Sci. 1995;50:B337–B341. doi: 10.1093/gerona/50a.6.b337. [DOI] [PubMed] [Google Scholar]
- Cefalu WT, Wagner JD, Wang ZQ, Bell-Farrow AD, Collins J, Haskell D, Bechtold R, Morgan T. A study of caloric restriction and cardiovascular aging in cynomolgus monkeys (Macaca fascicularis): a potential model for aging research. J. Gerontol. Biol. Sci. 1997;52A:B10–B19. doi: 10.1093/gerona/52a.1.b10. [DOI] [PubMed] [Google Scholar]
- Chapman T, Partridge L. Female fitness in Drosophila melanogaster and interaction between the effect of nutrition and of encounter rate with males. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1996;263:755–759. doi: 10.1098/rspb.1996.0113. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stacker H, Hafen E, Leevers SJ, Partridge L. Extension of life span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Coffer P. OutFOXing the grim reaper: novel mechanisms regulating longevity by forkhead transcription factors. Sci. 2003 doi: 10.1126/stke.2003.201.pe39. STKE PE39. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Combs TP, Berg AH, Rajala MW, Klebanov S, Iyengar P, Jimenez-Chillaron JC, Patti ME, Klein SL, Weinstein RS, Scherer PE. Sexual differentiation, pregnancy, calorie restriction, and aging affect the adipocyte-specific secretory protein adiponectin. Diabetes. 2003;52:268–276. doi: 10.2337/diabetes.52.2.268. [DOI] [PubMed] [Google Scholar]
- Corton JC, Apte U, Anderson SP, Limaye P, Yoon L, Latendresse J, Dunn C, Everitt JI, Voss KA, Swanson C, Kimbrough C, Wong JS, Gill SS, Chandraratna RAS, Kwak M-K, Kensler TW, Stulnig TM, Steffensen KR, Gustafsson J-A, Mehendale HA. Mimetics of caloric restriction include agonists of lipid-activated nuclear receptors. J. Biol. Chem. 2004;279:46204–46212. doi: 10.1074/jbc.M406739200. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J. Mol. Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. U.S.A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Lee J, Guo Z, Mattson MP. Dietary restriction stimulates BDNF production in the brain and thereby protects neurons against excitotoxic injury. J. Mol. Neurosci. 2001;16:1–12. doi: 10.1385/JMN:16:1:1. [DOI] [PubMed] [Google Scholar]
- Duan W, Mattson MP. Dietary restriction and 2-deoxyglucose administration improve behavioral outcome and reduce degeneration of dopaminergic neurons in models of Parkinson's disease. J. Neurosci. Res. 1999;57:195–206. doi: 10.1002/(SICI)1097-4547(19990715)57:2<195::AID-JNR5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey A, Forster MJ, Lal H, Sohal RS. Effect of age and caloric intake on protein oxidation in different brain regions and on behavioral functions of the mouse. Arch. Biochem. Biophys. 1996;333:189–397. doi: 10.1006/abbi.1996.0380. [DOI] [PubMed] [Google Scholar]
- Dunn SE, Kari FW, French J, Leininger JR, Travlos G, Wilson R, Barrett JC. Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res. 1997;57:4667–4672. [PubMed] [Google Scholar]
- Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142:4195–4202. doi: 10.1210/endo.142.10.8458. [DOI] [PubMed] [Google Scholar]
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J. Biol. Chem. 1993;268:26055–26058. [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Ann. Rev. Biochem. 2001;70:603–647. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Daitoku H, Hatta M, Matsuzaki H, Umemura S, Fukamizu A. Negative regulation of forkhead transcription factor AFX (Foxo4) by CBP-induced acetylation. Int. J. Mol. Med. 2003;12:503–508. [PubMed] [Google Scholar]
- Furukawa K, Barger SW, Blalock EM, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid precursor protein. Nature. 1996;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J. Biol. Chem. 2002;277:26729–26732. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003;375:365–371. doi: 10.1042/BJ20030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ. Role and regulation of the ER chaperone BiP. Semin. Cell. Dev. Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 2004;305:361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- Gilbert DL, Pyzik PL, Freeman JM. The ketogenic diet: seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. J. Child Neurol. 2000;15:787–790. doi: 10.1177/088307380001501203. [DOI] [PubMed] [Google Scholar]
- Goodrick CL, Ingram DK, Reynolds MA, Freeman JR, Cider NL. Effects of intermittent feeding upon growth and life span in rats. Gerontology. 1982;28:233–241. doi: 10.1159/000212538. [DOI] [PubMed] [Google Scholar]
- Gremlich S, Nolan C, Roduit R, Burcelin R, Peyot M-L, Delghingaro-Augusto V, Desvergne B, Michalik L, Prentki M, Wahli W. Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor atranscriptional up-regulation of fatty acid oxidation. Endocrinology. 2005;146:375–382. doi: 10.1210/en.2004-0667. [DOI] [PubMed] [Google Scholar]
- Griffin WS. Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 2006;83:470–474. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature. 2000;408:255–262. doi: 10.1038/35041700. [DOI] [PubMed] [Google Scholar]
- Hart RW, Turturro A. Evolution and dietary restriction. Exp. Gerontol. 1998;33:53–60. doi: 10.1016/s0531-5565(97)00063-6. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. Daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Houthoofd K, Braeckman BP, Lenaerts I, Brys K, De Vreese A, V an Eygen S, Vanfleteren JR. Axonic growth up-regulates mass-specific metabolic rate, stress resistance, and extends life-span in Caenorhabditis elegans. Exp. Gerontol. 2002;37:1371–1378. doi: 10.1016/s0531-5565(02)00173-0. [DOI] [PubMed] [Google Scholar]
- Hursting SD, Perkins SN, Donehower LA, Davis BJ. Cancer prevention studies in p53-deficient mice. Toxicol. Pathol. 2001;29:137–141. doi: 10.1080/019262301301418946. [DOI] [PubMed] [Google Scholar]
- Imae M, Fu Z, Yoshida A, Noguchi T, Kato H. Nutritional and hormonal factors control the gene expression of FoxOs, the mammalian homologues of DAF-16. J. Mol. Endocrinol. 2003;30:253–262. doi: 10.1677/jme.0.0300253. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Improta T, Salvatore AM, Di Luzio A, Romeo G, Coccia EM, Calissano P. IFN-gamma facilitates NGF-induced neuronal differentiation in PC12 cells. Exp. Cell. Res. 1988;179:1–9. doi: 10.1016/0014-4827(88)90342-4. [DOI] [PubMed] [Google Scholar]
- Ingram DK, Reynolds MA. The relationship of bodyweight to longevity within laboratory rodent species. Basic Life Sci. 1987;42:247–282. doi: 10.1007/978-1-4613-1939-9_18. [DOI] [PubMed] [Google Scholar]
- Ishida A, Furukawa K, Keller JN, Mattson MP. Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD, and enhances LTP in hippocampal slices. Neuroreport. 1997;8:2133–2137. doi: 10.1097/00001756-199707070-00009. [DOI] [PubMed] [Google Scholar]
- Johnson TS, Murray S, Young JB, Landsberg L. Restricted food intake limits brown adipose tissue hypertrophy in cold exposure. Life Sci. 1982;30:1423–1426. doi: 10.1016/0024-3205(82)90555-0. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y, Ohizumi H, Fujitani Y, Nemoto T, Tanaka T, Takahashi N, Kawada T, Miyoshi M, Ezaki O, Kakizuka A. PPARgamma coactivator 1beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc. Natl. Acad. Sci. U.S.A. 2003;100:12378–12383. doi: 10.1073/pnas.2135217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. D-beta-hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5440–5444. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayo T, Allison DB, Weindruch R, Prolla TA. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5093–5098. doi: 10.1073/pnas.081061898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemnitz W, Roecker EB, Weindruch R, Elson DF, Baum ST, Bergmann RN. Dietary restriction increases insulin sensitivity and lowers blood glucose in rhesus monkeys. Am. J. Physiol. 1994;266:E540–E547. doi: 10.1152/ajpendo.1994.266.4.E540. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch A, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim KW, Yu BP, Chung HY. The effect of age on cyclooxygenase-2 gene expression: NF-κB activation and IκBα degradation. Free Radic. Biol. Med. 2000;28:683–692. doi: 10.1016/s0891-5849(99)00274-9. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Jung KJ, Yu BP, Cho CG, Choi JS, Chung HY. Modulation of redox-sensitive transcription factors by calorie restriction during aging. Mech. Ageing Dev. 2002;123:1589–1595. doi: 10.1016/s0047-6374(02)00094-5. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Kressler D, Schreiber SN, Knutti D, Kralli A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor alpha. J. Biol. Chem. 2002;277:13918–13925. doi: 10.1074/jbc.M201134200. [DOI] [PubMed] [Google Scholar]
- Kroetz DL, Yook P, Costet P, Bianchi P, Pineau T. Peroxisome proliferator-activated receptor alpha controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J. Biol. Chem. 1998;273:31581–31589. doi: 10.1074/jbc.273.47.31581. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Baer DJ, Rumpler WV, Weindruch R, Ingram DK, Tilmont EM, Cutler RG, Roth GS. Calorie restriction lowers body temperature in rhesus monkeys, consistent with a postulated anti-aging mechanism in rodents. Proc. Natl. Acad. Sci. U.S.A. 1996;93:4159–4164. doi: 10.1073/pnas.93.9.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Ball SS, Ingram DK, Cutler RG, Engel J, Read V, Roth GS. Diet restriction in rhesus monkeys lowers fasting glucose-stimulated glucoregulatory end points. Am. J. Physiol. 1995;268:E941–E948. doi: 10.1152/ajpendo.1995.268.5.E941. [DOI] [PubMed] [Google Scholar]
- Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000a;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- Lee J, Bruce-Keller AJ, Kruman Y, Chan SL, Mattson MP. 2-deoxy-d-glucose protects hippocampal neurons against excitotoxic and oxidative injury: evidence for the involvement of stress proteins. J. Neurosci. Res. 1999;57:48–61. doi: 10.1002/(SICI)1097-4547(19990701)57:1<48::AID-JNR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Lee J, Herman JP, Mattson MP. Dietary restriction selectively decreases glucocorticoid receptor expression in the hippocampus and cerebral cortex of rats. Exp. Neurol. 2000b;166:435–441. doi: 10.1006/exnr.2000.7512. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002;82:1367–1375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim SJ, Son TG, Chan SL, Mattson MP. Interferon-gamma is up-regulated in the hippocampus in response to intermittent fasting and protects hippocampal neurons against excitotoxicity. J. Neurosci. Res. 2006 doi: 10.1002/jnr.20831. E-publication. [DOI] [PubMed] [Google Scholar]
- Legradi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1997;138:2569–2576. doi: 10.1210/endo.138.6.5209. [DOI] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 2002a;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, Cui L, Manieri M, Donovan MX, Wu Z, Cooper MP, Fan MC, Rohas LM, Zavacki AM, Cinti S, Shulman GI, Lowell BB, Krainc D, Spiegelman BM. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004a;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes. Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck-Nielsen H, Groop L, Vaag A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J. Clin. Invest. 2004;114:1518–1526. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein DH, Chan PH, Miles MF. The stress protein response in cultured neurons: characterization and evidence for a protective role in excitotoxicity. Neuron. 1991;7:1053–1060. doi: 10.1016/0896-6273(91)90349-5. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Mascarucci P, Taub D, Saccani S, Paloma MA, Dawson H, Roth GS, Lane MA, Ingram DK. Cytokine responses in young and old rhesus monkeys: effect of caloric restriction. J. Interferon. Cytokine Res. 2002;22:565–571. doi: 10.1089/10799900252982043. [DOI] [PubMed] [Google Scholar]
- Masoro EJ, McCarter RJM, Katz MS, McMahan CA. Dietary restriction alters the characteristics of glucose fuel use. J. Gerontol. Biol. Sci. 1992;47:B202–B208. doi: 10.1093/geronj/47.6.b202. [DOI] [PubMed] [Google Scholar]
- Maswood N, Young J, Tilmont E, Zhang Z, Gash DM, Gerhardt GA, Grondin R, Roth GS, Mattison J, Lane MA, Carson RE, Cohen RM, Mouton PR, Quigley C, Mattson MP, Ingram DK. Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 2004;101:18171–18176. doi: 10.1073/pnas.0405831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Experimental models of Alzheimer's disease. Sci. Med. 1998 March/April;:16–25. [Google Scholar]
- McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of life-span and upon the ultimate body size. J. Nutr. 1935;10:63–79. [PubMed] [Google Scholar]
- Means LW, Higgins JL, Fernandez TJ. Midlife onset of dietary restriction extends life and prolongs cognitive-functioning. Physiol. Behav. 1993;54:503–508. doi: 10.1016/0031-9384(93)90243-9. [DOI] [PubMed] [Google Scholar]
- Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin. Chem. 2004;50:1511–1525. doi: 10.1373/clinchem.2004.032482. [DOI] [PubMed] [Google Scholar]
- Mitchell GA, Kassovska-Bratinova S, Boukaftane Y, Robert MF, Wang SP, Ashmarina L, Lambert M, Lapierre P, Potier E. Medical aspects of ketone body metabolism. Clin. Invest. Med. 1995;18:193–216. [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Munch G, Thome J, Foley P, Schinzel R, Riederer P. Advanced glycation end products in ageing and Alzheimer's disease. Brain Res. Rev. 2000;23:134–143. doi: 10.1016/s0165-0173(96)00016-1. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Musi N, Hayashi T, Fujii N, Hirshman MF, Witters LA, Goodyear LJ. AMP-activated protein kinase activity and glucose uptake in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001;280:677–684. doi: 10.1152/ajpendo.2001.280.5.E677. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajvani UB, Scherer PE. Adiponectin: systemic contributor to insulin sensitivity. Curr. Diab. Rep. 2003;3:207–213. doi: 10.1007/s11892-003-0065-2. [DOI] [PubMed] [Google Scholar]
- Papaioannou N, Tooten PC, van Ederen AM, Bohl JR, Rofina J, Tsangaris T, Gruys E. Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid. 2001;8:11–21. doi: 10.3109/13506120108993810. [DOI] [PubMed] [Google Scholar]
- Parker JA, Arango M, Abderrahmane S, Lambert E, Tourette C, Catoire H, Neri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poynter ME, Daynes RA. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998;273:32833–32841. doi: 10.1074/jbc.273.49.32833. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Dauga E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell. Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML ANS HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial Sirtuin deacetylase, regulates mitochondrial function and thermogenesis in Brown adipocytes. J. Biol. Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Hollander W, Moss MB, Rosene DL, Abraham CR. Increased microglial activation and protein nitration in white matter of the aging monkey. Neurobiol. Aging. 1999;20:395–405. doi: 10.1016/s0197-4580(99)00066-4. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Agarwal S, Candas M, Forster MJ, Lal H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech. Ageing. Dev. 1994;76:215–224. doi: 10.1016/0047-6374(94)91595-4. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag WE, Lynch CD, Cefalu WT, Ingram RL, Bennett SA, Thornton PL, Khan AS. Pleiotropic effects of growth hormone and insulin-like growth factor (IGF)-1 on biological aging: inferences from moderate caloric-restricted animals. J. Gerontol. Biol. Sci. 1999;54:521–538. doi: 10.1093/gerona/54.12.b521. [DOI] [PubMed] [Google Scholar]
- Sprott RL. Diet and calorie restriction. Exp. Gerontol. 1997;32:205–214. doi: 10.1016/s0531-5565(96)00065-4. [DOI] [PubMed] [Google Scholar]
- Stephens JM, Lee J, Pilch PF. Tumor necrosis factor-α-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J. Biol. Chem. 1997;272:971–976. doi: 10.1074/jbc.272.2.971. [DOI] [PubMed] [Google Scholar]
- Sugden MC, Bulmer K, Gibbons GF, Holness MJ. Role of peroxisome proliferator-activated receptor-alpha in the mechanism underlying changes in renal pyruvate dehydrogenase kinase isoform 4 protein expression in starvation and after refeeding. Arch. Biochem. Biophys. 2001;395:246–252. doi: 10.1006/abbi.2001.2586. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPARγ 2: tissue-specific regulator of an adipocyte enhancer. Genes. Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr., DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Tsuchiya T, Dhahbi JM, Cui X, Mote PL, Bartke A, Spindler SR. Additive regulation of hepatic gene expression by dwarfism and caloric restriction. Physiol. Genom. 2004;17:307–315. doi: 10.1152/physiolgenomics.00039.2004. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee Park S, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J. Biol. Chem. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Vazquez JA, Morse EL, Adibi SA. Effect of dietary fat, carbohydrate, and protein on branched-chain amino acid catabolism during caloric restriction. J. Clin. Invest. 1985;76:737–743. doi: 10.1172/JCI112029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikman KS, Owe-Larsson B, Brask J, Kristensson KS, Hill RH. Interferon-gamma-induced changes in synaptic activity and AMPA receptor clustering in hippocampal cultures. Brain Res. 2001;896:18–29. doi: 10.1016/s0006-8993(00)03238-8. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- Wan R, Camandola S, Mattson MP. Intermittent food deprivation improves cardiovascular and neuroendocrine responses to stress in rats. J. Nutr. 2003;133:1921–1929. doi: 10.1093/jn/133.6.1921. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev. Cell. 2003;5:811–816. doi: 10.1016/s1534-5807(03)00323-x. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Weindruch R, Kayo T, Lee CK, Prolla TA. Gene expression profiling of aging using DNA microarrays. Mech. Ageing Dev. 2002;123:177–193. doi: 10.1016/s0047-6374(01)00344-x. [DOI] [PubMed] [Google Scholar]
- Weindruch R, Sohal RS. Seminars in medicine of the Beth Israel Deaconess Medical Center: caloric intake and aging. N. Engl. J. Med. 1997;337:986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL. In: The Retardation of Aging and Disease by Dietary Restriction. Thomas, Charles C, editors. Springfield, IL: 1988. [Google Scholar]
- Wheeler MD, Smutney OM, Check JF, Rusyn I, Schulte-Hermann R, Thurman RG. Impaired Ras membrane association and activation in PPARalpha knockout mice after partial hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;284:G302–G312. doi: 10.1152/ajpgi.00175.2002. [DOI] [PubMed] [Google Scholar]
- Wolkow CA, Kimura KD, Lee M-S, Ruvkun G. Regulation of C. elegans life span by insulin-like signaling in the nervous system. Science. 2000;290:147–150. doi: 10.1126/science.290.5489.147. [DOI] [PubMed] [Google Scholar]
- Wong G, Goldshmit Y, Turnley AM. Interferon-gamma but not TNF alpha promotes neuronal differentiation and neurite outgrowth of murine adult neural stem cells. Exp. Neurol. 2004;187:171–177. doi: 10.1016/j.expneurol.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ. Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes. 2003;52:207–213. doi: 10.2337/diabetes.52.6.1355. [DOI] [PubMed] [Google Scholar]
- Yeagley D, Guo S, Unterman T, Quinn PG. Gene- and activation-specific mechanisms for insulin inhibition of basal and glucocorticoid-induced insulin-like growth factor binding protein-1 and phosphoenolpyruvate carboxykinase transcription. Roles of forkhead and insulin response sequences. J. Biol. Chem. 2001;276:33705–33710. doi: 10.1074/jbc.M101215200. [DOI] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Jang Y, You-Ten AI, Okada H, Liepa J, Wakeham A, Zaugg K, Mak TW. p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc. Natl. Acad. Sci. U.S.A. 2004;101:14057–14062. doi: 10.1073/pnas.0406286101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Luo W, Fu W, Mattson MP. The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp. Neurol. 1999;155:302–314. doi: 10.1006/exnr.1998.7002. [DOI] [PubMed] [Google Scholar]
- Yu ZF, Mattson MP. Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: evidence for a preconditioning mechanism. J. Neurosci. Res. 1999;57:830–839. [PubMed] [Google Scholar]
- Zhu M, de Cabo R, Lane MA, Ingram DK. Caloric restriction modulates early events in insulin signaling in liver and skeletal muscle of rat. Ann. N. Y. Acad. Sci. 2004;19:448–452. doi: 10.1196/annals.1297.082. [DOI] [PubMed] [Google Scholar]