

Abstract
A series of S-alkylated derivatives of homocysteine were synthesized and characterized as inhibitors of human recombinant betaine-homocysteine S-methyltransferase (BHMT). Some of these compounds inhibit BHMT with IC50 values in the nanomolar range. BHMT is very sensitive to the structure of substituents on the sulfur atom of homocysteine. The S-Carboxybutyl and S-carboxypentyl derivatives make the most potent inhibitors, and an additional sulfur atom in the alkyl chain is well tolerated. The respective (R,S)-5-(3-amino-3-carboxy-propylsulfanyl)-pentanoic, (R,S)-6-(3-amino-3-carboxy-propylsulfanyl)-hexanoic and (R,S)-2-amino-4-(2-carboxymethylsulfanyl-ethylsulfanyl)-butyric acids are very potent inhibitors and are the strongest ever reported. We determined that (R,S)-5-(3-amino-3-carboxy-propylsulfanyl)-pentanoic acid displays competitive inhibition with respect to betaine binding with a Kiapp of 12 nM. Some of these compounds are currently being tested in mice to study the influence of BHMT on the metabolism of sulfur amino acids in vivo.
Keywords: Inhibitor, BHMT, zinc, transition-state, homocysteine derivatives
INTRODUCTION
Betaine-homocysteine S-methyltransferase (BHMT, EC 2.1.1.5) is a cytosolic enzyme that catalyzes the transfer of a methyl group from betaine to L-homocysteine forming dimethylglycine and L-methionine (Met). The reaction follows an ordered bi-bi mechanism; homocysteine is the first substrate to bind, and Met is the last product off.1 BHMT contains a zinc atom2 that is tetrahedrally coordinated by three cysteines (Cys217, Cys299 and Cys300)3,4 and one tyrosine (Tyr160).5 The Zn2+ ion is absolutely essential for catalysis because it is required for the activation of the homocysteine thiol to the thiolate anion.6,7 The crystal structures of BHMT indicate that it is a homotetramer.4,5 The monomeric subunit has a molecular weight of 45 kDa and oligomerization appears to be essential for activity.8 The enzyme is abundant in human liver and kidney, but absent from other major organs.9
BHMT probably has a critical role in betaine, homocysteine, methionine and S-adenosylmethionine (AdoMet) homeostasis. Betaine is a intermediate of choline oxidation, and in addition to its role as a methyl donor it functions also as an organic osmolyte that is kept or released by the cell in response to osmotic changes in the kidney and liver.10–12 It was recently shown that the expression of BHMT in liver and kidney is dramatically down-regulated in salt loaded guinea pigs, suggesting that BHMT has a significant role modulating tissue betaine concentrations.13
An imbalance between homocysteine formation and catabolism can result in the elevation of plasma total homocysteine (tHcy), a condition known as hyperhomocysteinemia. The most common causes of hyperhomocysteinemia are suboptimal vitamin nutrition (folate, cobalamin, and/or vitamin B6) and/or genetic mutations that cause deficiencies of enzymes required for the synthesis of methylcobalaminn, or deficiencies of methylenetetrahydrofolate reductase or cystathionine-β-synthase activities. During the last couple of decades many studies have shown that hyperhomocysteinemia represents a risk factor for the development of vascular diseases and thrombosis14–16, and can result in pregnancy complications.17,18 Homocysteine also has been reported to be neurotoxic19 and to be associated with an increased risk for Alzheimer's disease.20 High levels of tHcy are found also in connection with chronic renal failure.21 Since an in vitro simulation of liver metabolism suggested that half of the conversion of homocysteine to methionine was BHMT-dependent22, it is certainly possible that a genetic defect that results in reduced BHMT activity could result in hyperhomocysteinemia and confer increased risk for homocysteine-related diseases.
If it is indeed true that half of the methionine produced in liver is BHMT-dependent22, and that the vast majority of AdoMet synthesis and utilization (perhaps ≥ 85%) occur in liver23, then it is reasonable to suggest that a dramatic reduction of BHMT activity also could result in reduced Met and AdoMet availability in liver, and perhaps other organs as well. The consequences of reduced Met and AdoMet biosynthesis could be many, including a reduction in transmethylation reactions (e.g. reduced DNA Methylation), but also a reduction in spermidine and spermine synthesis since the amino propyl moieties of these compounds are derived from AdoMet. Polyamines have a key role in cell growth and differentiation, and it is known that cancer cells have very high demands for AdoMet for both transmethylation reactions and polyamine synthesis.24,25 Additionally, it is known that about half of the cysteine that is used for glutathione synthesis comes from AdoMet26, and since cysteine is a limiting reagent for glutathione synthesis, a significant reduction in BHMT activity could reduce tissue glutathione levels.
As discussed above, there is a lack of information regarding the physiological role BHMT has regulating betaine, homocysteine, methionine and AdoMet metabolism. There has been no report of a human lacking BHMT activity, nor has a BHMT knockout mouse been generated to date. In addition, there has been no report describing the use of a BHMT inhibitor in vivo to investigate the biochemical and physiological consequences of such inhibition. To study BHMT function in vivo, it would be useful to have potent, selective and metabolically stable inhibitors. Not only would such inhibitors be useful to determine whether a reduction in BHMT activity affects tHcy levels supporting or refuting whether a BHMT-related link to hyperhomocysteinemia exists, but they could be clinically useful as well. For example, it possible that transiently inhibiting BHMT would reduce betaine degradation as a mechanism to restore osmotic balance during unwanted diuresis. Or, it is possible that a BHMT inhibitor could deplete the liver of methionine and AdoMet and be part of a combined strategy to treat some forms of cancer.
To date, only a few compounds have been synthesized that inhibit BHMT in vitro, and none have been tested in vivo. The first series of BHMT inhibitors were synthesized by Awad et al27 in 1983. These sulfur-containing compounds were designed as transition-state 1 mimicking analogues. In Awad’s study27, the most potent bi-substrate analogue (R,S)-5-(3-amino-3-carboxy-propylsulfanyl)-pentanoic acid 2 (or S-(4-carboxybutyl)-D,L-homocysteine, CBHcy) inhibited human liver BHMT with a Kiapp towards betaine of 6.5 µM. Recently, the crystal structure of human BHMT in complex with inhibitor 2 revealed4 that the sulfur of compound 2 became the fourth ligand to the zinc atom, confirming the biochemical evidence of a homocysteine-S-Zn interaction. In 2004, using changes in intrinsic fluorescence of BHMT, we determined Kd of compound 2 towards the enzyme to be about 280 nM.28 This high-affinity interaction was surprising considering the relatively high Ki (6.5 µM) reported by Awad et al27, which will be discussed later in further detail. In 1992, a series of boronic acid based analogues of betaine was published29. The most potent compound, N,N,N-trimethylammonium-methylboronate 3 was a competitive inhibitor at the betaine binding site of rat liver BHMT with Ki of about 45 µM. Recently, we reported30–34 very selective affinity purification of rat BHMT using immobilized phosphinic pseudopeptide 4. We determined Kiapp′s of compound 4 towards both substrates, D,L-homocysteine and betaine, to be about 10 µM and we found that the type of inhibition towards both substrates is noncompetitive (Collinsova M., Jiracek J., unpublished results).

RESULTS AND DISCUSSION
We synthesized a series of S-alkyl derivatives of homocysteine and one S-alkyl derivative of cysteine. These compounds are shown in Table 1. The syntheses were performed in aqueous solutions under alkaline conditions by (i) alkylation of unprotected D,L-homocysteine (5) or L-cysteine (6) with different alkylating agents (Method A used for inhibitors 2, 8, 9, 12, 16–20, 22–25), and by (ii) alkylation of respective thiols with (R,S)-2-amino-4-bromo-butyrate (7) (Method B used for inhibitors 13–15). Compounds 10 and 11 were prepared by the oxidation of compound 2. Compounds 26 and 28 (Table 1) are commercially available cystathionine and homocystine, respectively. Compound 27 was prepared by the oxidation of cystathionine. Table 2 shows the structures of the alkylating agents (7, 29–31 and 35–42) and thiols (5, 6, 32–34) used to produce each product and each reaction yield.
Table 1.
Inhibition (relative) of human BHMT by S-substituted derivatives of homocysteine. The percentage inhibition of each compound was determined at 20 and 1 µM. See Experimental for details.
| Compound | % of Inhibitiona (0.25 mM betaine, 0.1 mM D,L-homocysteine) | IC50 (µM)b (2 mM betaine, 1 mM D,L-homocysteine) | ||
|---|---|---|---|---|
| 20 µM | 1 µM | |||
| 2 | 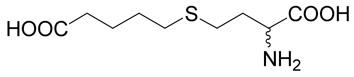 |
100 | 98.3 | 0.087 |
| 8 | 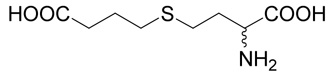 |
20.1 | nd | nd |
| 9 | 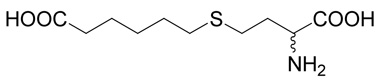 |
100 | 99.9 | 0.2 |
| 10 | 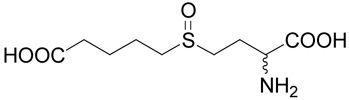 |
97.3 | 67.8 | 5 |
| 11 | 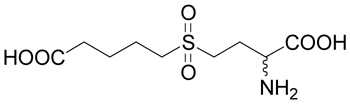 |
28.8 | nd | nd |
| 12 | 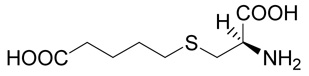 |
0 | nd | nd |
| 13 | 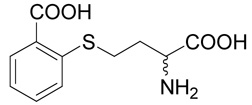 |
42.5 | nd | nd |
| 14 | 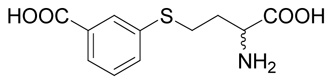 |
12.8 | nd | nd |
| 15 | 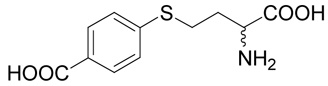 |
98.6 | 84.8 | 7 |
| 16 | 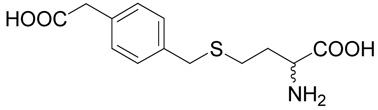 |
10.1 | nd | nd |
| 17 | 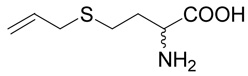 |
0 | nd | nd |
| 18 | 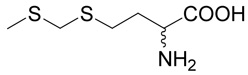 |
20.5 | nd | nd |
| 19 | 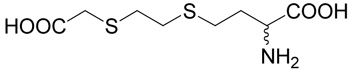 |
100 | 96.8 | 0.096 |
| 20 | 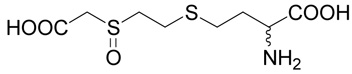 |
89.8 | 55.3 | nd |
| 21 | 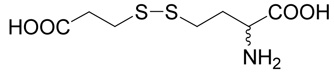 |
30 | 0 | nd |
| 22 | 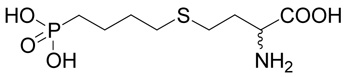 |
97.8 | 74 | 5.7 |
| 23 | 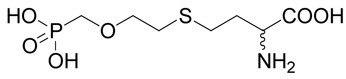 |
25.5 | nd | nd |
| 24 | 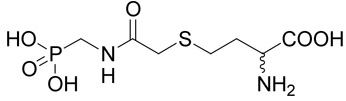 |
8.8 | nd | nd |
| 25 | 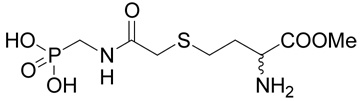 |
0 | nd | nd |
| 26 | 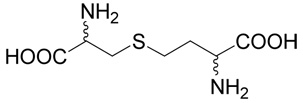 |
18.8 | nd | nd |
| 27 | 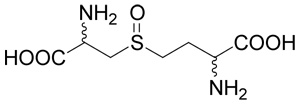 |
9.5 | nd | nd |
| 28 | 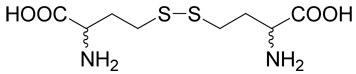 |
0 | nd | nd |
All assays were done in triplicates and the data obtained were reproducible within ± 15%.
All data points for IC50 values were derived from assays performed in duplicates, and the values obtained from 3 different assays were reproducible within ± 10%. nd means not-determined.
Table 2.
Structures of alkylating agents and thiols used for the synthesis of BHMT inhibitors. The yields are reported after HPLC purification. For details see Experimental. The structures of products are shown in Table 1.
| Alkylating agent | Thiol | Reaction Product | Yield (%) | ||
|---|---|---|---|---|---|
| 29 | 5 | 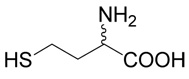 |
2 | 31a, 76c | |
| 30 | 5 | 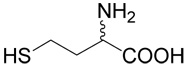 |
8 | 20a | |
| 31 | 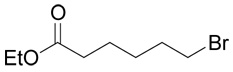 |
5 | 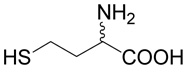 |
9 | 27a |
| 29 | 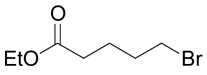 |
6 | 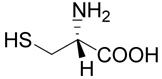 |
12 | 36a |
| 7 | 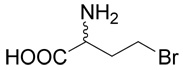 |
32 | 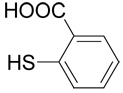 |
13 | 36b |
| 7 | 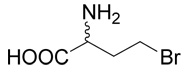 |
33 | 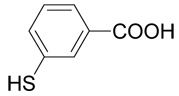 |
14 | 28b |
| 7 | 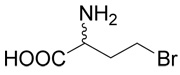 |
34 | 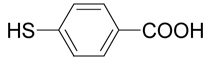 |
15 | 19b |
| 35 | 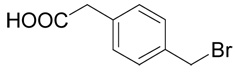 |
5 | 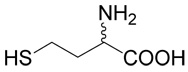 |
16 | 15a |
| 36 | 5 | 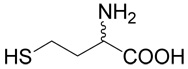 |
17 | 69a | |
| 37 | 5 | 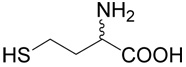 |
18 | 2a | |
| 38 | 5 | 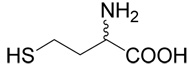 |
19 | 53a | |
| 39 | 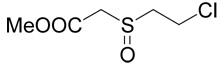 |
5 | 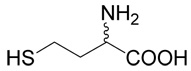 |
20 | 21a |
| 40 | 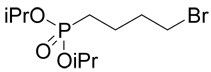 |
5 | 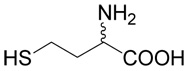 |
22 | 46a |
| 41 | 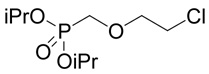 |
5 | 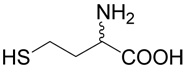 |
23 | 8a |
| 42 | 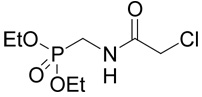 |
5 | 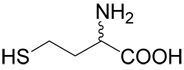 |
24 | 52a |
Product was prepared according to the Method A.
Product was prepared according to the Method B.
Product was prepared according to Method C.
The average yields of Methods A and B were around 30%. The lowest yield was 2 % for compound 18, probably due to the instability of its dithioformal moiety under acidic treatment, and the highest yield was 69 % for compound 17. The average yield was relatively low, even considering the products were purified by RP-HPLC. To investigate the possibility that our low yields were due to the oxidation of homocysteine, we monitored the ratio of homocysteine/homocystine in a reaction mixture (Method A) for 48 h using capillary electrophoresis (data not shown). We found that after 24 h the ratio was 89/11, and after 48 h it was 20/80. Since we used 3 equivalents of homocysteine to 1 equivalent of alkylating agent in our reactions, the consumption of the S-alkylating agent in aqueous alkaline media rather than the oxidation of homocysteine was responsible for the relatively low yields. In another experiment (Method C), we prepared compound 2 by a reaction involving the reduction of homocystine by sodium in liquid ammonia. This procedure improved our yield of compound 2 to 76 %, and represents a suitable method for the preparation of our S-alkylated homocysteine derivatives in higher yields. However, we used Methods A or B because they allowed us to rapidly prepare our target compounds in quantities sufficient for testing.
At first, we determined the percentage inhibition of BHMT using the test compounds at 20 µM. For the most potent compounds, we then determined the percentage inhibition at 1 µM, and also their IC50 values. The percentages of inhibition were measured at relatively low concentrations of substrates, 0.25 mM betaine and 100 µM D,L-homocysteine (Km of BHMT for betaine and D,L-homocysteine is 2 mM and 8 µM, respectively), in order to maximize our ability to detect inhibition. In contrast, we measured IC50 values of the most potent inhibitors at higher concentrations of substrates (2 mM betaine and 1 mM D,L-homocysteine) so that we could determine IC50 values in measurable concentrations and more accurately. The results of these inhibition experiments are summarized in Table 1.
Compound 2, originally designed by Awad et al27, inhibited BHMT very strongly (IC50 ~ 0.087 µM). This was surprising since Awad et al reported a Ki value of only 6.5 µM. To investigate this discrepancy we decided to re-evaluate the Kiappof this compound, which proved to be difficult because of the low kcat of the BHMT reaction and the high affinity of compound 2 for the enzyme. However, using a very low concentration of enzyme with high specific activity 14C-betaine, we determined that inhibitor 2 shows competitive inhibition relative to betaine and that is has a Kiappof 12 ± 0.9 nM (Figure 1). Although our estimate of Kiapp is much lower than the value reported by Awad et al27, it is in better agreement with the Kd of the BHMT-compound 2 complex recently measured using intrinsic fluorescence28. Why is compound 2 in our hands much more potent inhibitor of BHMT than previously published by Awad et al?27 The discrepancy could be that our compound was more pure. We purified compound 2 by RP-HPLC whereas they used a combination of DEAE-cellulose and Sephadex G-10 chromatography. The NMR and mass spectrometry data are not available. Therefore, it cannot be excluded that compound 2, as prepared by Awad et al, was not pure despite analyses by TLC, electrophoresis and amino acid analyzer. In addition, we used recombinant enzyme whereas Awad et al used enzyme isolated from human liver, and so it is possible that there are unknown differences in the kinetic properties of these enzymes.
Figure 1.

Determination of Kiapp of inhibitor 2 for human BHMT towards betaine. The curves were measured at fixed concentration of D,L-homocysteine (100 µM) and four different concentrations of betaine (a, 0.5 mM; b, 1 mM; c, 2 mM; d, 4 mM). The intersection point of curves gives Kiapp of about 12.0 ± 0.9 nM. For details see Experimental.
The IC50 value (87 nM) obtained for compound 2 is less than half of the concentration of BHMT (200 nM) used in the assays. There is no evidence to suggest that this discrepancy can be explained by an allosteric interaction of the inhibitor with the enzyme. It is possible that the amount of active enzyme used in these reactions were overestimated since the Bradford35 procedure cannot discern active from inactive protein. The loss of Zn2+ or enzyme denaturation could be factors that weren't corrected for. Additionally, although it is accepted that BHMT is a tetramer composed of identical subunits best described as a dimer of dimers, it is not known whether a tetramer can catalyze four reactions simultaneously. Although kinetically there is no evidence of subunit interaction, it has been shown that residues from both monomers within a dimer pair are required to form an active site8, and that some structural elements of one monomer undergoes movement when the active site of its partner becomes occupied with ligand(s).28 Hence, it is possible that only 1 of the 2 active sites that make up a dimer can be active at any given instant.
Compounds 8 and 9 are analogues of inhibitor 2 with shorter and longer alkyl chains, respectively. Compound 8 has been already prepared by Awad et al27 and we confirmed that the shortening of carboxybutyl chain results in a drastic loss of affinity towards BHMT. On the other hand, compound 9 with carboxypentyl chain is still an excellent inhibitor of BHMT with the IC50 value only slightly weaker that that of compound 2. Compounds 10 and 11 are oxidation products of inhibitor 2. Both these compounds are much weaker inhibitors than the parent compound 2, decreasing in potency with increasing degree of oxidation at the sulfur atom. However, it is interesting that sulfoxide 10 is still much better tolerated than the shortened compound 8. We also synthesized compound 12, which is the “cysteine” analogue of inhibitor 2. The absolute lack of inhibition of BHMT by this compound, even at 20 µM concentration, underlines the selectivity of BHMT for homocysteine.
The crystal structure of BHMT in a complex with inhibitor 2 revealed that the carboxybutyl chain of inhibitor 2 is surrounded by a series of aromatic residues4. Therefore we investigated compounds 13–15, which have a 2-, 3- or 4-carboxyphenyl group instead of the carboxybutyl moiety of inhibitor 2. Only derivative 15 with the 4-carboxyphenyl group replacing the carboxybutyl group in 2 is a relatively potent BHMT inhibitor with an IC50 about 7 µM. Inhibitor 16, with two extra methylene groups located both between the carboxyl group and phenyl ring, and the sulfur atom and phenyl ring, exhibited a marginal affinity.
Compound 17, which has the allyl group, is inactive. However, the inhibitor 18 of the same length as compound 17 but having dithioformal moiety in the chain still retains some activity. We hypothesize that the second sulfur atom of compound 18 could also interact with the Zn2+ of BHMT and that the analogue of 2, S-(2-carboxyethylthiomethyl)homocysteine, containing dithioformal moiety could be a very potent inhibitor of BHMT. Unfortunately, our attempts to prepare this inhibitor failed, possibly due to the low stability of S-CH2-S moiety under acidic conditions. This hypothesis seems to be supported by the potency we observed for compound 19, which is of the same length as compound 2, includes the terminal carboxylate but contains the S-CH2-CH2-S moiety. Inhibitor 19 has about the same high affinity for BHMT as the “reference” compound 2. Sulfoxide 20 is a much weaker inhibitor of BHMT. Compound 21 is the mixed disulfide of D,L-homocysteine and mercaptopropionic acid. This compound is slightly more active than inhibitor 8, which has the same length.
According to Evans et al4, the carboxyl group of the carboxybutyl moiety of inhibitor 2 forms two hydrogen bonds with side chains of Tyr77 and Trp44 of BHMT. We replaced the carboxyl by the more acidic phosphonate moiety and introduced two different structural alterations into the butyl chain. Of the phosphonate analogues 22–24, only compound 22 retains significant affinity to BHMT, but it remains much weaker than compound 2. Introduction of the oxygen atom or amide bond (analogues 23 and 24) into the butyl chain of respective phosphonate analogues results only in further decrease of binding affinity. Compound 25 is the methyl ester of analogue 24 and the lack of affinity confirms the crucial importance of free carboxyl of the homocysteine moiety of these inhibitors.
Derivatives 26–28 are naturally occurring compounds; cystathionine, its sulfoxide and homocystine, respectively. All these compounds contain a homocysteine moiety but differ in substituents on sulfur atom. Since these compounds participate in the metabolism of sulfur amino acids and could influence activity of BHMT in vivo, we decided to test them as inhibitors of BHMT. However, none of these compounds inhibit BHMT to any significant degree. We believe that cystathionine and homocystine are not inhibitors of BHMT in vivo.
CONCLUSIONS
We synthesized a series of S-substituted derivatives of homocysteine and evaluated them as potential inhibitors of human recombinant BHMT. Some of these compounds are very potent inhibitors, having IC50 values in the nanomolar range. We found that compound 2, (R,S)-5-(3-amino-3-carboxy-propylsulfanyl)-pentanoic acid, is a much more potent inhibitor of BHMT than previously reported. We determined its Kiapp towards betaine to be about 12 nM. We found that BHMT is very sensitive to any modification in the structure of inhibitor 2 since most analogues were less active than this parent compound. Nevertheless, we found that elongating the alkyl chain by one methylene group leads to the very potent inhibitor 9, and that an additional sulfur atom in the otherwise alkyl chain is well tolerated (inhibitor 19). Compounds 9 and 19 are of similar potency towards BHMT as inhibitor 2. These compounds are the most potent inhibitors of BHMT prepared to date. All these compounds were prepared as mixtures of enantiomers. Evans et al4 found that only S-enantiomer binds to the active site of BHMT. We assume that only S-enantiomers of our compounds inhibit BHMT and that respective IC50′s or Kiapp′s will be lower than reported in this study. Compound 2 is also a very selective inhibitor of BHMT because it does not inhibit other enzymes involved in sulfur metabolism, such as methionine synthase, cystathionine-β-synthase and cystathionase (data not shown). Our inhibitors are currently being tested in vivo in mice to study the influence of BHMT on the metabolism of sulfur amino acids (Collinsova, Strakova, Jiracek and Garrow, manuscript submitted).
EXPERIMENTAL
Chemistry. General
Unless otherwise stated, materials were obtained from commercial suppliers (Sigma-Aldrich, Fluka, Merck) and used without purification. 4-Mercaprobenzoic acid (34) was purchased from TCI America (Portland, OR). Compound 26, R,S,R,S-2-amino-4-(2-amino-2-carboxy-ethylsulfanyl)-butyric acid (cystathionine) was purchased from Sigma-Aldrich. Compound 28, R,S,R,S-2-amino-4-(3-amino-3-carboxy-propyldisulfanyl)-butyric acid (D,L-homocystine) was purchased from Fluka. Column chromatography was performed on silica gel 60 (70–230 mesh). Preparative RP-HPLC was performed using Vydac (218TP510, 25 × 1 cm; Columbia, MD, USA) or Phenomenex (Luna C-18, 5µm, 25 × 2.12 cm, Torrance, CA, USA) columns. Analytical RP-HPLC was performed using a Watrex (Nucleosil 120, 5µm, C18, 25 × 0.46 cm; Prague, Czech Republic) column. For gradient RP-HPLC analysis, a Waters LC 625 System (Milford, MA, USA) was used. Different gradients acetonitrile (1–80 %) in water containing 0.1% (v/v) of TFA were used for the elution of compounds. Anion-exchange analytical HPLC was performed using a AS11-HC column (0.2 × 25 cm, Dionex Corporation, Sunnyvale, CA) with a BioLC system (GP50 gradient pump, ED50 electrochemical detector) from Dionex Corporation (Sunnyvale, CA). Mass spectroscopy was performed using a ZAB-EQ spectrometer with BEQQ geometry (VG Analytical; Manchester, UK). NMR spectra were recorded on Bruker AVANCE-500 and Varian UNITY-500 (1H at 500 MHz; 13C at 125.7 MHz) in CDCl3, DMSO-d6 or D2O solutions. Chemical shifts are given in ppm (referenced to tetramethylsilane) and coupling constants in Hz.
Method A for the preparation of S-alkylated derivatives of homocysteine (used for compounds 2, 8, 9, 12, 16–20, 22–25)
Respective halogenated agent (1 mmole; compounds 29–31, 35–39 and 40–42 shown in Table 2) was added to D,L-homocysteine (5) or L-cysteine (6) (3 mmoles) in 10% sodium carbonate in 50% aqueous ethanol (6 ml) and stirred under argon at room temperature. After 48 h, if needed, sodium hydroxide was added to 1 M concentration. After one h the reaction mixture was applied to Dowex 50W (H+), the resin was washed with water and the compound was eluted with 2.5% ammonia. After evaporation, the product was purified by RP-HPLC.
Method B for the preparation of S-alkylated derivatives of homocysteine (used for compounds 13–15)
Respective mercapto-benzoic acid (1 mmole; compounds 32–34 shown in Table 2) was dissolved in 50% aqueous ethanol (10 ml) containing sodium carbonate (528 mg, 5 mmoles). Then, the hydrobromide of compound 7 (210 mg, 0.8 mmole) was added. The reaction was stirred under argon at room temperature overnight. The reaction mixture was applied to Dowex 50W (H+), the resin was washed with water and the compound was eluted with 2.5% ammonia. After evaporation, the product was purified by RP-HPLC.
(R,S)-5-(3-Amino-3-carboxy-propylsulfanyl)-pentanoic acid (2)
was first prepared according to Method A using ethyl-5-bromo-pentanoate (29; 1.2 mmoles, 251 mg). The yield was 88 mg (31%). 1H NMR (DMSO): δ 1.55 (m, 4H), 1.80 (m, 1H), 1.96 (m, 1H), 2.22 (t, J = 7.0 Hz, 2H), 2.48 (t, J = 7.0 Hz, 1H), 2.56 (m, 2H), 3.27 (dd, J = 7.2 and 5.6 Hz, 1H); 13C NMR (DMSO): δ 23.87, 27.42, 28.69, 30.47,31.45, 33.53, 53.28, 174.64, 174.65. HR-MS (FAB) calculated for C9H18NO4S (MH+) 236.0957, found 236.0961.
Compound 2 was also prepared according to the modified procedure (Method C) as follows. D,L-homocystine (1 mmole, 0.236 g) was dissolved in liquid ammonia (about 30 ml) in a cooled flask and sodium (4.3 mmoles, 0.1 g) was slowly added in small pieces until the reaction mixture turned blue. Ethyl-5-bromo-pentanoate acid (29; 2.2 mmoles, 0.46 g) was then added and the reaction proceeded without cooling until the ammonia was completely evaporated. The dry residue was dissolved in 30 ml of water, and sodium hydroxide was added to 1 M (final). After one h, the reaction mixture was applied to Dowex 50W (H+), and the resin was washed with water and the compound eluted with 2.5% ammonia. After evaporation, the product was purified by RP-HPLC. The yield was 0.358 g (76 %). The quality of the product was verified with MS and NMR.
Hydrobromide of (R,S)-2-amino-4-bromo-butyric acid (7)
The title compound was prepared according to Farrington at al36 with several modifications. (R,S)-2-Amino-4-butyrolactone hydrobromide (1 g, 5.5 mmoles) was heated to 60–65°C for 48 h in a sealed tube with 20 ml of hydrobromic acid (33%) in acetic acid. The reaction mixture was evaporated to dryness and the residue was treated with diethyl ether. White crystals of the hydrobromide of (R,S)-2-amino-4-bromo-butyric acid (7), were washed with diethyl ether. The yield was 1.42 g (99%). 1H NMR (D2O): δ 2.43 (m, J = 15.2, 7.3, 7.2, 6.1 Hz, 1H), 2.59 (m, J = 15.2, 7.3, 7.2, 6.1 Hz, 1H), 3.62 (ddd, J = 10.8, 7.2, 6.1 Hz, 1H), 3.67 (ddd, J = 10.8, 7.2, 6.1 Hz, 1H), 4.26 (t, J = 7.3, 7.3 Hz, 1H); 13C NMR (D2O): δ 29.92, 34.68, 53.44, 173.30. MS (FAB) calculated for C4H9NO2Br (MH+) 183.9796 and 181.9817, found 183.9796 and 181.9818.
(R,S)-2-Amino-4-(3-carboxy-propylsulfanyl)-butyric acid (8)
The title compound was prepared from ethyl-4-bromo-butyrate (30; 195 mg, 1 mmole) according to Method A. The yield was 49 mg (20 %). 1H NMR (D2O): δ 1.90 (p, J = 7.5 Hz, 2H), 2.16 (m, J = 15.0, 7.2, 7.2 and 6.8 Hz, 1H), 2.26 (m, J = 15.0, 7.2, 7.2 and 5.9 Hz, 1H), 2.50 (t, J = 7.5 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H), 2.71 (t, J = 7.5 Hz, 2H), 4.13 (dd, J = 6.8 and 5.9 Hz, 1H); 13C NMR (D2O): δ 26.80, 28.94, 32.37, 32.66, 35.92, 55.03, 175.07, 180.90; HR-MS (FAB) calculated for C8H16NO4S (MH+) 222.0800, found 222.0806.
(R,S)-6-(3-Amino-3-carboxy-propylsulfanyl)-hexanoic acid (9)
The title compound was prepared from ethyl-6-bromo-hexanoate (31; 223 mg, 1 mmole) according to Method A. The yield was 68 mg (27 %). 1H NMR (DMSO): δ 1.34 (m, 2H), 1.51 (m, 4H), 1.90 (m, 1H), 2.00 (m, 1H), 2.20 (t, J = 7.4 Hz, 2H), 2.47 (t, J = 7.3 Hz, 2H), 2.57 (ddd, J = 13.5, 9.6 and 5.6 Hz, 1H), 2.60 (ddd, J = 13.5, 9.5 and 6.5 Hz, 1H), 3.63 (dd, J = 6.5 and 5.6 Hz, 1H); 13C NMR (DMSO): δ 24.27, 26.91, 27.92, 28.90, 30.67, 31.01, 33.85, 52.34, 170.58, 174.66; HR-MS (FAB) calculated for C10H20NO4S (MH+) 250.1035, found 250.1059.
(R,S)-5-(3-Amino-3-carboxy-propylsulfinyl)-pentanoic acid (10)
Compound 2 (10 mg, 42 µmoles) was suspended in 100 µl of water containing 46 µmoles of hydrochloric acid and 50 µmoles of hydrogen peroxide. The suspension was stirred at room temperature overnight. The product was purified by RP-HPLC. The yield was 9 mg (85 %). 1H NMR (D2O): δ 1.70–1.80 (m, 4H), 2.25–2.37 (m, 4H), 2.90–3.15 (m, 4H); 13C NMR (D2O): mixture of diastereoisomers (1:1) leads to doubling of some carbon signals (shown in brackets), δ 23.94 (23.96), 26.10 (26.15), 26.79, 38.65, 48.38 (48.40), 52.44 (52.52), 55.56 (55.74), 175.40, 184.65; HR-MS (FAB) calculated for C9H18NO5S (MH+) 252.0906, found 252.0895.
(R,S)-5-(3-Amino-3-carboxy-propylsulfonyl)-pentanoic acid (11)
Compound 2 (10 mg, 42 µmoles) was suspended in 100 µl of water containing 450 µmoles of hydrochloric acid and 200 µmoles of hydrogen peroxide. The suspension was stirred at room temperature overnight. The product was purified by RP-HPLC. The yield was 7.3 mg (65 %). 1H NMR (D2O): δ 1.77 (m, 2H), 1.87 (m, 2H), 2.37 (m, 2H), 2.45 (t, J = 7.3 Hz, 2H), 3.32 (m, 2H), 3.36 (ddd, J = 14.0, 9.8 and 6.2 Hz, 1H), 3.44 (ddd, J = 14.0, 9.6 and 6.6 Hz, 1H), 3.89 (t, J = 6.2 Hz, 1H); 13C NMR (D2O): δ 22.27, 24.49, 24.80, 35.00, 49.94, 53.44, 54.65, 174.33, 179.86; HR-MS (FAB) calculated for C9H18NO6S (MH+) 268.0840, found 268.0855.
(R)-5-(2-Amino-2-carboxy-ethylsulfanyl)-pentanoic acid (12)
The title compound was prepared from ethyl-6-bromo-pentanoate (29; 209 mg, 1 mmole) according to Method A. The yield was 80 mg (36 %). 1H NMR (D2O + NaOD): δ 1.58 (m, 2H), 1.62 (m, 2H), 2.19 (t, J = 7.2 Hz, 2H), 2.59 (t, J = 7.1 Hz, 2H), 2.76 (dd, J = 13.4 and 6.8 Hz, 1H), 2.85 (dd, J = 13.4 and 5.2 Hz, 1H), 3.40 (dd, J = 6.8 and 5.2 Hz, 1H); 13C NMR (D2O + NaOD): δ 27.85, 31.54, 34.14, 39.69, 39.86, 57.95, 183.91, 186.38; HR-MS (FAB) calculated for C8H16NO4S (MH+) 222.0800, found 222.0804.
(R,S)-2-(3-Amino-3-carboxy-propylsulfanyl)-benzoic acid (13)
The title compound was prepared from 2-mercapto-benzoic acid (32; 106 mg, 0.69 mmole) according to the Method B. The yield was 52 mg (36 %). 1H NMR (DMSO): δ 2.09 (m, 2H), 3.02 (ddd, J = 13.0, 9.3 and 5.8 Hz, 1H), 3.08 (ddd, J = 13.0, 9.6 and 6.4 Hz, 1H), 4.00 (t, J = 6.2 Hz, 2H), 7.24 (ddd, J = 7.8, 7.5 and 0.9 Hz, 1H), 7.40 (bd, J = 7.8 Hz, 1H), 7.54 (ddd, J = 7.8, 7.5 and 1.7 Hz, 1H), 7.89 (dd, J = 7.8 and 1.7 Hz, 1H), 8.30 (b, 2H), 13.20 (vb, 1H); 13C NMR (DMSO): δ 26.64, 29.28, 51.74, 124.38, 125.55, 128.67, 131.26, 132.58, 139.86, 167.62, 170.69; HR-MS (FAB) calculated for C11H14NO4S (MH+) 256.0644, found 256.0649.
(R,S)-3-(3-Amino-3-carboxy-propylsulfanyl)-benzoic acid (14)
The title compound was prepared from 3-mercapto-benzoic acid (33; 120 mg, 0.78 mmole) according to the Method B. The yield was 46 mg (28 %). 1H NMR (DMSO): δ 2.04 (m, 2H), 3.14 (m, 2H), 3.93 (t, J = 6.4 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.59 (ddd, J = 7.8, 2.1 and 1.1 Hz, 1H), 7.77 (ddd, J = 7.8, 1.6 and 1.1 Hz, 1H), 7.85 (dd, J = 2.1 and 1.6 Hz, 1H); 8.30 (b, 2H), 13.20 (vb, 1H); 13C NMR (DMSO): δ 28.06, 29.91, 51.57, 127.04, 128.91, 129.62, 131.96, 132.53, 136.23, 166.96, 170.56; HR-MS (FAB) calculated for C11H14NO4S (MH+) 256.0644, found 256.0651.
(R,S)-4-(3-Amino-3-carboxy-propylsulfanyl)-benzoic acid (15)
The title compound was prepared from 4-mercapto-benzoic acid (34; 185 mg, 1.2 mmole) according to the Method B. The yield was 47 mg (19 %). 1H NMR (DMSO + AcOD): δ 1.99 (m, 1H), 2.07 (m, 1H), 3.14 (ddd, J = 13.5, 9.7 and 5.7 Hz, 1H), 3.20 (ddd, J = 13.5, 9.7 and 6.0 Hz, 1H), 3.61 (t, J = 6.1 Hz, 1H), 7.39 (m, 2H), 7.85 (m, 2H); 13C NMR (DMSO + AcOD): δ 27.17, 30.40, 52.62, 126.42 (2C), 127.59, 130.14 (2C), 142.83, 167.18, 170.52; HR-MS (FAB) calculated for C11H14NO4S (MH+) 256.0644, found 256.0650.
(R,S)-2-Amino-4-(4-carboxymethyl-benzylsulfanyl)-butyric acid (16)
The title compound was prepared from (4-bromomethyl-phenyl)-acetic acid (35; 229 mg, 1 mmole) according to Method A. The yield was 43 mg (15 %). 1H NMR (DMSO + D2O): δ 1.93 (m, 1H), 2.03 (m, 1H), 2.48 (m, 2H), 3.52 (s, 2H), 3.58 (t, J = 6.0 Hz, 1H), 3.68 (s, 2H), 7.17 (m, 2H), 7.23 (m, 2H); 13C NMR (DMSO + D2O): δ 27.16, 30.80, 34.66, 40.51, 52.66, 128.77 (2C), 129.48 (2C), 133.68, 136.92, 170.93, 173.56; HR-MS (FAB) calculated for C13H18NO4S (MH+) 284.0878, found 284.0899.
(R,S)-4-Allylsulfanyl-2-amino-butyric acid (17)
The title compound was prepared from 3-bromo-propene (36; 120 mg, 1 mmole) according to Method A. The yield was 121 mg (69 %). 1H NMR (DMSO): δ 1.99 (m, 2H), 2.54 (m, 2H), 3.15 (ddd, J = 7.2, 1.3 and 0.9 Hz, 2H), 3.86 (t, J = 6.3 Hz, 1H), 5.08 (m, J = 10.0, 1.8, 0.9 and 0.9 Hz, 1H), 5.13 (m, J = 17.1, 1.8, 1.3 and 1.3 Hz, 1H), 5.76 (m, J = 17.1, 10.0, 7.2 and 7.2 Hz, 1H), 8.27 (bs, 1H); 13C NMR (DMSO): δ 25.63, 30.27, 33.49, 51.59, 117.46, 134.51, 170.84; HR-MS (FAB) calculated for C7H14NO2S (MH+) 176.0745, found 176.0749.
(R,S)-2-Amino-4-methylsulfanylmethylsulfanyl-butyric acid (18)
The title compound was prepared from chloro-methylsulfanyl-methane (37; 96 mg, 1 mmole) according to Method A. The yield 14 was 4 mg (2 %). 1H NMR (DMSO): δ 1.85 (m, J = 14.4, 7.8, 7.8 and 7.0 Hz, 1H), 2.01 (m, J = 14.4, 7.8, 7.8 and 5.4 Hz, 1H), 2.10 (s, 3H), 2.68 (t, J = 7.8 Hz, 2H), 3.36 (m, 1H), 3.73 (s, 2H), 7.86 (vb, 2H); 13C NMR (DMSO): δ 14.19, 26.89, 30.93, 36.68, 52.99, 169.75; HR-MS (FAB) calculated for C6H14NO2S2 (MH+) 196.0466, found 196.0429.
(R,S)-2-Amino-4-(2-carboxymethylsulfanyl-ethylsulfanyl)-butyric acid (19)
Methyl-mercapto-acetate (1.485 mg, 14 mmoles) was added drop-wise, on ice and under argon atmosphere, to triethylamine (1.413 g, 14 mmoles) in dichloroethane (15 ml). After 18 h at room temperature, the reaction mixture was evaporated and the product, methyl-(2-chloro-ethylsulfanyl)-acetate (38), was purified on the column of silica gel using a linear gradient of ethyl acetate in toluene (0–5%) with the yield of 1.1 g (47%). The quality of the product was verified with MS and NMR. The title compound (19) was prepared from methyl (2-chloro-ethylsulfanyl)-acetate (38, prepared as described above; 150 mg, 0.89 mmole) according to Method A. The yield was 120 mg (53 %). 1H NMR (DMSO): δ 1.89 (m, 1H), 2.01 (m, 1H), 2.60 (m, 2H), 2.70–2.80 (m, 4H), 3.25 (d, J = 14.5 Hz, 1H), 3.28 (d, J = 14.5 Hz, 1H), 3.58 (m, 1H); 13C NMR (DMSO): δ 26.94, 30.55, 31.12, 32.11, 33.65, 52.65, 170.74, 172.14; HR-MS (FAB) calculated for C8H16NO4S2 (MH+) 254.0521, found 254.0533.
(R,S)-2-Amino-4-(2-carboxymethylsulfinyl-ethylsulfanyl)-butyric acid (20)
Methyl-(2-chloro-ethylsulfanyl)-acetate (38, prepared as described above; 0.336 g, 2 mmoles) was oxidized with hydrochloric acid and hydrogen peroxide as described for compound 10 Yield 0.206 g (56%). The resulting (2-chloro-ethanesulfinyl)-acetic acid methyl ester (39; 0.17 g, 0.92 mmole) was reacted with D,L-homocysteine according to Method A. The yield was 53 mg (21 %). 1H NMR (D2O): δ 2.21 (m, 1H), 2.30 (m, 1H), 2.80 (m, 2H), 2.98 (m, 1H), 3.08 (m, 1H), 3.30 (m, 2H), 3.92 (d, J = 15.0 Hz, 1H), 4.04 (d, J = 15.0 Hz, 1H), 4.14 (t, J = 6.4 Hz, 1H); 13C NMR (D2O): mixture of diastereoisomers (1:1) leads to doubling of some carbon signals (shown in brackets), δ 25.83 (25.91), 28.71 (28.82), 31.72 (31.75), 52.67 (52.70), 54.40 (54.42), 57.38, 171.60, 174.54; HR-MS (FAB) calculated for C8H16NO5S2 (MH+) 270.0470, found 270.0466.
(R,S)-2-Amino-4-(2-carboxy-ethyldisulfanyl)-butyric acid (21)
D,L-homocysteine (5; 202 mg, 1.5 mmole) and methyl-mercapto-acetate (178 mg, 1.5 mmole) were treated with hydrogen peroxide (169 µl, 1.65 mmole) dissolved in 20% aqueous ethanol (12 ml) at room temperature overnight. The reaction mixture was applied to Dowex 50W (H+), the resin was washed with water and the products eluted with 2.5 % ammonia. After evaporation, the product was purified with RP-HPLC. The yield was 42 mg (11 %). 1H NMR (D2O): δ 2.32 (m, J = 14.8, 7.5, 7.0 and 6.9 Hz, 1H), 2.40 (m, J = 14.8, 7.5, 7.5 and 6.0 Hz, 1H), 2.83 (m, 2H), 2.87 (m, 2H), 2.98 (m, 2H), 4.16 (dd, J = 6.9 and 6.0 Hz, 1H); 13C NMR (D2O): δ 31.99, 35.13, 35.26, 36.29, 54.59, 174.74, 179.28; HR-MS (FAB) calculated for C7H14NO4S2 (MH+) 240.0364, found 240.0370.
(R,S)-2-Amino-4-(4-phosphono-butylsulfanyl)-butyric acid (22)
The reaction of diisopropyl-(4-bromo-butyl)-phosphonate37 (40; 0.3 g, 1 mmole) with D,L-homocysteine (5) according to Method A afforded (R,S)-2-amino-4-[4-(diisopropoxy-phosphoryl)-butylsulfanyl]-butyrate in the yield of 0.164 g (46 %). 1H NMR (DMSO): δ 1.227 (d, J = 6.2 Hz, 6H), 1.230 (d, J = 6.2 Hz, 6H), 1.54 (m, 2H), 1.59 (m, 2H), 1.67 (m, 2H), 2.01 (m, 2H), 2.51 (t, J = 7.0 Hz, 2H), 2.55 (ddd, J = 13.5, 9.1 and 6.0 Hz, 1H), 2.62 (ddd, J = 13.5, 9.2 and 6.5 Hz, 1H), 3.99 (bt, J ~ 6 Hz, 1H), 4.53 (dh, 3J(H,P) = 8.0, J(H,H) = 6.2 Hz, 1H), 4.54 (h, J = 6.2 Hz, 1H), 8.28 (b, 2H); 13C NMR (DMSO): δ 21.53 (d, 3J(C,P) = 4.9 Hz), 23.98 (d, 3J(C,P) = 4.4 Hz, 4C), 25.57 (d, 1J(C,P) = 140.6 Hz), 26.40, 29.52, 30.19, 30.33, 51.24, 69.22 (d, 2J(C,P) = 6.3 Hz), 170.90; HR-MS (FAB) calculated for C14H31NO5PS (MH+) 356.1661, found 356.1651. This compound (0.156 g, 0.44 mmole) was treated with bromotrimethylsilane (0.456 g, 3 mmoles) in dry DMF (3 ml) under argon atmosphere at 50°C for 2 h. Then, 5 ml of methanol was added and the reaction was warmed up at 50°C for an additional h. The reaction mixture was evaporated and the residue was partioned between water and ethyl acetate. The water layer was evaporated to dryness and the product, compound 22, was purified by RP-HPLC. The yield was 76 mg (64%). 1H NMR (D2O): δ 1.61–1.89 (m, 6H), 2.18 (m, 1H), 2.28 (m, 1H), 2.62 (t, J = 7.0 Hz, 2H); 2.72 (t, J = 7.5 Hz, 2H), 4.16 (dd, J = 6.7 and 6.0 Hz, 1H); 13C NMR (D2O): δ 23.63 (d, 3J(C,P) = 4.7 Hz), 28.21 (d, 1J(C,P) = 134.2 Hz), 28.47, 31.59 (d, 2J(C,P) = 16.6 Hz), 31.75, 32.31, 54.29, 174.30; HR-MS (FAB) calculated for C8H19NO5PS (MH+) 272.0722, found 272.0734.
(R,S)-2-Amino-4-(2-phosphonomethoxy-ethylsulfanyl)-butyrate (23)
The reaction of diisopropyl-(2-chloro-ethoxymethyl)-phosphonate38 (41; 0.259 g, 1 mmole) with D,L-homocysteine (5) according to Method A afforded (R,S)-2-amino-4-[2-(diisopropoxy-phosphorylmethoxy)-ethylsulfanyl]-butyrate in the yield of 21 mg (8 %). 1H NMR (DMSO): δ 1.97 (m, 1H), 2.04 (m, 1H), 2.64 (m, 2H), 2.68 (t, J = 6.6 Hz, 2H), 3.66 (t, J = 6.6 Hz, 2H), 3.76 (d, J(H,P) = 8.4 Hz, 2H), 3.92 (dd, J = 6.6 and 5.9 Hz, 1H), 4.60 (dh, J(H,P) = 7.7 and J(H,H) = 6.2 Hz, 2H), 8.22 (vb, 2H); 13C NMR (DMSO): δ 23.93 (d, 3J(C,P) = 4.5 Hz, 2C), 24.03 (d, 3J(C,P) = 3.8 Hz, 2C), 27.04, 30.06, 30.52, 51.35, 64.77 (d, 1J(C,P) = 164.9 Hz), 70.39 (d, 2J(C,P) = 6.3 Hz), 72.08 (d, 3J(C,P) = 12.1 Hz), 170.85; HR-MS (FAB) calculated for C13H29NO6PS (MH+) 358.1453, found 358.1434. This compound (20 mg, 56 µmoles) was treated with bromotrimethylsilane (0.106 g, 0.7 mmole) in dry DMF (1 ml) under argon atmosphere at room temperature overnight. The reaction mixture was concentrated in vacuo and the residue was co-evaporated with 10% triethylamine in acetonitrile (3 × 1 ml) and then with water. The product 23 was purified by RP-HPLC. The yield was 15 mg (31%). 1H NMR (D2O): δ 2.21 (m, 1H), 2.30 (m, 1H), 2.78 (t, J = 7.5 Hz, 2H); 2.82 (t, J = 6.1 Hz, 2H), 3.70 (d, J(H,P) = 8.8 Hz, 2H), 3.79 (t, J = 6.0 Hz, 2H), 4.16 (dd, J = 6.6 and 6.0 Hz, 1H); 13C NMR (D2O): δ 27.60, 30.43, 31.03, 52.92, 67.08 (d, 1J(C,P) = 167.1 Hz), 72.59 (d, 3J(C,P) = 10.8 Hz), 172.94; HR-MS (FAB) calculated for C7H17NO6PS (MH+) 274.0516, found 274.0523
(R,S)-2-Amino-4-[(phosphonomethyl-carbamoyl)-methylsulfanyl]-butyrate (24)
The reaction of diethyl-[(2-chloro-acetylamino)-methyl]-phosphonate39 (42; 0.125 g, 0.51 mmole) with D,L-homocysteine (5) according to Method A afforded (R,S)-2-amino-4-{[(diethoxy-phosphorylmethyl)-carbamoyl]-methylsulfanyl}-butyrate in the yield of 92 mg (52%). 1H NMR (DMSO): δ 1.23 (t, J = 7.0 Hz, 6H), 2.00 (m, 1H), 2.08 (m, 1H), 2.68 (ddd, J = 13.5, 8.4 and 6.2 Hz, 1H), 2.73 (ddd, J = 13.5, 8.6 and 6.8 Hz, 1H), 3.19 (s, 2H), 3.58 (dd, J(H,P) = 11.6 and J(H,H) = 6.0 Hz, 2H), 3.96 (bdd, J = 6.8 and 6.2 Hz, 1H), 4.02 (m, 4H), 8.30 (vb, 2H), 8.43 (bt, J = 6.0 Hz, 1H); 13C NMR (DMSO): δ 16.41 (d, 3J(C,P) = 5.4 Hz, 2C), 27.37, 29.92, 33.68, 34.31 (d, 1J(C,P) = 157.1 Hz), 51.20, 62.00 (d, 2J(C,P) = 5.9 Hz, 2C), 169.17 (d, 3J(C,P) = 4.3 Hz), 170.87; HR-MS (FAB) calculated for C11H24N2O6PS (MH+) 343.11093, found 343.1097. This compound (19 mg, 55 µmoles) was treated with bromotrimethylsilane (61 mg, 450 µmoles) in dry DMF (1 ml) under argon atmosphere at 50°C overnight. The reaction mixture was concentrated in vacuo and the residue was co-evaporated with 10% triethylamine in acetonitrile (3 × 1 ml) and then with water. The product 24 was purified by RP-HPLC. The yield was 10 mg (62%). 1H NMR (D2O): δ 2.18 (m, 1H), 2.27 (m, 1H), 2.76 (m, 2H), 3.36 (d, J(H,P) = 0.6 Hz, 2H); 3.50 (d, J(H,P) = 12.4 Hz, 2H), 4.12 (t, J = 6.4 Hz, 1H); 13C NMR (D2O): δ 28.11, 30.23, 35.58, 38.26 (d, 1J(C,P) = 171.5 Hz), 52.92, 172.58, 172.77; HR-MS (FAB) calculated for C7H16N2O6PS (MH+) 287.0467, found 287.0459.
In a parallel experiment, (R,S)-2-amino-4-{[(diethoxy-phosphorylmethyl)-carbamoyl]- methylsulfanyl}-butyrate (19 mg, 55 µmoles) was treated with bromo trimethylsilane by the same manner as described above for compound 24. However, after completing of the deprotection, the reaction mixture was evaporated to dryness and then warmed up at 50°C for 1 h with methanol (5 ml). After evaporation, the product, methyl (R,S)-2-amino-4-[(phosphonomethyl-carbamoyl)-methylsulfanyl]-butyric acid ester (25), was purified by RP-HPLC. The yield was 5 mg (29%). 1H NMR (D2O): δ 2.21 (m, 1H), 2.31 (m, 1H), 2.78 (m, 2H), 3.36 (s, 2H), 3.51 (d, J(H,P) = 12.4 Hz, 2H), 3.86 (s, 3H), 4.30 (t, J = 6.5 Hz, 1H); 13C NMR (D2O): δ 29.52, 31.43, 37.05, 39.76 (d, 1J(C,P) = 146.5 Hz), 53.76, 55.82, 172.60, 174.30; MS (FAB) calculated for C8H18N2O6PS (MH+) 301.05, found 301.00.
(R,S,R,S)-2-Amino-4-(2-amino-2-carboxy-ethylsulfinyl)-butyric acid (27)
This oxidized derivative was prepared starting from commercial cystathionine (26; 0.1 mg, 0.45 mmoles) using the same procedure as for compound 10. The yield was 67 mg (63%). The presence of four diastereoisomers leads to the observation up to four signals for individual hydrogens and carbon atoms. 1H NMR (D2O): δ 2.35–2.51 (m, 2H), 3.09–3.33 (m, 2H), 3.51–4.03 (m, 2H), 4.15–4.20 (m, 1H), 4.43–4.47 (m, 1H); 13C NMR (D2O): δ 23.83, 23.89, 24.05 and 24.10 (CH2), 47.66, 47.77, 48.28 and 48.38 (S-CH2-), 50.83, 51.14, 51.18 and 52.61 (CH2-S), 50.00 and 50.45 (CH-N), 52.61 and 52.76 (CH-N), 171.00, 171.99; HR-MS (FAB) calculated for C7H15N2O5S (MH+) 239.0702, found 239.0694.
BHMT Inhibition Assays
Human recombinant BHMT was prepared as described previously3 N-methyl-14C-betaine (57 mCi/mmol) was prepared and supplied by Moravek Biochemicals (Brea, CA). Compounds were tested for their ability to inhibit BHMT activity using an assay procedure we have described previously in detail40 with only several modifications. Briefly, D,L-homocysteine was freshly prepared by dissolving D,L-homocysteine thiolactone hydrochloride (15.4 mg) in 400 µl of 2 M NaOH. The solution was allowed to stand for 5 min at room temperature. The solution was then neutralized by the addition of 600 µl of a saturated solution of KH2PO4 and immediately used in BHMT assay.
The standard BHMT assay (500 µl) used to determine the percent inhibition contained 0.2 µM BHMT, different concentrations of inhibitor (20 µM or 1 µM), 100 µM D,L-homocysteine, 250 µM betaine (0.05 µCi), 10 mM β-mercaptoethanol and 50 mM K-phosphate buffer pH 7.5. Human recombinant BHMT was first mixed with inhibitor(s), then the substrates were added and the mixture incubated at 37°C for 30 minutes. The reaction was stopped by transferring the reaction tubes into ice water and by adding 2.5 ml of ice-cold water. The samples were applied to a Dowex 1×4 (200–400 mesh) and the non-reacted betaine was washed from the column with water. Dimethylglycine and methionine were eluted into scintillation vials with 1.5 ml of 1.5 M HCl and then 10 ml of scintillation mixture were added into each vial and counted. Blanks contained all the reaction components except enzyme and their values were subtracted from the sample values. All samples were assayed in triplicates and results (reproducible within ± 15%) are expressed relative (%) to a sample containing no inhibitor.
Inhibition curves for the determination of IC50 values were measured using the conditions described above except the concentrations of substrates used were 1 mM D,L-homocysteine and 2 mM betaine (0.15 µCi). The inhibition at ten different inhibitor concentrations was determined for each curve. The data were analyzed by nonlinear regression fit using program GraphPad Prism3.02.
The apparent inhibition constant (Kiapp, cit.41–43) of inhibitor 2 towards betaine was measured at fixed concentration of D,L-homocysteine (100 µM) and varied concentrations of betaine (0.5, 1, 2 and 4 mM) and inhibitor (0, 15, 25, 50, 75 and 100 nM). The reaction proceeded in a total volume of 250 µl and contained 15 nM BHMT and 1 µCi of 14C-betaine. The enzyme was pre-incubated with inhibitor for 15 min at room temperature, and then the substrates were added and the reaction was allowed to proceed for 1 h at 37°C. The data were analyzed by Dixon plot.44
All points for determination of IC50′s or Kiapp′s were measured in duplicates and the values from 3 different assays are reproducible within ± 10%.
Supplementary Material
HPLC purity data and NMR spectra records for compounds 2, 9 and 19. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgement
This work was supported by a grant from the Grant Agency of the Academy of Sciences of the Czech Republic (A4055302, J.J.), Research Project of the Academy of Sciences of the Czech Republic (Z40550506, J.J.) and by grants from the National Institutes of Health (DK52501, T.A.G.) and the Illinois Agricultural Research Station (50–352, T.A.G.) and by NIH Research Grant funded by the Fogarty International Center (R01 TW0052501, T.A.G. and J.J.). We thank to Dr. Vaclav Kasicka (IOCB, Prague) for performing capillary electrophoreses.
References
- 1.Finkelstein JD, Harris BJ, Kyle WE. Methionine metabolism in mammals: kinetic study of betaine-homocysteine methyltransferase. Arch. Biochem. Biophys. 1972;153:320–324. doi: 10.1016/0003-9861(72)90451-1. [DOI] [PubMed] [Google Scholar]
- 2.Millian NS, Garrow TA. Human betaine-homocysteine methyltransferase is a zinc metalloenzyme. Arch. Biochem. Biophys. 1998;356:93–98. doi: 10.1006/abbi.1998.0757. [DOI] [PubMed] [Google Scholar]
- 3.Breksa AP, III, Garrow TA. Recombinant human liver betaine-homocysteine S-methyltransferase: identification of three cysteine residues critical for zinc binding. Biochemistry. 1999;38:13991–13998. doi: 10.1021/bi991003v. [DOI] [PubMed] [Google Scholar]
- 4.Evans JC, Huddler DP, Jiracek J, Castro C, Millian NS, Garrow TA, Ludwig ML. Betaine-homocysteine methyltransferase. Zinc in a distorted barrel. Structure. 2002;10:1159–1071. doi: 10.1016/s0969-2126(02)00796-7. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez B, Pajares MA, Martinez-Ripoll M, Blundell TL, Sanz-Aparicio J. Crystal structure of rat liver betaine homocysteine S-methyltransferase reveals new oligomerization features and conformational changes upon substrate binding. J. Mol.Biol. 2004;338:771–782. doi: 10.1016/j.jmb.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Peariso K, Goulding CW, Huang S, Matthews RG, Penner-Hahn JE. Characterization of the zinc binding site in methionine synthase enzymes of Escherichia coli: The role of zinc in the methylation of homocysteine. J. Am. Chem. Soc. 1998;120:8410–8416. [Google Scholar]
- 7.Peariso K, Zhou ZS, Smith AE, Matthews RG, Penner-Hahn JE. Characterization of the zinc sites in cobalamin-independent and cobalamin-dependent methionine synthase using zinc and selenium X-ray absorption spectroscopy. Biochemistry. 2001;40:987–993. doi: 10.1021/bi001711c. [DOI] [PubMed] [Google Scholar]
- 8.Szegedi SS, Garrow TA. Oligomerization is required for betaine-homocysteine S-methyltransferase function. Arch. Biochem. Biophys. 2004;426:32–42. doi: 10.1016/j.abb.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 9.McKeever MP, Weir DG, Molloy A, Scott JM. Betaine-homocysteine methyltransferase: organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. (Colch. ) 1991;81:551–556. doi: 10.1042/cs0810551. [DOI] [PubMed] [Google Scholar]
- 10.Kempson SA, Montrose MH. Osmotic regulation of renal betaine transport: transcription and beyond. Pflugers Arch. 2004;449:227–234. doi: 10.1007/s00424-004-1338-6. [DOI] [PubMed] [Google Scholar]
- 11.Wettstein M, Weik C, Holneicher C, Häussinger D. Betaine as an osmolyte in rat liver: metabolism and cell-to-cell interactions. Hepatology. 1998;27:787–793. doi: 10.1002/hep.510270321. [DOI] [PubMed] [Google Scholar]
- 12.Haussinger D. Neural control of hepatic osmolytes and parenchymal cell hydration. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004;280:893–900. doi: 10.1002/ar.a.20094. [DOI] [PubMed] [Google Scholar]
- 13.Delgado-Reyes CV, Garrow TA. High sodium chloride intake decreases betaine-homocysteine methyltransferase expression in guinea pig liver and kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R182–R187. doi: 10.1152/ajpregu.00406.2004. [DOI] [PubMed] [Google Scholar]
- 14.Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- 15.Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA. 2002;288:2015–2022. doi: 10.1001/jama.288.16.2015. [DOI] [PubMed] [Google Scholar]
- 16.Ray JG. Meta-analysis of hyperhomocysteinemia as a risk factor for venous thromboem bolic disease. Arch. Intern. Med. 1998;158:2101–2106. doi: 10.1001/archinte.158.19.2101. [DOI] [PubMed] [Google Scholar]
- 17.Vollset SE, Refsum H, Irgens LM, Emblem BM, Tverdal A, Gjessing HK, Monsen AL, Ueland PM. Plasma total homocysteine, pregnancy complications, and adverse pregnancy outcomes: the Hordaland homocysteine study. Am. J. Clin. Nutr. 2000;71:962–968. doi: 10.1093/ajcn/71.4.962. [DOI] [PubMed] [Google Scholar]
- 18.Ray JG, Laskin CA. Folic acid and homocyst(e)ine metabolic defects and the risk of placental abruption, pre-eclampsia and spontaneous pregnancy loss: A systematic review. Placenta. 1999;20:519–529. doi: 10.1053/plac.1999.0417. [DOI] [PubMed] [Google Scholar]
- 19.Bottiglieri T. Folate, vitamin B-12, and neuropsychiatric disorders. Nutr. Rev. 1996;54:382–390. doi: 10.1111/j.1753-4887.1996.tb03851.x. [DOI] [PubMed] [Google Scholar]
- 20.Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D'Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N. Engl. J. Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 21.Chauveau P, Chadefaux B, Coude M, Aupetit J, Hannedouche T, Kamoun P, Jungers P. Hyperhomocysteinemia, a risk factor for atherosclerosis in chronic uremic patients. Kidney Int. Suppl. 1993;41:S72–S77. [PubMed] [Google Scholar]
- 22.Finkelstein JD, Martin JJ. Methionine metabolism in mammals. Distribution of homocysteine between competing pathways. J. Biol. Chem. 1984;259:9508–9513. [PubMed] [Google Scholar]
- 23.Mato JM, Corrales FJ, Lu SC, Avila MA. S-adenosylmethionine: a control switch that regulates liver function. Faseb J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 24.Duranton B, Freund JN, Galluser M, Schleiffer R, Gosse F, Bergmann C, Hasselmann R, Raul F. Promotion of intestinal carcinogenesis by dietary methionine. Carcinogenesis. 1999;20:493–497. doi: 10.1093/carcin/20.3.493. [DOI] [PubMed] [Google Scholar]
- 25.Pavillard V, Nicolaou A, Double JA, Phillips RM. Methionine dependence of tumours: A biochemical strategy for optimizing paclitaxel chemosensitivity in vitro. Biochem. Pharmacol. 2006;71:772–778. doi: 10.1016/j.bcp.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 26.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 27.Awad WM, Jr, Whitney PL, Skiba WE, Mangum JH, Wells MS. Evidence for direct methyl transfer in betaine: homocysteine S-methyl- transferase. J. Biol. Chem. 1983;258:12790–12792. [PubMed] [Google Scholar]
- 28.Castro C, Gratson AA, Evans JC, Jiracek J, Collinsova M, Ludwig ML, Garrow TA. Dissecting the catalytic mechanism of betaine-homocysteine S-methyltransferase by use of intrinsic tryptophan fluorescence and site-directed mutagenesis. Biochemistry. 2004;43:5341–5351. doi: 10.1021/bi049821x. [DOI] [PubMed] [Google Scholar]
- 29.Lee KH, Cava M, Amiri P, Ottoboni T, Lindquist RN. Betaine:homocysteine methyltransferase from rat liver: Purification and inhibition by a boronic acid substrate analog. Arch. Biochem. Biophys. 1992;292:77–86. doi: 10.1016/0003-9861(92)90053-y. [DOI] [PubMed] [Google Scholar]
- 30.Collinsova M, Castro C, Garrow TA, Yiotakis A, Dive V, Jiracek J. Combining combinatorial chemistry and affinity chromatography: highly selective inhibitors of human betaine:homocysteine S-methyltransferase. Chem. Biol. 2003;10:113–122. doi: 10.1016/s1074-5521(03)00008-5. [DOI] [PubMed] [Google Scholar]
- 31.Koval D, Kasicka V, Jiracek J, Collinsova M. Separation of diastereomers of phosphinic pseudopeptides by capillary zone electrophoresis and reverse phase high-performance liquid chromatography. J. Sep. Sci. 2003;26:653–660. [Google Scholar]
- 32.Koval D, Kasicka V, Jiracek J, Collinsova M. Physicochemical characterization of phosphinic pseudopeptides by capillary zone electrophoresis in highly acid background electrolytes. Electrophoresis. 2003;24:774–781. doi: 10.1002/elps.200390097. [DOI] [PubMed] [Google Scholar]
- 33.Koval D, Kasicka V, Jiracek J, Collinsova M, Garrow TA. Determination of dissociation constant of phosphinate group in phosphinic pseudopeptides by capillary zone electrophoresis. J. Chromatogr. B. 2002;770:145–154. doi: 10.1016/s1570-0232(01)00595-5. [DOI] [PubMed] [Google Scholar]
- 34.Koval D, Kasicka V, Jiracek J, Collinsova M, Garrow TA. Analysis and characterization of phosphinic pseudopeptides by capillary zone electrophoresis. Electrophoresis. 2002;23:215–222. doi: 10.1002/1522-2683(200202)23:2<215::AID-ELPS215>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 35.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 36.Farrington GK, Kumar A, Wedler FC. Design and synthesis of phosphonate inhibitors of glutamine synthetase. J. Med. Chem. 1987;30:2062–2067. doi: 10.1021/jm00394a022. [DOI] [PubMed] [Google Scholar]
- 37.Viornery C, Pechy P, Boegli M, Aronsson BO, Descouts P, Gratzel M. Synthesis of new polyphosphonic acids. Phosphorus Sulfur Silicon Relat. Elem. 2002;177:231–241. [Google Scholar]
- 38.Rejman D, Masojidkova M, De Clercq E, Rosenberg I. 2 '-C-alkoxy and 2 '-C-aryloxy derivatives of N-(2-phosphonomethoxyethyl)purines and -pyrimidines: Synthesis and biological activity. Nucleosides Nucleotides & Nucleic Acids. 2001;20:1497–1522. doi: 10.1081/NCN-100105244. [DOI] [PubMed] [Google Scholar]
- 39.Arbuzov BA, Vinogradova VS, Novoselskaja AD. Reaction of N-hydroxymethylchloroacetamide with triethyl phosphite and some dialkyl chlorophosphites. J. Gen. Chem. USSR (Engl. Transl. ) 1967;37:2061–2065. [Google Scholar]
- 40.Garrow TA. Purification, kinetic properties, and cDNA cloning of mammalian betaine-homocysteine methyltransferase. J. Biol. Chem. 1996;271:22831–22838. doi: 10.1074/jbc.271.37.22831. [DOI] [PubMed] [Google Scholar]
- 41.Todhunter JA. Reversible Enzyme Inhibition. In: Purich DL, editor. Enzyme Kinetics and Mechanism. New York: Academic Press; 1979. pp. 383–411. [Google Scholar]
- 42.Min K-L, Steghens J-P, Henry R, Doutheau A, Collombel C. N-Dibenzylhospho-N'-3-(2,6-dichlorophenyl)propyl-guanidine is a bisubstrate-analog for creatine kinase. Biochim. Biophys. Acta. 1997;1342:83–89. doi: 10.1016/s0167-4838(97)00088-5. [DOI] [PubMed] [Google Scholar]
- 43.Segel IH. Enzyme Kinetics. Behaviour and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: John Wiley & Sons, Inc.; 1993. [Google Scholar]
- 44.Dixon M. The graphical determination of Km and Ki. Biochem. J. 1972;129:197–202. doi: 10.1042/bj1290197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HPLC purity data and NMR spectra records for compounds 2, 9 and 19. This material is available free of charge via the Internet at http://pubs.acs.org.