

Abstract
The cholesterol-dependent cytolysins (CDCs) are a large family of pore-forming toxins that often exhibit distinct structural changes that modify their pore-forming activity. A soluble platelet aggregation factor from Streptococcus mitis (Sm-hPAF) was characterized and shown to be a functional CDC with an amino-terminal fucose-binding lectin domain. Sm-hPAF, or lectinolysin (LLY) as renamed herein, is most closely related to CDCs from Streptococcus intermedius (ILY) and Streptococcus pneumoniae (pneumolysin or PLY). The LLY gene was identified in strains of S. mitis, S. pneumoniae, and Streptococcus pseudopneumoniae. LLY induces pore-dependent changes in the light scattering properties of the platelets that mimic those induced by platelet aggregation but does not induce platelet aggregation. LLY monomers form the typical large homooligomeric membrane pore complex observed for the CDCs. The pore-forming activity of LLY on platelets is modulated by the amino-terminal lectin domain, a structure that is not present in other CDCs. Glycan microarray analysis showed the lectin domain is specific for difucosylated glycans within Lewis b (Leb) and Lewis y (Ley) antigens. The glycan-binding site is occluded in the soluble monomer of LLY but is apparently exposed after cell binding, since it significantly increases LLY pore-forming activity in a glycan-dependent manner. Hence, LLY represents a new class of CDC whose pore-forming mechanism is modulated by a glycan-binding domain.
The viridans streptococci, which include Streptococcus mitis, Streptococcus mutans, Streptococcus salivarius, and Streptococcus sanguis, are normally found as members of the normal flora of the oropharyx. These organisms can escape this niche, however, and cause a variety of diseases that include infective endocarditis, bacteremia, and septicemia. S. mitis is one of the more common isolates, and its antibiotic resistance is increasing, making treatment of its infections difficult (1–4). S. mitis was recently associated with a large outbreak of a toxic shock-like syndrome that affected thousands of individuals in China (5). S. mitis is typically not considered to be toxigenic, and it is characterized by an α-hemolytic phenotype. Jeffries et al. (6), however, recently showed that some S. mitis strains express a cholesterol-dependent cytolysin (CDC), designated mitilysin (MLY), which is nearly identical to Streptococcus pneumoniae pneumolysin. Another potential S. mitis pathogenesis factor is human platelet aggregation factor (Sm-hPAF)1 that was purified from S. mitis isolated from a patient with Kawasaki disease (7). This protein was shown to aggregate human platelets and was designated S. mitis human platelet aggregation factor or Sm-hPAF. More recently, Ohkuni et al. (8) showed that antibody titers to a Sm-hPAF-derived peptide were significantly elevated in children with Kawasaki disease, a disease that is often associated with platelet aggregation and coronary artery thrombosis.
The peptide sequence reported by Ohkuni et al. (8) was derived from the sequence for a Sm-hPAF gene from S. mitis Nm-65 recently reported in GenBank (accession number AB051299.1). The sequence of Sm-hPAF encodes a predicted CDC structure that is most closely related to inter-medilysin (ILY), the CDC from Streptococcus intermedius (9). The DNA-derived primary structure of Sm-hPAF exhibits an extra amino-terminal domain of about 162 amino acids. Based on the DNA-derived primary structure, this domain is related to the European eel Anguilla anguilla agglutinin (AAA), a fucose-binding lectin that preferentially binds glycans containing fucosylated structures found in the blood group antigens A, B, H, and Lewis a (Lea) (10, 11).
Herein we describe the properties of purified recombinant Sm-hPAF (referred to as lectinolysin or LLY hereafter) from S. mitis SK597. These studies show that LLY is a pore-forming CDC with a functional glycan-binding domain. LLY does not trigger platelet aggregation but does form pores in platelet membranes. The isolated fucose-binding lectin domain of LLY exhibits specificity for glycans containing the Lewis y (Ley) and b (Leb) antigens. The lectin domain of LLY appears to modulate the pore-forming activity of LLY in a glycan-dependent manner after LLY binds to the cell surface.
METHODS
Bacterial Strains, Plasmids, and Chemicals
The full-length gene for LLY was cloned by PCR from the chromosomal DNA of S. mitis strain SK597 using the primer ATGAATCAA-GAAAAACGTTTGCATCGCTTTGTCAAAAAG, and the reverse primer sequence was 5′-TTACTCATTCACAATTTTTTCAT-CAACTTTAGGGTTTAG. For purposes of expression the cloned LLY gene was amplified with the primers 5′-TCG-GATCCGAGCAAGGGAATCGTCCAGTTG that introduced a 5′ BamHI endonuclease site and 5′-CTGAATTCTTACT-CATTCACAATTTTTTCATCAACTTTAGGG that placed a 3′ EcoRI endonuclease after the stop codon at the 3′ end of the gene. These primers removed the signal peptide and placed the gene for the secreted LLY in-frame in the pTrcHisA vector (Invitrogen). ILY and PFO were previously cloned into pTrcHisA as described (12, 13). All mutations were made in the native LLY (naturally cysteine-less), ILY (naturally cysteine-less), or cysteine-less PFO (PFOC459A) background. All chemicals and enzymes were obtained from Sigma, VWR, or Research Organics. All fluorescent probes were obtained from Molecular Probes (Invitrogen).
Generation and Purification of Recombinant LLY, ILY, and PFO and Their Derivatives
Using PCR QuikChange mutagenesis (Stratagene), various amino acid substitutions were introduced into native LLY, ILY, or PFOC459A. The Oklahoma Medical Research Foundation Core DNA Sequencing Facility performed all DNA sequence analysis. The expression and purification of recombinant LLY, ILY, PFO, and their derivatives from Escherichia coli were performed as described (13, 14). Purified protein was dialyzed into buffer [300 mM NaCl, 10 mM MES, 1 mM EDTA (pH 6.5)] overnight at 4 °C, made 5 mM in dithiothreitol (DTT) and 10% (v/v) sterile glycerol, and stored at −80 °C. For mutants that contained an engineered disulfide bridge the DTT was left out of the storage buffer.
PCR Screening Streptococci for the LLY Gene
Primers were used in the PCR amplification at a 20 pmol final concentration. These primers were designed to amplify out the entire coding sequence for the LLY gene. The forward primer sequence was 5′-ATGAATCAAGAAAAACGTTTG-CATCGCTTTGTCAAAAAG, and the reverse primer sequence was 5′-TTACTCATTCACAATTTTTTCATCAACTT-TAGGGTTTAG. Chromosomal DNA from each streptococcal strain was prepared using the Invitrogen Easy DNA kit, and 100 ng was added to the PCR reaction. An additional 2 μL of 25 mM MgCl2 was added to Qiagen Master Mix for a final PCR volume of 25 μL. Each PCR product was separated on a 1% agarose gel.
Chemical Modification of LLY and Its Derivatives with Sulfhydryl-Specific Fluorescent Probes
The cysteine derivatives of LLY and LLYLec were modified with Alexa Fluor 488 C5-maleimide (Invitrogen) via the sulfhydryl group as previously described (12). Modified protein stocks were adjusted to 10% (v/v) sterile glycerol, quick frozen in liquid nitrogen, and stored at −80 °C. Proteins were labeled at an efficiency of 75% or greater.
Kd Determination
Fluorescein-labeled glycans were prepared as previously described using free reducing oligosaccharides (15). The Kd of the interaction of LLYLec with Ley was determined by fluorescence anisotropy. Fluorescence measurements were carried out using an SLM 8100 fluorometer equipped with excitation and emission calcite polarizers. The excitation and emission monochromator wavelengths were set to 480 and 520 nm, respectively. The excitation and emission bandpass was set to 4 nm. Sample volume was maintained at 300 μL in a Starna microcell with a total capacity of 560 μL. Temperature was maintained at 23 °C. The fluorescein-labeled Ley was maintained at 167 nM, and the LLYLec was varied from 300 nM to 120 μM. The anisotropy (r) was calculated at each concentration of LLY from the equation:
I is intensity, V is vertically polarized light, and H is horizontally polarized light. The G factor (G = IHV/IHH) was calculated for each sample. The Δr was plotted versus the concentration of LLY, and the dissociation constant (Kd) was calculated using the equation Y = BmaxX/(Kd + X) for one site binding, where Bmax is the maximal binding and the Kd is the concentration of ligand (in this case the LLY) necessary to achieve half-maximal binding. Data analysis was carried out using Graphpad Prism software.
Platelet Aggregation Assay
Platelet-rich plasma (PRP) was prepared from normal human donors as described previously (16). Blood donors were free of aspirin or other antiplatelet medications for at lease 7 days prior to donation. Aggregation experiments were performed with a Model PAP-4 aggregometer from Bio/Data Corp. Total reaction volume was 200 μL. Convulxin-induced aggregation (500 ng/mL final concentration (16)) was used as a positive control. LLY was assayed at 71 nM, LLYCDC at 95 nM, and LLYLec at 280 nM.
Calcein Release Assay
PRP was diluted 1:100 with HEPES/saline assay buffer (2 mM CaCl2, 1 mM MgCl2, 150 mM NaCl, 10 mM HEPES, pH 7.5) and incubated with 2 μM calcein acetoxy methyl ester (Molecular Probes, Eugene, OR) for 10 min at room temperature as previously described (17). LLY (0.7–2.9 nM) was incubated with the calcein-loaded PRP for 10 min at room temperature. Samples were then diluted with HEPES/saline and analyzed on a FACS-Calibur flow cytometer (Becton-Dickinson) using settings as described previously (17). Cells losing calcein fluorescence were quantified as a percentage of total platelets.
The EC50 values for LLY- and LLYCDC-dependent calcein release were calculated using a nonlinear sigmoidal dose–response curve fit of the data (Prism Software).
Glycan Microarray Binding Assay and Scanning
Glycan microarrays were prepared as described previously (33, 37) and obtained from the NIH/NIGMS-funded Consortium for Functional Glycomics (see http://www.functionalglycomics.org/static/index.shtml). Before assay, the slides were re-hydrated for 5 min in TSM buffer (20 mM Tris-HCl, 150 mM sodium chloride (NaCl), 0.2 mM calcium chloride (CaCl2), and 0.2 mM magnesium chloride (MgCl2)). Alexa-488-labeled LLYQ190C and its derivatives were used in the binding assay (560–640 nM), and the bound proteins were detected directly via the fluorescent tag.
The slides were scanned with a Perkin-Elmer ProScanarray MicroArray scanner (Waltham, MA) using an excitation wavelength of 488 nm. ImaGene software (BioDiscovery, Inc., El Segundo, CA) was used to quantify fluorescence. The data are reported as average relative fluorescence units (RFU) from four of the six replicates (after removal of the highest and lowest values) for each glycan represented on the array. Versions 2.1 and 3.0 of the printed array were used in these studies, and a complete list of glycans printed on these arrays can be found at the Functional Glycomics Gateway Web site: www.functionalglycomics.org/static/consortium/resources/resourcecoreh10.shtml.
SDS–Agarose Gel Electrophoresis (SDS–AGE)
SDS–AGE of monomer and oligomer species of the various CDCs was carried out as previously described (18).
Liposome Preparation
Cholesterol–phosphatidylcholine (55/45 mol %) liposomes were prepared as previously described (13). Lipids and cholesterol were obtained from Avanti Polar Lipids.
RESULTS
The Primary Structure of LLY
The LLY gene was cloned from S. mitis SK597, which exhibits characteristics of both S. mitis and Streptococcus oralis (Hollingshead, unpublished data). The gene sequence (see GenBank accession number EU597013) exhibits the typical AT-rich (60% A–T) sequence of streptococcal genes. The deduced primary structure of LLY is nearly identical to the GenBank sequence for Sm-hPAF from S. mitis Nm-65 (accession number AB051299.1); 12 mostly conservative amino acid differences were identified (Figure 1a). These differences did not result from PCR-generated errors in the LLY sequence reported herein; the same sequence was derived from the cloned products of two separate PCR amplification experiments. Hence, the primary structure of LLY can apparently vary to some extent. The open reading frame encodes a protein with a mass of 73780.3 Da. Based on the previously published amino-terminal sequence of purified protein from the supernatant of S. mitis NM-65 (7) the amino terminus of secreted LLY is located at or near Thr-37, which is preceded by a 36-residue peptide that exhibits a typical type II signal peptide structure. The primary structure of LLY from residue 199 to 665 (Figure 1a) is most closely related to CDCs from S. intermedius (ILY) and S. pneumoniae (PLY). The CDC structure of LLY also contains an undecapeptide sequence (EKTGLVWEP-WR) that is similar to the hallmark consensus sequence (ECTGLAWEWWR) found in most CDCs but contains substitutions at positions similar to those found in ILY (GATGLAWEPWR).
Figure 1.

Primary structure of LLY. (a) The primary structure of LLY and its comparison with the primary structures of ILY and PLY. Comparison of the primary structure of LLYLec with that of the (b) A. anguilla agglutinin (AAA) (10) and (c) the glycan binding domain of the family 98 glycoside hydrolases from S. pneumoniae (SP2159) (19). The letters above the LLY sequence in panel a represent the amino acid differences with the GenBank sequence of Sm-hPAF (accession number AB051299.1). Homology comparisons were carried out using CLC Free Workbench version 4.6 (CLCBio). The conserved undecapeptide sequence of the CDCs is boxed in panel a. The conserved active site residues of AAA are bolded and underlined in panels b and c. Conserved residues (*), conservative substitutions (:).
Unlike other characterized CDCs LLY contains an extra 162-residue peptide fused to its amino terminus (excluding the signal peptide) (Figure 1a). This peptide region exhibits similarity with the A. anguilla (European eel) agglutinin (AAA) (33% identity) (10) (Figure 1b) and the carboxy-terminal carbohydrate-binding module of a putative member of the family 98 glycoside hydrolases from S. pneumoniae (47% identity) (Figure 1c) (19). Both proteins are fucose-binding proteins that exhibit a preference for glycans containing the H-antigen structure (Fucα1–2Galβ1–3(4)-GlcNAc-R).
Distribution of the LLY Gene
A collection of 165 strains of mostly mitis group streptococci was examined for the presence of the LLY gene by PCR using primers that encompassed its complete coding region. From this collection, twelve S. mitis strains, one S. pneumoniae, four Streptococcus pseudopneumoniae, and three S. mitis/oralis strains carried the gene. The PCR product size did not vary in size for any of the positive strains and comigrated with the 2 kb marker, consistent with its 1998 base pair sequence (including the stop codon) (data not shown). We confirmed expression and secretion of LLY in the SK597 strain (data not shown) that served as the source of the gene for our studies but have not confirmed its expression in the other species and strains that appear to carry the gene.
Hemolytic Activity of LLY and Derivatives
The coding region for LLY was cloned, expressed in E. coli, and purified (see Methods). Shown in Table 1 is the HD50 (dose of toxin required for 50% hemolysis under standard conditions; see Methods) for purified LLY, LLYCDC (LLY lacking the amino-terminal lectin domain), PFO, and ILY. In Table 1 are the HD50 values, using human erythrocytes, for LLY and LLYCDC and, for comparison, those of PFO and ILY. All of the CDCs exhibit picomolar HD50 values, but it appears LLY and LLYCDC are 4–5-fold less active than PFO and ILY. These values are within the typical range for these toxins and can vary depending on the cell type and species of origin. As described below, however, the amino-terminal lectin domain appears to exert a glycan-dependent enhancement of LLY activity with platelets.
Table 1.
Hemolytic Activity of LLY and Related CDCs Used in This Studya
| CDC | HD50 (pM) |
|---|---|
| ILY | 1.1 |
| PFO | 0.8 |
| LLY | 4.1 |
| LLYCDC | 4.4 |
The relative HD50 (hemolytic dose for 50% lysis of a standard erythrocyte suspension; see Methods) for each CDC used in these studies is provided.
Similar to other CDCs LLY and LLYCDC also formed large oligomeric complexes on cholesterol-rich liposomes (Figure 2). LLYCDC monomer (52147 Da) and oligomer migrated similarly to PFO monomer (52672 Da) and oligomer. The similarity in the mass of both proteins suggests that both form oligomers with similar numbers of monomers. In contrast to PFO and LLYCDC the migration of the LLY oligomer was significantly retarded. The slower migration of LLY was likely due to the additional mass contributed by the amino-terminal lectin domain (17751 Da). The CDCs typically form oligomers of 35–40 monomers (20); thus the oligomer mass of LLY would be at least 600 kDa greater than that of LLYCDC or PFO, consistent with its slower migration on the gel.
Figure 2.

Oligomer formation by LLY. Purified PFO, LLY, and LLYCDC were incubated in the absence and presence cholesterol-rich liposomes, and the monomer and oligomer species were separated by SDS–AGE. Shown is the Coomassie stained gel. The smearing of the oligomer bands results from the presence of the liposome-derived lipids in the oligomer samples.
LLY and Other CDCs Do Not Aggregate Platelets
Ohkuni et al. (7) reported that LLY (Sm-hPAF) stimulated platelet aggregation in human platelet-rich plasma (PRP). Qualitative analysis of platelet aggregation is commonly determined using an aggregometer, an instrument that measures the decrease in the light scattering properties of PRP as platelets aggregate into large complexes. Aggregometer analysis herein also showed apparent LLY-dependent platelet aggregation resulting in the decreased light scatter by the platelets (Figure 3a). Similar results were observed with purified LLYCDC, demonstrating that the predicted amino-terminal fucose-binding lectin domain was not required for this activity (Figure 3a). Purified lectin domain (LLYLec) did not induce any change in the light scattering properties of the platelets (Figure 3a). To confirm the aggregometer measurements reflected platelet aggregation by LLY and LLYCDC, we performed a microscopic examination of the platelets after treatment. Surprisingly, we could not identify any aggregated platelets (Figure 3b). By comparison, we observed large platelet aggregates in PRP treated with convulxin, a nonenzymatic glycoprotein from venom of Crotalus durissus terrificus that induces platelet aggregation. Although the aggregometer readings indicated that LLY and LLYCDC could aggregate platelets, the microscopic examination did not support this interpretation. Although unlikely, it was possible that we simply missed the aggregates formed by LLY. Therefore, we quantified the relative levels of platelets in each preparation.
Figure 3.

Platelet aggregation by LLY. Shown in (a) are the aggregometer recordings for platelets treated with LLY (71 nM), LLYCDC (95 nM), and LLYLec (280 nM) and the positive control convulxin (500 ng/mL). Shown in (b) are confocal microscopic images of untreated, LLY-treated, and convulxin-treated platelets. These fields are representative of 12 separate fields.
To measure the relative levels of platelets present in these mixtures, we utilized a flow cytometry based approach. PRP was either left untreated or treated in the aggregometer with 140 nM LLY or 500 ng/mL convulxin for 3 min at 37 °C with stirring. Aliquots of treated PRP were then incubated with FITC-labeled antiglycoprotein IIb/IIIa antibody, and platelet concentration was determined by flow cytometry. The relative platelet concentration in each sample was determined by the time required to accumulate 5000 events on the cytometer. If platelets aggregated into larger complexes, it would take longer to accumulate 5000 counts. If no significant aggregation occurs, then the time required for 5000 counts should be similar to untreated platelets. The LLY-treated and control (untreated) samples had nearly identical platelet concentrations (5000 counts/34 s and 5000 counts/31 s, respectively). In contrast, the convulxin-treated sample was significantly depleted of individual platelets (5000 counts/84 s). Hence, both convulxin and LLY induce changes in the light scattering properties of the platelets, but it is clear that LLY does not induce platelet aggregation. How then did LLY affect the light scattering properties of the platelets without aggregation?
The light scattering properties of cells often result from shape change and/or loss of cytoplasmic contents. For example, erythrocytes lose their light scattering properties upon pore formation by the CDC PFO (21). Pore formation in platelets was determined by monitoring calcein release from LLY-treated platelets. As shown in Figure 4, calcein is efficiently released in a dose-dependent manner from the platelets treated with LLY or with LLYCDC, suggesting that each formed a membrane pore. The relative EC50 for calcein release by both proteins was quantified below in the studies described in Figure 7, where we show that LLY exhibits variable activity on platelets from different donors.
Figure 4.

Calcein release from LLY-treated platelets. Calcein-loaded platelets were assayed by flow cytometry before (shaded peak) and after treatment with 0.7 nM (dashed line) or 1.4 nM (solid line) LLY (a) or LLYCDC (b).
Figure 7.

The glycan-binding domain of LLY modulates pore-forming activity. LLY-dependent calcein release from calcein-loaded PRP was measured by flow cytometry. (a) LLY-dependent calcein release from platelets of high (donor 1) and low responding donors (donor 3) which were preincubated without (solid lines) or with (dashed lines) LLYLec (56 nM). No change is observed with LLYLec alone (not shown). (b) Calcein release from platelets treated with LLY or LLYCDC. (c) Same as the experiments shown in (a) with donor 1 platelets except that LLYCDC was substituted for LLY.
To assess whether the observed change in light scattering properties of LLY in the aggregometer was dependent on pore formation, a mutant of LLY was trapped in the prepore complex by the introduction of an engineered disulfide, but which could be converted to the pore-forming oligomer by reduction of this bond. We previously showed a disulfide bridge between domains 2 and 3 of the CDC structure prevents membrane insertion of the transmembrane β-hairpins but not binding and assembly of the prepore oligomer (22). When the disulfide bridge was reduced, the transmembrane β-hairpins inserted into the membrane and formed the pore.
A prepore-locked mutant of LLY was generated by introducing cysteines for Gly-222 and Asn-351 (LLYG222C/N351C), residues analogous to Gly-83 and Ser-217 of ILY and to Gly-57 and Ser-190 of PFO, which were shown previously to form a disulfide and trap both toxins in prepore complexes (ref 22 and Soltani and Tweten, unpublished data). LLYG222C/N351C functioned as expected; when the disulfide bond was oxidized, it was not hemolytic, whereas reduction of the disulfide restored near native hemolytic activity (data not shown). Treatment of PRP with oxidized LLYG222C/N351C did not elicit a change in the light scattering properties of the platelets (Figure 5a), but reduction of the disulfide bond restored this activity. Similarly, the oxidized disulfide-locked variants of ILY (Figure 5b) and PFO (Figure 5c) did not change the light scattering properties of the platelets, whereas the reduced forms of each triggered significant changes. These data show that the changes in light scattering properties of platelets are dependent on the pore-forming properties of LLY, PFO, and ILY and are not the result of platelet aggregation.
Figure 5.

Platelet lysis requires pore formation. Shown are aggregometer readings for platelets treated with prepore locked mutants of LLY (a), ILY (b), and PFO (c) in their oxidized (LLYppl-ox, ILYppl-ox, and PFOppl-ox) and reduced forms (LLYppl-red, ILYppl-red, and PFOppl-red). Conditions were similar to those in Figure 3a; all toxins were added at approximately 70 nM. The reduction of the disulfide allows each prepore-locked mutant to convert to the pore complex.
Glycan Specificity and Affinity of the LLY Lectin Domain
We next determined if the putative fucose-binding lectin domain of LLY was functional. A glycan microarray of vertebrate-type glycans (see Methods for a link to the complete list of glycans on this array) was probed with fluorescently tagged LLY. The probe consisted of the cysteine-substituted mutant LLYQ190C labeled with the maleimide derivative of Alexa-488 via the cysteine sulfhydryl (LLYQ190C-Alexa). Gln-190 was chosen for substitution with cysteine since it is located at the junction of the CDC and lectin domains. When the glycan microarray was probed with LLYQ190C-Alexa, no significant binding was detected (Figure 6a). This result suggested several possibilities: the lectin domain might bind to glycans that were not represented in the array, the lectin domain was not functional, or the glycan-binding site was sterically occluded or somehow inhibited in the soluble monomer of LLY.
Figure 6.

Glycan binding by LLY, LLYLec, and LLYLec-R112A. Version 3.0 of the printed glycan microarray, containing 320 eukaryotic-derived glycans (see Methods for a link to the full list of glycans on this array), was probed with fluorescently tagged (Alexa-488) versions of (a) LLY (670 nM), (b) LLYLec (560 nM), and (c) LLYLec-R112A (560 nM) that had been labeled at the cysteine-substituted Gln-190. Standard error of the mean is shown as gray error bars. Note: The microarray analyses herein were carried out using the updated version 3.0 of the glycan array whereas the results in Table 2 were obtained using version 2.1. This was a result of a change in the microarray version during the course of these studies. There are only five differences in fucose-containing glycans in the two versions. Only two of these glycans, numbers 290 and 301 on the version 3.0 array, contain the H-antigen structure and were shown to be bound by LLYLec whereas the other three glycans were not (data not shown). In panel d the Kd of the Ley–LLYLec interaction was determined by measuring changes in the anisotropy (Δr) of Ley (kept constant at 167 nm) in the presence of LLYLec that was varied from 300 nM to 120 μM. Lex incubated with the highest concentration of LLY (120 μM) exhibited a Δr of ≈10% of that observed for Ley.
We first tested the latter possibility, primarily because of the amino-terminal location of the lectin domain. This location positions the lectin domain distal to the carboxy-terminal domain 4, which contains the membrane-binding site of the CDCs (23–26). If the glycan-binding site was exposed in the monomer, it could bind LLY to cells in an unfavorable orientation, preventing critical domain 4 membrane interactions that initiate a cascade of ordered structural changes in the CDCs that lead to pore formation (reviewed in ref 27). Therefore, in this scenario occlusion or inhibition of the glycan-binding site may be necessary until after LLY has bound to the membrane. If correct, then expression of the lectin domain alone should restore its binding activity. A pair of stop codons was introduced after residue 190 of LLYQ190C to eliminate the translation of the downstream CDC structure. This construct was designated LLYLec-Q190C.
Purified LLYLec-Q190C was fluorescently labeled with Alexa-488 and used to probe the glycan microarray. As shown in Figure 6b, we observed significant binding to several glycans on the microarray. We also determined whether binding to the glycans on the glycan microarray was lost if we changed a residue that has been shown to be conserved in the fucose-binding site of related lectins. The AAA glycan binding site contains three key residues that make polar contacts with the fucose molecule. The Nε of His-52 contacts O-5 of the fucose ring whereas the guanidinium groups of Arg-79 and Arg-86 make contact with 3-OH and 4-OH ring hydroxyls of fucose (10). These residues are also conserved in the fucose-binding site of the family 98 glycoside hydrolases from S. pneumoniae (SP2159) (19) and correspond to residues His-85, Arg-112, and Arg-120 of LLY, respectively (Figure 1b,c). Arg-112 of LLYLec was changed to Ala in LLYLec-Q190C. This mutant (LLYLec-Q190C/R112A) was labeled with Alexa-488 at Cys190 and used to probe the glycan microarray. This mutation eliminated the glycan binding activity of LLYLec (Figure 6c), consistent with the conservation of this active site residue.
We further investigated the binding of LLYLec to the glycan microarray to determine its glycan specificity. We probe the microarray with three different concentrations of LLYLec that spanned a 100-fold range of concentration from 5.6 to 560 nM. These results (Table 2) are shown for all glycans bound at the highest concentration of LLYLec and selected glycans representative of those not bound by LLYLec at the highest concentration of LLYLec (representative of most glycans on the array). When the array was probed at 560 nM LLYLec, it bound to glycans containing the H-antigen structure (Fucα1–2Galβ1–3(4)GlcNAc-R) as found within the A, B, O, Ley, and Leb blood group antigens (Table 2, glycan IDs 1–24) and showed little affinity for the nonfucosylated type 1 and type 2 oligosaccharides (Table 2, glycan IDs 33–38), fucosylated glycans that did not contain the H-antigen, including Lea and Lex containing structures (Table 2, glycan IDs 27–32), and the difucosylated structures (Table 2, glycan IDs 42–50). At this high concentration of LLYLec, we observed some weak binding to irrelevant glycans (Table 1, glycan IDs 39–41). However, when the microarray was probed with 10-fold (56 mM) and 100-fold lower (5.6 nM) concentrations of LLYLec, we observed a marked shift in binding preference for glycans containing Ley and Leb antigens (Table 2, glycan IDs 1–7). These results demonstrate that LLYLec has a unique recognition of difucosylated glycans Fucα1–2Galβ1–3/4(Fucα1–4/3)GlcNAc-R) as found in Leb and Ley blood group antigens. Interestingly, glycans 6 and 7 are such difucosylated glycans, which also contain the blood group B and A determinants, respectively. However, as noted above, when the array is probed at the lowest concentration of LLYLec, it binds weakly or not at all to blood group B (Galα1–3[Fucα1–2]Galβ1–3/4R), blood group A (GaNAclα1–3[Fucα1–2]Galβ1–3/4R), or blood group H (Fucα1–2Galβ) determinants that lack the necessary difucosylated structures.
Table 2.
LLYLec Preferentially Binds Ley and Leb Glycansa
| Glycan Family | Glycan # | Glycan Structure | 560 nM | 56 nM | 5.6 nM | |||
|---|---|---|---|---|---|---|---|---|
| AVG RFU | %CV | AVG RFU | %CV | AVG RFU | %CV | |||
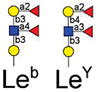
|
1 | Fucα1–2Galβ1–4(Fucα1–3)GlcNAcβ-Sp8 | 42035 | 1004 | 50113 | 18 | 16748 | 3 |
| 2 | Fucα1–2Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 52303 | 8 | 40704 | 6 | 15357 | 11 | |
| 3 | Fucα1–2Galβ1–3(Fucα1–4)GlcNAcβ-Sp8 | 44801 | 7 | 44960 | 9 | 11763 | 5 | |
| 4 | Fucα1–2Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 42253 | 4 | 54171 | 10 | 9445 | 10 | |
| 5 | Fucα1–2Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 49653 | 11 | 54594 | 11 | 9201 | 3 | |
| 6 | Galα1–3(Fucα1–2)Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 43236 | 3 | 39872 | 2 | 9022 | 5 | |
| 7 | GalNAcα1–3(Fucα1–2)Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 45556 | 4 | 37850 | 1 | 5506 | 2 | |
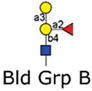
|
8 | Galα1–3(Fucα1–2)Galβ-Sp8 | 42263 | 30 | 28788 | 6 | 2936 | 5 |
| 9 | Galα1–3(Fucα1–2)Galβ1–4Glcβ-Sp0 | 55729 | 9 | 11208 | 14 | 846 | 26 | |
| 10 | Galα1–3(Fucα1–2)Galβ1–3GlcNAcβ-Sp0 | 35310 | 5 | 9312 | 19 | 743 | 17 | |
| 11 | Galα1–3(Fucα1–2)Galβ1–4GlcNAc-Sp0 | 9081 | 8 | 2701 | 9 | 338 | 26 | |
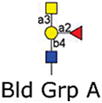
|
12 | GalNAcα1–3(Fucα1–2)Galβ-Sp8 | 13806 | 9 | 3635 | 14 | 282 | 18 |
| 13 | GalNAcα1–3(Fucα1–2)Galβ1–3GlcNAcβ-Sp0 | 203 | 18 | 247 | 34 | 304 | 59 | |
| 14 | GalNAcα1–3(Fucα1–2)Galβ1–4Glcβ-Sp0 | 617 | 11 | 302 | 36 | 97 | 12 | |
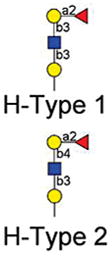
|
15 | Fucα1–2Galβ-Sp8 | 51057 | 11 | 17066 | 19 | 1443 | 13 |
| 16 | Fucα1–2[6OSO3]Galβ1–4[6OSO3]Glc-Sp0 | 35842 | 10 | 12584 | 14 | 486 | 47 | |
| 17 | Fucα1–2-Galβ1–4[6OSO3]Glc-Sp0 | 23604 | 8 | 9152 | 9 | 176 | 65 | |
| 18 | Fucα1–2Galβ1–4Glcβ-Sp0 | 17933 | 1 | 5716 | 12 | 537 | 14 | |
| 19 | Fucα1–2Galβ1–4GlcNAcβ-Sp0 | 13896 | 12 | 3452 | 17 | 327 | 13 | |
| 20 | Fucα1–2-(6OSO3)-Galβ1–4Glc-Sp0 | 13295 | 15 | 2345 | 90 | 56 | 42 | |
| 21 | Fucα1–2Galβ1–3GlcNAcβ-Sp0 | 8226 | 10 | 1727 | 19 | 214 | 8 | |
| 22 | Fucα1–2Galβ1–4GlcNAcβ-Sp8 | 7311 | 8 | 2133 | 14 | 164 | 73 | |
| 23 | Fucα1–2Galβ1–3GlcNAcβ-Sp8 | 5908 | 16 | 1845 | 25 | 276 | 22 | |
| 24 | Fucα1–2Galβ1–4[6OSO3]GlcNAc-Sp8 | 5045 | 10 | 1175 | 13 | 147 | 36 | |
| 25 | Fucα1–2Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAc-Sp0 | 135 | 27 | 478 | 40 | 208 | 40 | |
| 26 | Fucα1–2Galβ1–3GlcNAcβ1–3Galβ1–4Glcβ-Sp8 | 215 | 42 | 163 | 33 | 135 | 33 | |
|
27 | Fucα1–4GlcNAcβ-Sp8 | 189 | 134 | 211 | 80 | 78 | 37 |
| 28 | Galβ1–3(Fucα1–4)GlcNAoβ-Sp8 | 237 | 61 | 101 | 15 | 144 | 88 | |
| 29 | Neu5Acα2-3Galβ1–3(Fucα1–4)GlcNAcβ-Sp8 | 116 | 41 | 212 | 26 | 112 | 37 | |
| 30 | Fucα1–3GlcNAcβ-Sp8 | 169 | 52 | 164 | 38 | 232 | 26 | |
| 31 | Galβ1–4(Fucα1–3)GlcNAcβ-Sp8 | 91 | 79 | 184 | 8 | 194 | 26 | |
| 32 | Neu5Acα2–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ-Sp8 | 187 | 75 | 98 | 61 | 89 | 36 | |
|
33 | Neu5Acα2–3Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAcβ-Sp0 | 281 | 49 | 169 | 27 | 111 | 39 |
| 34 | Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAcβ-Sp0 | 217 | 37 | 189 | 50 | 152 | 60 | |
| 35 | Galβ1–4GlcNAcβ1–3Galβ1–4GlcNAcβ-Sp0 | 183 | 26 | 120 | 35 | 170 | 48 | |
| 36 | NeuAcα2–3Galβ1-SGIcNAcβ1–3Galβ1–4GlcNAcβ-Sp0 | 194 | 33 | 147 | 49 | 117 | 49 | |
| 37 | Galβ1-SGIcNAcβ1–3Galβ1–4Glcβ-Sp10 | 340 | 77 | 170 | 60 | 172 | 34 | |
| 38 | Galβ1–3GlcNAcβ1–3Galβ1–4GlcNAcβ-Sp0 | 333 | 123 | 123 | 37 | 79 | 79 | |
| Other | 39 | α-L-Fuc-Sp8 | 3038 | 14 | 1435 | 36 | 326 | 28 |
| 40 | [3OSO3]Galβ1–4(6OSO3)Glcβ-Sp8 | 2745 | 28 | 848 | 11 | 397 | 17 | |
| 41 | Transferrin | 4066 | 31 | 1679 | 34 | 905 | 20 | |
| Difucosylated, lacking Blood Group H | 42 | Galβ1–4(Fucα1–3)GlcNAcβ1–4Galβ1–4(Fucα1–3)GlcNAcβ1–4Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 522 | 68 | 138 | 16 | 216 | 48 |
| 43 | NeuAcα2-3Galβ1–3(Fucα1–4)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβSp0 | 407 | 28 | 171 | 25 | 122 | 51 | |
| 44 | Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–3(Fucα1–4)GlcNAcβ-Sp0 | 649 | 97 | 490 | 46 | 169 | 42 | |
| 45 | Galβ1–3(Fucα1–4)GlcNAcβ1–3Galβ1–3(Fucα1–4)GlcNAcβ-Sp0 | 254 | 111 | 215 | 46 | 155 | 67 | |
| 46 | Galβ1–3(Fucα1–4)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 223 | 40 | 167 | 49 | 197 | 27 | |
| 47 | Galβ1–4(Fucα1–3)GlcNAoβ1–4Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 234 | 31 | 418 | 24 | 229 | 37 | |
| 48 | Neu5Acα2-6Galβ1–4GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 422 | 104 | 158 | 18 | 93 | 32 | |
| 49 | Galβ1–4GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 105 | 34 | 166 | 31 | 137 | 29 | |
| 50 | Neu5Acα2-3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ1–3Galβ1–4(Fucα1–3)GlcNAcβ-Sp0 | 23 | 517 | 287 | 58 | 170 | 15 | |
Version 2.1 of the printed glycan microarray (Consortium for Functional Glycomics) was probed with Alexa-488 labeled LLYLec-Q190C at 560, 56, and 5.6 nM. To demonstrate specificity of binding by LLYLec, all glycans that were bound at the highest LLYLec concentration are shown as well as related glycans that were not bound or weakly bound. The average RFU (from four or six analysis with high and low values eliminated) for each bound glycan was determined as described in Methods and the % CV (100 × standard deviation ÷ average) provides an indication of the precision of each measurement. All other glycans on the version 2.1 array probed at the highest concentration of ILYLec did not exhibit signals above background. AVG RFU is average relative fluorescence units; SP, spacer arm or linker between glycan and surface of the array; SP0, –CH2CH2NH2; and SP8, –CH2CH2CH2NH2. Key: yellow circles, galactose; yellow squares, GalNac; red triangles, fucose; blue squares, GlcNAc.
The affinity of LLYLec for Ley was determined by measuring changes in fluorescence anisotropy (Δr) of fluorescently tagged Ley as the concentration of LLYLec was varied. From this analysis the Kd of the LLYLec-Ley interaction was determined to be approximately 38 μM (Figure 6d). As expected from the microarray data, Lex antigen, which lacks the H-antigen structure, was not bound significantly (<10% of Ley) at the highest concentration of LLYLec.
The Lectin Domain of LLY Modulates Its Pore-Forming Activity
Ohkuni et al. (7) previously showed that platelets of some donors were unresponsive to Sm-hPAF-mediated aggregation (shown herein to be due to pore-dependent changes in platelet light scattering properties). This observation suggested that different donor platelets might exhibit different susceptibilities to the pore-forming activity of LLY. Consistent with this prediction, we observed that platelets from different donors exhibited differential susceptibility to LLY (Dale and Friese, unpublished observations). Shown in Figure 7a is the EC50 for LLY pore formation determined on high (donor 1) and low (donor 3) sensitivity platelets. The platelets from donor 1 are nearly 6-fold more sensitive to LLY (EC50 = 4.890 × 10−11) than those from donor 3 (EC50 = 2.88 × 10−10). These results suggested that a feature of donor 1 platelets significantly enhanced their susceptibility to LLY.
We hypothesized that enhancement of LLY activity resulted from a glycan-dependent interaction of the lectin domain after monomer bound the cell surface. If correct, then preincubating platelets with LLYLec should prevent interaction of the lectin domain of LLY with surface glycans and block the enhancement of LLY activity on the highly sensitive donor 1 platelets. The EC50 for calcein release by LLY from platelets pretreated with LLYLec (EC50 = 3.8 × 10−10) was approximately 8-fold higher than the EC50 for LLY on platelets that were not pretreated with LLYLec (EC50= 4.890 × 10−11) (Figure 7a). Similar results were observed if the platelets were incubated with 50 mM fucose instead of LLYLec but not with 50 mM sucrose as a control (data not shown). Furthermore, pretreatment of the low sensitivity donor 3 platelets with LLYLec had little impact on the activity of LLY, suggesting that the glycan(s) was largely absent from this donor’s platelets. The results show the lectin domain increases LLY specific activity, apparently by interacting with one or more glycans on the cell surface of platelets.
These data also predict LLYCDC, which lacks the lectin domain, should be less active than LLY on donor 1’s platelets. To test this prediction, the EC50 for calcein release by LLY and LLYCDC was determined. As is shown in Figure 7b the EC50 for LLYCDC on donor 1 platelets was 7-fold higher than the EC50 for LLY, consistent with the change observed in LLY activity on LLYLec-pretreated donor 1 platelets. Furthermore, LLYCDC platelet pore-forming activity was not affected by preincubating the platelets with LLYLec (Figure 7c).
DISCUSSION
The studies herein show LLY is a functional CDC with an amino-terminal fucose-binding lectin domain. The LLY gene was detected in various isolates of the mitis group of the streptococci. This was not surprising, since there is evidence for the extensive exchange of genetic material between these species (28) and recently it has been shown that some S. mitis strains carry a gene encoding a CDC nearly identical to pneumolysin (6). Hence, it is perhaps not unexpected that the LLY gene appears to be present in one isolate of S. pneumoniae. LLY exhibits the typical large oligomeric membrane complexes and pore-forming activity of the CDCs. Pore formation in the platelet membrane was shown herein to be responsible for changes in their light scattering properties rather than the previously reported aggregation (7). The primary structure of LLY is distinct from other CDCs due to the presence of an amino-terminal fucose-binding lectin domain. The isolated lectin domain binds to glycans that contain H-antigen structure (Fucα1–2Galβ1–3(4)GlcNAc-R), with a marked preference for the difucosylated Ley and Leb antigens. The glycan binding site is occluded in the soluble LLY monomer but is apparently exposed after the monomer binds to the cell surface since it enhances LLY-dependent pore-forming activity through a glycan-dependent mechanism. To date, LLY is the only characterized CDC to exhibit a glycan-binding lectin domain.
The glycan-binding site is occluded in the soluble LLY monomer, presumably to prevent nonproductive interactions that would inhibit the assembly of the oligomeric pore complex. Previous studies showed that the CDC monomer is bound in a perpendicular orientation to the membrane surface via loops at the tip of domain 4, an interaction that initiates ordered conformational changes in the CDC structure necessary for pore formation (reviewed in ref 27). The amino-terminal location of the lectin domain places it next to domain 1 of the CDC structure and distal to the membrane-binding site in domain 4 (Figure 8). If the glycan-binding site was exposed in the soluble monomer, its interaction with a surface glycan could bind LLY to the surface in a nonproductive orientation that would prevent the assembly of the oligomeric pore complex. Yet, as described below, the glycan-binding site must be exposed after LLY binds to the membrane surface, since it enhances pore-forming activity.
Figure 8.

Molecular models of LLYCDC and LLYLec. Shown are the ribbon representations of the LLYCDC (cyan) and LLYLec (pink) molecular models generated by Swiss Model (swissmodel.expasy-.org). The LLYCDC model was generated using the ILY crystal structure (47) as the template whereas the LLYlec was modeled based on the structure of the Ley-binding module of a S. pneumoniae virulence factor of the family 98 glycoside hydrolases (19). The locations are shown for the fucose-binding residues [His-85 (blue), Arg-112 (yellow), and Arg-120 (green)] and the C-terminal residue (space filled, magenta) of LLYLec and the amino-terminal residue of LLYCDC (space filled, gray). The L1-L3 loops and undecapeptide at the tip of domain 4 are shown in gold. The red-colored regions of the LLYCDC structural model exhibited a nonfavorable energy environment when analyzed with ANOLEA (29). Molecular structures were drawn with VMD (48).
Although caution must be used in extrapolating conclusions from molecular models, they suggest a possible explanation for the occlusion of the glycan-binding site in LLY. The structural model of the LLYLec, based on the crystal structure of the Ley-binding module of S. pneumoniae family 98 glycoside hydrolase (19), positions the C-terminus of LLYLec on the same face as Arg-112, a residue we showed herein is conserved and essential for glycan binding. In LLY the carboxy-terminal lectin domain would be fused to the amino terminus of the CDC, orienting the binding site toward the upper surface of domain 1 of the CDC structure, potentially occluding the glycan-binding site. An assessment of the packing quality of the LLYCDC structural model using ANOLEA (29) shows that residues in domain 1 (Figure 8), near the predicted site of interaction between the amino-terminal fucolectin domain and domain 1 of the CDC, exhibit a nonfavorable energy environment. This suggests that these residues do not pack similarly to the analogous residues in ILY. Hence, the residues in this region may adopt a different conformation in LLY so that they can specifically interact with the glycan-binding surface of the lectin domain in the soluble monomer.
Our studies revealed that platelets from different donors responded differently to LLY-dependent pore formation. Ohkuni et al. also observed a similar phenomenon in their aggregation experiments (7) (which we now know to be due to platelet lysis). The EC50 for LLY on the platelets of a high responder donor was about six times lower than that observed when the platelets from a low responder when they were treated with LLY. This difference was abolished if both sets of platelets were treated with the lectin-deficient LLYCDC or if high responder platelets were first treated with LLYLec or L-fucose before adding ILY. When the high responder platelets were pretreated with LLYLec to block its binding sites before the addition of LLY, we observed an approximately 8-fold increase in the EC50. Hence, LLY exhibits an intrinsic pore-forming activity that is enhanced by the presence of the lectin domain, apparently in a glycan-dependent manner. The most likely explanation for the differences in platelet sensitivity is that the levels of glycan recognized by the lectin domain differ in platelets from various donors. Based on the array analysis Ley and/or Leb antigens are likely candidates for these differences, but we cannot rule out the possibility that other fucose-containing glycans mediate this interaction. The mechanistic stage of the LLY pore-forming mechanism that is modulated by the lectin domain is not yet known but apparently occurs after the initial domain 4-mediated cell binding. Most toxins that have a lectin-like domain use it to bind to glycan-containing receptors (30–36), whereas the LLY lectin domain does not appear to participate in receptor recognition.
The glycan microarray analysis showed Ley and Leb are preferred ligands of the LLY lectin, with the H-antigen itself being a weaker ligand. The H-antigen is expressed in many cells and tissues of all individuals, but expression of Ley and Leb antigens is more restricted in adults. Besides being on erythrocytes and in body secretions (e.g., saliva), they are expressed throughout the gastrointestinal tract but at levels that can differ based on location (37, 38). Expression of both antigens may be highly upregulated on epithelial tumors of the GI tract (39, 40). In normal tissue, Leb is highly expressed on mucosal surfaces of the fetal colon, but in adults its expression is restricted to the proximal colon (41). Ley is expressed strongly in proximal regions of the terminal ileum and cecum and weakly in the ascending colon and beyond (42). In addition, both Ley and Leb can be found weakly expressed in gastric mucosa (43). Ley is also expressed by some Helicobacter pylori isolates (44).
Interestingly, Ley is expressed at low levels in circulating human peripheral blood granulocytes but is upregulated upon their activation (45). Ley-expressing neutrophils are also found in the synovial fluid of patients with arthritic joint disease (46). The mitis group of streptococci is part of the normal flora of the oropharynx and nasopharyx, but as indicated in the introduction they are increasingly associated with antibiotic-resistant bacteremia or septicemia. The lectin-dependent enhancement of LLY activity may increase its activity toward Ley-expressing activated neutrophils, thus potentially blunting one of the early responses to infection.
In summary, these studies show that LLY is a member of the CDC family of pore-forming toxins and is the only identified CDC that has a glycan-binding lectin domain. The lectin domain exhibits a strong preference for Leb and Ley and modulates LLY pore-forming activity in a glycan-dependent manner after LLY has bound to the membrane. The mechanistic basis for this effect remains unknown. Finally, several species of streptococci appear to carry the gene for LLY. One S. pneumoniae isolate appeared to carry the gene for LLY, suggesting that there may be a subpopulation of this important pathogen that expresses LLY in association with PLY or it has been substituted for PLY.
Acknowledgments
The excellent technical assistance of P. Coan is appreciated. The authors also acknowledge The Consortium for Functional Glycomics for support of the glycan array analysis and for providing the amino-derivatized Ley and Lex glycans for fluorescent derivatization.
Footnotes
This work was supported by a grant from the National Institute of Allergies and Infectious Diseases (AI037657) and in part by NIGMS–The Consortium for Functional Glycomics (GM62116).
Abbreviations: ILY, intermedilysin; PLY, pneumolysin; PFO, perfringolysin; Sm-hPAF, Streptococcus mitis human platelet aggregation factor; EC50, half-maximal effective concentration.
References
- 1.Kennedy MJ, Jackson MA, Kearns GL. Delayed diagnosis of penicillin-resistant Streptococcus mitis endocarditis following single-dose amoxicillin prophylaxis in a child. Clin Pediatr (Philadelphia) 2004;43:773–776. doi: 10.1177/000992280404300814. [DOI] [PubMed] [Google Scholar]
- 2.Gowda RM, Ansari AW, Khan IA. Complete endocardial cushion defect (complete atrioventricular canal) manifested in adult life by Streptococcus mitis endocarditis of the common atrioventricular valve. Int J Cardiol. 2003;89:109–110. doi: 10.1016/s0167-5273(02)00459-x. [DOI] [PubMed] [Google Scholar]
- 3.Hall GE, Baddour LM. Apparent failure of endocarditis prophylaxis caused by penicillin-resistant Streptococcus mitis. Am J Med Sci. 2002;324:51–53. doi: 10.1097/00000441-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Huang IF, Chiou CC, Liu YC, Hsieh KS. Endocarditis caused by penicillin-resistant Streptococcus mitis in a 12-year-old boy, J. Microbiol. Immunol, Infect (China) 2002;35:129–132. [PubMed] [Google Scholar]
- 5.Lu HZ, Weng XH, Zhu B, Li H, Yin YK, Zhang YX, Haas DW, Tang YW. Major outbreak of toxic shock-like syndrome caused by Streptococcus mitis. J Clin Microbiol. 2003;41:3051–3055. doi: 10.1128/JCM.41.7.3051-3055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jefferies J, Nieminen L, Kirkham LA, Johnston C, Smith A, Mitchell TJ. Identification of a secreted cholesterol-dependent cytolysin (mitilysin) from Streptococcus mitis. J Bacteriol. 2007;189:627–632. doi: 10.1128/JB.01092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohkuni H, Todome Y, Okibayashi F, Watanabe Y, Ohtani N, Ishikawa T, Asano G, Kotani S. Purification and partial characterization of a novel human platelet aggregation factor in the extracellular products of Streptococcus mitis, strain Nm-65. FEMS Immunol Med Microbiol. 1997;17:121–129. doi: 10.1111/j.1574-695X.1997.tb01004.x. [DOI] [PubMed] [Google Scholar]
- 8.Ohkuni H, Todome Y, Takahashi H, Nagamune H, Abe J, Ohtsuka H, Hatakeyama H. Antibody titers to Streptococus mitis-derived human platelet aggregation factor (Sm-hPAF) in the sera of patients with Kawasaki disease. In: Sriprakash KS, editor. Proceedings of the XVIth Lancefield International Symposium on Streptococci and Streptococcal Diseases. Elsevier B.V.; Palm Cove, Australia: 2006. pp. 71–74. [Google Scholar]
- 9.Nagamune H, Whiley RA, Goto T, Inai Y, Maeda T, Hardie JM, Kourai H. Distribution of the intermedilysin gene among the anginosus group streptococci and correlation between intermedilysin production and deep-seated infection with Streptococcus intermedius. J Clin Microbiol. 2000;38:220–226. doi: 10.1128/jcm.38.1.220-226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bianchet MA, Odom EW, Vasta GR, Amzel LM. A novel fucose recognition fold involved in innate immunity. Nat Struct Biol. 2002;9:628–634. doi: 10.1038/nsb817. [DOI] [PubMed] [Google Scholar]
- 11.Majumder S, Roy A, Mandal C. Prediction of 3-D structures of fucose-binding proteins and structural analysis of their interaction with ligands. Glycoconjugate J. 2004;20:545–550. doi: 10.1023/B:GLYC.0000043291.42999.98. [DOI] [PubMed] [Google Scholar]
- 12.Giddings KS, Johnson AE, Tweten RK. Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc Natl Acad Sci USA. 2003;100:11315–11320. doi: 10.1073/pnas.2033520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry. 1998;37:14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 14.Soltani CE, Hotze EM, Johnson AE, Tweten RK. Specific protein-membrane contacts are required for prepore and pore assembly by a cholesterol-dependent cytolysin. J Biol Chem. 2007;282:15709–15716. doi: 10.1074/jbc.M701173200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song X, Xia B, Lasanajak Y, Smith DF, Cummings RD. Quantifiable fluorescent glycan microarrays. Glyco-conjugate J. 2008;25:15–25. doi: 10.1007/s10719-007-9066-8. [DOI] [PubMed] [Google Scholar]
- 16.Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. 2000;95:1694–1702. [PubMed] [Google Scholar]
- 17.Remenyi G, Szasz R, Friese P, Dale GL. Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscl, Thromb Vasc Biol. 2005;25:467–471. doi: 10.1161/01.ATV.0000152726.49229.bf. [DOI] [PubMed] [Google Scholar]
- 18.Shepard LA, Shatursky O, Johnson AE, Tweten RK. The mechanism of assembly and insertion of the membrane complex of the cholesterol-dependent cytolysin perfringolysin O: Formation of a large prepore complex. Biochemistry. 2000;39:10284–10293. doi: 10.1021/bi000436r. [DOI] [PubMed] [Google Scholar]
- 19.Boraston AB, Wang D, Burke RD. Blood group antigen recognition by a Streptococcus pneumoniae virulence factor. J Biol Chem. 2006;281:35263–35271. doi: 10.1074/jbc.M607620200. [DOI] [PubMed] [Google Scholar]
- 20.Czajkowsky DM, Hotze EM, Shao Z, Tweten RK. Vertical collapse of a cytolysin prepore moves its trans-membrane β-hairpins to the membrane. EMBO J. 2004;23:3206–3215. doi: 10.1038/sj.emboj.7600350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris RW, Sims PJ, Tweten RK. Kinetic aspects of the aggregation of Clostridium perfringens theta toxin on erythrocyte membranes: A fluorescence energy transfer study. J Biol Chem. 1991;266:6936–6941. [PubMed] [Google Scholar]
- 22.Hotze EM, Wilson-Kubalek EM, Rossjohn J, Parker MW, Johnson AE, Tweten RK. Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane beta-sheet from a prepore intermediate. J Biol Chem. 2001;276:8261–8268. doi: 10.1074/jbc.M009865200. [DOI] [PubMed] [Google Scholar]
- 23.Giddings KS, Zhao J, Sims PJ, Tweten RK. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat Struct Mol Biol. 2004;12:1173–1178. doi: 10.1038/nsmb862. [DOI] [PubMed] [Google Scholar]
- 24.Heuck AP, Tweten RK, Johnson AE. Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J Biol Chem. 2003;278:31218–31225. doi: 10.1074/jbc.M303151200. [DOI] [PubMed] [Google Scholar]
- 25.Ramachandran R, Heuck AP, Tweten RK, Johnson AE. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat Struct Biol. 2002;9:823–827. doi: 10.1038/nsb855. [DOI] [PubMed] [Google Scholar]
- 26.Ramachandran R, Tweten RK, Johnson AE. The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc Natl Acad Sci USA. 2005;102:7139–7144. doi: 10.1073/pnas.0500556102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tweten RK. The cholesterol-dependent cytolysins; a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawamura Y, Hou XG, Sultana F, Miura H, Ezaki T. Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int J Syst Bacteriol. 1995;45:406–408. doi: 10.1099/00207713-45-2-406. [DOI] [PubMed] [Google Scholar]
- 29.Melo F, Feytmans E. Assessing protein structures with a non-local atomic interaction energy. J Mol Biol. 1998;277:1141–1152. doi: 10.1006/jmbi.1998.1665. [DOI] [PubMed] [Google Scholar]
- 30.Merritt EA, Sixma TK, Kalk KH, van Zanten BA, Hol WG. Galactose-binding site in Escherichia coli heat-labile enterotoxin (LT) and cholera toxin (CT) Mol Microbiol. 1994;13:745–753. doi: 10.1111/j.1365-2958.1994.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 31.Stein PE, Boodhoo A, Armstrong GD, Cockle SA, Klein MH, Read RJ. The crystal structure of pertussis toxin. Structure. 1994;2:45–57. doi: 10.1016/s0969-2126(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 32.Olson R, Gouaux E. Crystal structure of the Vibrio cholerae cytolysin (VCC) pro-toxin and its assembly into a heptameric transmembrane pore. J Mol Biol. 2005;350:997–1016. doi: 10.1016/j.jmb.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 33.Clark GF, Krivan HC, Wilkins TD, Smith DF. Toxin A from Clostridium difficile binds to rabbit erythrocyte glycolipids with terminal Gal alpha 1–3Gal beta 1–4GlcNAc sequences. Arch Biochem Biophys. 1987;257:217–229. doi: 10.1016/0003-9861(87)90561-3. [DOI] [PubMed] [Google Scholar]
- 34.Nesic D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- 35.Tateno H, Goldstein IJ. Molecular cloning, expression, and characterization of novel hemolytic lectins from the mushroom Laetiporus sulphureus, which show homology to bacterial toxins. J Biol Chem. 2003;278:40455–40463. doi: 10.1074/jbc.M306836200. [DOI] [PubMed] [Google Scholar]
- 36.Rossjohn J, Buckley JT, Hazes B, Murzin AG, Read RJ, Parker MW. Aerolysin and pertussis toxin share a common receptor-binding domain. EMBO J. 1997;16:3426–3434. doi: 10.1093/emboj/16.12.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hakomori SI, Siddiqui B. Isolation and characterization of glycosphingolipid from animal cells and their membranes. Methods Enzymol. 1974;32:345–367. doi: 10.1016/0076-6879(74)32036-8. [DOI] [PubMed] [Google Scholar]
- 38.Marcus DM. The ABO and Lewis blood-group system. Immunochemistry, genetics and relation to human disease. N Engl J Med. 1969;280:994–1006. doi: 10.1056/NEJM196905012801806. [DOI] [PubMed] [Google Scholar]
- 39.Inoue M, Nakayama M, Tanizawa O. Altered expression of Lewis blood group and related antigens in fetal, normal adult and malignant tissues of the uterine endometrium. Virchows Arch. 1990;416:221–228. doi: 10.1007/BF01678981. [DOI] [PubMed] [Google Scholar]
- 40.Heller DS, Thung SN. Expression of Lewis(x) and Lewis(y) blood group related antigens in fetal livers. Pediat Pathol. 1990;10:681–687. doi: 10.3109/15513819009064704. [DOI] [PubMed] [Google Scholar]
- 41.Jass JR, Roberton AM. Colorectal mucin histochemistry in health and disease: a critical review. Pathol Int. 1994;44:487–504. doi: 10.1111/j.1440-1827.1994.tb02599.x. [DOI] [PubMed] [Google Scholar]
- 42.Abe K, Hakomori S, Ohshiba S. Differential expression of difucosyl type 2 chain (LeY) defined by monoclonal antibody AH6 in different locations of colonic epithelia, various histological types of colonic polyps, and adenocarcinomas. Cancer Res. 1986;46:2639–2644. [PubMed] [Google Scholar]
- 43.Sakamoto S, Watanabe T, Tokumaru T, Takagi H, Nakazato H, Lloyd KO. Expression of Lewisa, Lewisb, Lewisx, Lewisy, siayl-Lewisa, and sialyl-Lewisx blood group antigens in human gastric carcinoma and in normal gastric tissue. Cancer Res. 1989;49:745–752. [PubMed] [Google Scholar]
- 44.Appelmelk BJ, Simoons-Smit I, Negrini R, Moran AP, Aspinall GO, Forte JG, DeVries T, Quan H, Verboom T, Maaskant JJ, Ghiara P, Kuipers EJ, Bloemena E, Tadema TM, Townsend RR, Tyagarajan K, Crothers JM, Jr, Monteiro MA, Savio A, DeGraaff J. Potential role of molecular mimicry between Helicobacter pylori lipopolysac-charide and host Lewis blood group antigens in autoimmunity. Infect Immun. 1996;64:2031–2040. doi: 10.1128/iai.64.6.2031-2040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dettke M, Palfi G, Loibner H. Activation-dependent expression of the blood group-related lewis Y antigen on peripheral blood granulocytes. J Leukocyte Biol. 2000;68:511–514. [PubMed] [Google Scholar]
- 46.Dettke M, Palfi G, Pursch E, Fischer MB, Loibner H. Increased expression of the blood group-related Lewis Y antigen on synovial fluid granulocytes of patients with arthritic joint diseases. Rheumatology (Oxford, England) 2001;40:1033–1037. doi: 10.1093/rheumatology/40.9.1033. [DOI] [PubMed] [Google Scholar]
- 47.Polekhina G, Giddings KS, Tweten RK, Parker MW. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc Natl Acad Sci USA. 2005;102:600–605. doi: 10.1073/pnas.0403229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]