

Abstract
Neisseria gonorrhoeae (GC) or Escherichia coli expressing phase-variable opacity (Opa) protein (Opa+ GC or Opa+ E. coli) adhere to human neutrophils and stimulate phagocytosis, whereas their counterparts not expressing Opa protein (Opa− GC or Opa− E. coli) do not. Opa+ GC or E. coli do not adhere to human lymphocytes and promyelocytic cell lines such as HL-60 cells. The adherence of Opa+ GC to the neutrophils can be enhanced dramatically if the neutrophils are preactivated. These data suggest that the components binding the Opa+ bacteria might exist in the granules. CGM1a antigen, a transmembrane protein of the carcinoembryonic antigen family, is exclusively expressed in the granulocytic lineage. The predicted molecular weight of CGM1a is ≈30 kDa. We observed specific binding of OpaI+ E. coli to a 30-kDa band of polymorphonuclear leukocytes lysates. To prove the hypothesis that the 30-kDa CGM1a antigen from neutrophils was the receptor of Opa+ bacteria, we showed that a HeLa cell line expressing human CGM1a antigen (HeLa-CGM1a) bound Opa+ E. coli and subsequently engulfed the bacteria. Monoclonal antibodies (COL-1) against CGM1 blocked the interaction between Opa+ E. coli and HeLa-CGM1a. These results demonstrate that HeLa cells when expressing the CGM1a antigens bind and internalize OpaI+ bacteria.
Neisseria gonorrhoeae (GC), the etiologic agent of gonorrhea, can adhere to and penetrate mucosal epithelial cells (1, 2) and attain access to submucosal sites. Much of the knowledge of these interactions are based the in vitro infection model employing human fallopian tubes (3, 4). In this system GC pili are required for the initial adherence (5). Subsequent human volunteer studies have confirmed that pili are essential for virulence (6). In the fallopian epithelial model the distant pili-mediated attachment converts over several hours to a close attachment that is believed to be dependent on a family of outer membrane proteins, the phase-variable opacity (Opa) proteins (4). Inoculation of volunteers has indicated that Opa+ gonococci are strongly positively selected in the experimental infection (7, 8). In gonococcal strain MS11, this family consists of 11 unlinked opa genes whose sequences are known (9). To study the role of the different Opa proteins seven Opa proteins genes have been cloned and expressed in Escherichia coli. In terms of cellular location, protein conformation and their interaction with human cells Opa+ E. coli closely mimic the Opa+ GC (10). Additionally, this also overcame the problem of antigenic variation and unstable expression of Opa proteins in GC. One distinct Opa protein, OpaA is correlated with adherence and subsequent internalization of GC by cultured epithelial cell lines notably Chang conjunctival cells (11–13). Moreover, it has been shown that the interaction of OpaA GC with epithelial cells involves binding to heparan sulfate on the cell surface, and that the interaction is heparin-inhibitable (14, 15). In the case of Chang cells GC-bearing Opa proteins other than OpaA do not invade the cells. However, this does not apply to all epithelial cell lines. The OpaG1 protein from strain F62 GC expressed in E. coli DH5α promotes attachment and invasion of ME180 cervical carcinoma cells (16). By dendrogram analysis of Opa proteins the OpaG1 protein does not belong to the same branch as MS11 OpaA (13). We have shown that OpaI expressed by E. coli HB101 (pEXI) also is able to adhere to ME180 cells, and that this interaction is not inhibitable with heparin (data not shown).
A major property of Opa proteins is the ability to stimulate adherence and nonopsonic phagocytosis of the Opa+ bacteria by polymorphonuclear leukocytes (PMN). This increased association with human neutrophils by Opa+ GC was first observed by Swanson et al. in 1975 (17, 18). Subsequently, several other groups demonstrated that the Opa protein mediated interaction to PMN in an opsonin-independent manner (19–21). Characteristically, some Opa proteins promote strong PMN phagocytosis such as OpaI in MS11, and other Opa proteins elicit intermediate interaction. However, OpaA GC do not stimulate PMN phagocytosis and behave like Opa− organisms (10, 13). Although Opa+ GC adhere to and stimulate phagocytosis by PMN, they do not adhere to human lymphocytes and HL-60 cells (22). Farrell et al. (22) also noted that the interaction of Opa+ GC with neutrophils could be enhanced dramatically if the PMN were preactivated with PMA and suggested that the receptors for Opa proteins were stored in secondary granules.
There has been little investigation of the biochemistry of the PMN–Opa protein interaction. It has been reported that Opa+ GC bind to a 19-kDa protein of unknown identity when PMN membrane or secondary granules are separated by SDS/PAGE and transferred to nitrocellulose (23). We have found that OpaI-expressing E. coli are able to bind to a 30-kDa surface protein of PMN. Acting on the possibility that this protein might be CGM1a, a protein of the carcinoembryonic antigen (CEA) family, which is in that molecular weight range and is expressed only by mature PMN, we tested transfected HeLa cells expressing this antigen and found that they permitted adherence of pEXI and that the bacteria were internalized.
MATERIALS AND METHODS
Bacterial Strains, mAbs, and Cell Lines.
Recombinant opa genes from N. gonorrhoeae MS11 were expressed in E. coli HB101 as described (10). The designations of Opa proteins of both GC and E. coli are based on papers of Swanson et al. (7) and Belland et al. (10). E. coli HB101 containing the vector pGEM-3Z is designated as pGEM. E. coli HB101 expressing OpaA, OpaB, OpaC, and OpaI proteins are designated as pEXA, pEXB, pEXC, and pEXI, respectively. Suspensions were prepared from bacteria grown for 16–20 h at 37°C on Luria–Bertani plates containing 50 μg/ml carbenicillin. E. coli strain HB101 does not express type I fimbriae. For the coinfection experiment a nalidixic acid resistant mutant of HB101 expressing the OpaA protein was employed.
COL-1 mAb, specific for CGM1 and CEA, was kindly donated by Zuorong Shi (Zymed). IB4 mAb, specific for CD18, was generously provided by Sam Wright (The Rockefeller University).
Chang conjunctival and HL-60 cell lines were purchased from the American Type Culture Collection. HeLa-CGM1a cells were constructed by transfecting HeLa cells with CGM1a cDNA, and selected for CGM1a antigen expression on the cell surface (24). HeLa-Neo cells are HeLa cells that were transfected with the neomycin-resistance gene only. These HeLa cell lines were kindly provided by Fritz Grunert (Institute for Immunobiology, Albert–Ludwigs University, Freiburg, Germany).
Adherence and Internalization Assays.
All cell lines were cultured in RPMI 1640 medium (GIBCO/BRL) with 10% fetal calf serum (HyClone). The HL-60 cell were maintained in RPMI 1640 medium containing 10% fetal calf serum, 16 μg/ml of serine, 8.4 μg/ml of asparagine, and 16.8 μg/ml of glutamine. For adherence assays, cells were grown to confluence (≈2 × 105 cells per well) in 24-well culture plates (Falcon), and washed twice with serum-free RPMI 1640 medium. E. coli were suspended in RPMI 1640 medium to the desired OD540. Bacterial suspension (0.5 ml) was added to each well. The plates were incubated at 37°C with 5% CO2 for 3–6 h. Experiments were terminated by washing with 1 ml of serum-free RPMI 1640 medium for 2 min on an orbital shaker at 110 rpm. The wash procedure was repeated three times. Adherent bacteria were counted by suspending the cells in PBS containing 0.5% saponin (Calbiochem) and plating dilutions on Luria–Bertani-agar medium containing 50 μg/ml of carbenicillin. The level of adherence of E. coli to cells was calculated by determining the colony-forming units associated with the host cell monolayers. Internalization assays were done in a similar fashion, but following the period of bacterial interaction with the cells, the monolayer was washed twice and incubated for 90 min with 1.5 ml of RPMI 1640 medium/fetal calf serum (5%) supplemented with gentamicin (GIBCO/BRL) at a final concentration of 100 μg/ml. This concentration was capable of killing >99.99% of either E. coli or GC in the absence of epithelial cells. For adherence inhibition assays, the bacteria were added as a suspension in RPMI 1640 medium containing desired concentrations of heparin (30 μg/ml) or antibodies (25 μg/ml). The experiments were performed in duplicate or triplicate.
Interaction with PMN.
PMN from 14 ml of whole human blood were purified by centrifugation through Polymorphprep (GIBCO/BRL). The purified PMN were suspended in dPBS (PBS containing 5 mM MgCl2 and 1 mM CaCl2) at concentration of 1 × 106 per ml. PMN suspensions (0.5 ml) were added to glass coverslips (10 mm diameter) in 24-well plates and preincubated at 37°C, with 5% CO2 for 45 min to allow the neutrophils to adhere to the glass surface (13). After washing once with dPBS to remove the nonadherent PMN, 500 μl of Opa+ E. coli suspensions (OD540 = 0.04) were added and allowed to incubate for 90 min at 37°C with CO2. Nonadherent neutrophils and E. coli were removed by washing three times with dPBS, and then the PMN monolayers were fixed with 1% glutaraldehyde in dPBS containing Giemsa stain. The number of bacteria (adherent and internalized) per PMN was determined by microscopy by counting the E. coli associated with 100 neutrophils.
Preparation of Biotinylated PMN and HL-60 Cell Lysate.
Purified PMN were suspended in 10 ml of dPBS at a concentration of 1 × 106 per ml, transferred to a 10-cm glass Petri dish, and incubated at 37°C with 5% CO2 for 15 min to allow attachment to the Petri dish. Thereafter, 1 mg of N-hydroxysuccinimide-LC-biotin (Pierce) and phorbol 12-myristate 13-acetate (100 ng/ml) (Calbiochem) were added to the Petri dish, and incubated at 37°C with CO2 for 45 min. The reaction was stopped by washing the PMN monolayers three times with buffer containing 50 mM Tris and 150 mM NaCl at pH 7.4. The attached PMN were extracted with 3.5 ml of 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 50 units/ml aprotinin, and 2% Triton X-100 for 30 min. The extracts were centrifuged for 5 min and the supernatants were stored at −20°C for future use. Since HL-60 cells do not bind to glass Petri dishes, the preparations of biotinylated HL-60 cells were performed in a 15-ml centrifuge tube in place of a glass Petri dish.
Binding of Specific Components from PMN By Opa+ E. coli.
Opa− or Opa+ E. coli (pGEM or pEXI) were suspended at OD540 = 0.8 in 1 ml of 0.05 M Tris/150 mM NaCl buffer containing 2% BSA, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin and 50 units/ml aprotinin. Fifty microliters of biotinylated supernatant of PMN or HL-60 was added the bacterial suspension, and incubated at 37°C for 120 min with gentle shaking. Bacterial suspensions were pelleted. The bacterial pellets were used for Western blot analysis.
SDS/PAGE and Western Blot Analysis.
Gel electrophoresis and Western blot analysis were as described (25). The Immobilon-P membrane (Millipore) with transferred biotinylated bacterial–PMN lysates was incubated in 0.05 M Tris at pH 7.4, 150 mM NaCl buffer containing 5% vitamin free-casein (Sigma) for 4 h at room temperature or overnight at 4°C. The blots were washed once with PBS-Tween (PBS containing 0.05% of Tween-20), and reacted with extravidin-conjugated peroxidase (Sigma) diluted 1:20,000 in PBS-Tween with 0.5% casein. After incubation at room temperature for 1 h, the blots were washed three times with PBS-Tween with shaking. Each wash was for 40 min. The biotinylated proteins were detected by enhanced chemiluminescence (Amersham).
RESULTS
Two Distinct Interactions of Opa+ E. coli with Epithelial Cells and PMN.
The OpaA protein expressed by N. gonorrhoeae MS11 promotes not only adherence but also internalization by epithelial cells (11, 12) and this interaction is inhibitable with heparin (14, 15). pEXA also adhere to Chang cells and the interaction is inhibited by heparin (Fig. 1A). pEXI adhere less well to Chang cells and this interaction is unaffected by heparin. In contrast, with PMN pEXA adhere poorly while pEXI adhere avidly and the interaction with pEXI is not inhibited by heparin (Fig. 1B). These data confirm that there are two distinct interaction mechanisms promoted by Opa proteins with epithelial cells and PMN.
Figure 1.
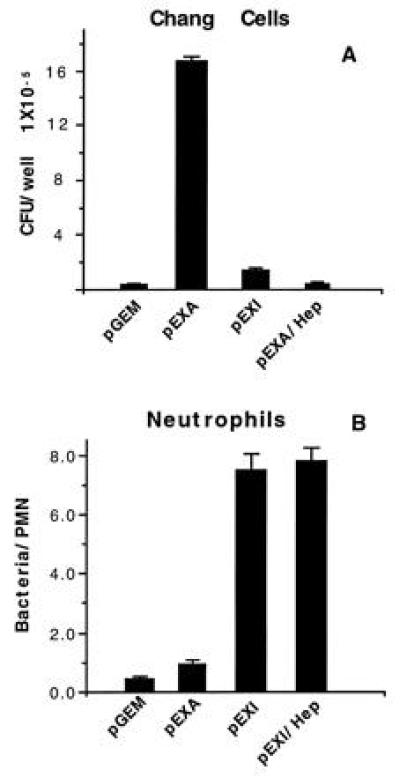
Interaction of Opa+ E. coli with epithelial cells and neutrophils. (A) Opa− or Opa+ E. coli were incubated with Chang conjunctival monolayers. pEXA adhere to the epithelial cells best. This interaction is inhibited by addition of heparin. pGEM and pEXI adhere at a negligible level. (B) Bacteria were incubated with PMN for 90 min. pEXI showed the highest level of adherence to PMN and this interaction was not inhibited by addition of heparin. Bars = SEM.
Opa+ E. coli Binds a 30-kDa Band from PMN Lysate.
Among the Opa proteins, OpaI, whether expressed by GC or by E. coli (pEXI), shows the strongest association with PMN (10). HL-60 cells are unable to support adherence by Opa+ GC (22) and were used as a negative control. We examined whether OpaI+ bacteria bind to a specific protein from PMN lysed with Triton X-100. Activated and surface-biotinylated PMN or HL-60 cells were lysed in 2% Triton X-100, and then Opa- E. coli (pGEM) and pEXI were used to extract components from the lysates. Fig. 2 shows that pEXI specifically bound a 30-kDa band that was not seen with the control strain pGEM. Neither pEXI or pGEM bound proteins in the HL-60 lysates.
Figure 2.
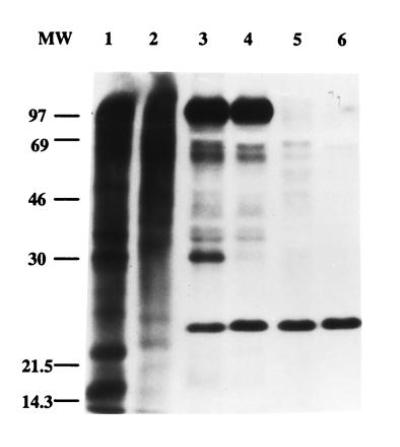
pGEM and pEXI reacted with surface biotinylated PMN and HL-60 cells. PMN or HL-60 cells were lysed with Triton X-100 and mixed with bacteria. After incubation the bacteria were recovered by centrifugation, lysed, the lysates subjected to SDS/PAGE and transferred to Immobilon-P membrane. Biotinylated proteins were detected by staining with extravidin-conjugated peroxidase and chemiluminescence. PMN total lysate (lane 1) and HL-60 cell total lysate (lane 2). Proteins recovered from PMN lysate with pEXI (lane 3) and pGEM (lane 4). Proteins recovered from HL-60 cells lysate with pEXI (lane 5) and pGEM (lane 6). pEXI bind a 30-kDa band specifically from PMN lysates (lane 3). The 23-kDa protein recognized by extravidin is probably the biotin carboxy carrier protein found in E. coli (26).
OpaI+ E. coli Adheres to HeLa Cells Expressing CGM1a Antigens, But Not the HeLa Control Cells.
Based on the molecular weight of about 30 kDa and the restricted expression in mature granulocytes we speculated that the component responsible for PMN and pEXI interaction might be CGM1a. This protein is a member of the CEA family, is restricted to the granulocytic line and is about 30 kDa in size. We used a stably transfected CGM1a HeLa cell line (HeLa-CGM1a) to test this hypothesis, and the HeLa cell line transfected only with the vector (HeLa-Neo) served as a control. As shown in Fig. 3A, there was no adherence of pGEM to the cell lines, but pEXA adhered to both cell lines. pEXI attached to HeLa-CGM1a cells only. Furthermore, the OpaA protein-mediated adherence could be blocked by soluble heparin in both cell lines, but the adherence of pEXI to HeLa-CGM1 was not influenced by heparin. Qualitatively similar results were obtained when the adherence of Opa−, OpaA+ and OpaI+ GC to the transfected HeLa cells was determined (Fig. 3B) CGM1a-HeLa also bound OpaB+ and OpaC+ E. coli (pEXB and pEXC) and their corresponding Opa+ GC to a lesser extent (data not shown).
Figure 3.
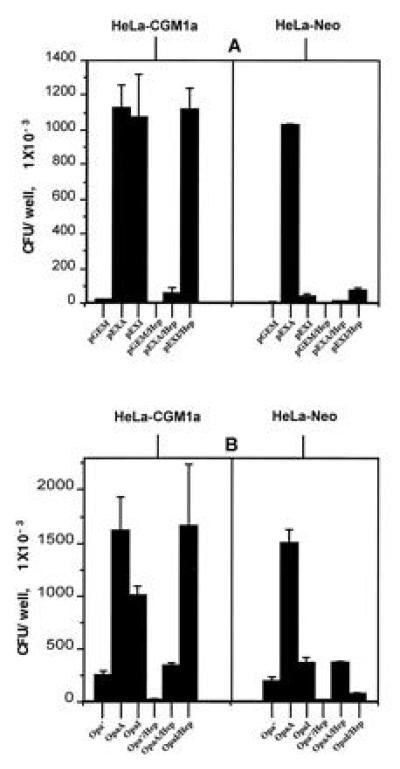
Interactions of Opa+ E. coli and GC with HeLa-CGM1a cells. Opa+ E. coli (A) and GC (B) were incubated with HeLa-CGM1a and HeLa-Neo cells in RPMI medium 1640 buffer with or without soluble heparin (Hep. 30 μg/ml). pEXI adhere to the CGM1a cells but not the control cells. OpaA mediated adherence is inhibited by heparin, but the adherence of pEXI to CGM1a is not influenced by soluble heparin. Similar results were observed with OpaA and OpaI GC.
The Interaction of OpaI+ E. coli to HeLa-CGM1a Cells Was Blocked by Anti-CGM1 mAb.
We investigated whether a specific mAb to CGM1 could inhibit this interaction. COL-1 mouse mAb (IgG2a) is specific for CGM1, and does not react with nonspecific crossreacting antigen (NCA), BGP, and CGM6 antigens which are expressed by PMN (27) (see Discussion for description of these antigens). Anti-CD18-specific mAb, IB4 (IgG2a), was employed as a control antibody. Only COL-1 antibody inhibited the interaction of OpaI+ E. coli (pEXI) to HeLa-CGM1a (Fig. 4). COL-1 mAb did not inhibit the OpaA mediated adherence to HeLa-CGM1a cells (data not shown). This antibody could not be examined for effects on the adherence of pEXI to PMN since it rapidly caused significant changes of morphology of PMN.
Figure 4.
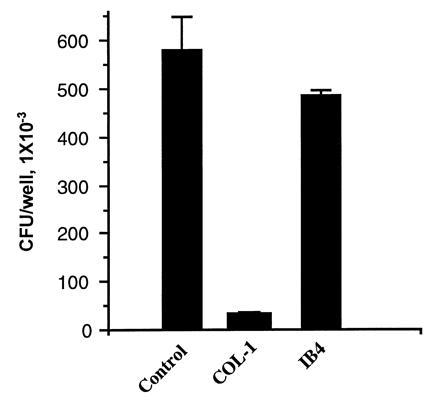
Inhibition of CGM1a mediated adherence by antibody. Addition of COL-1 antibody inhibited adherence of pEXI to HeLa-CGM1a. IB4 antibody had no effect when added to the same final concentrations (25 μg/ml). The interaction of pEXI with HeLa-CGM1a was for 2.5 h.
The Internalization of OpaI+ E. coli by HeLa-CGM1a.
Once CGM1a was shown to be the receptor of adherence for OpaI protein, we determined whether this interaction would promote invasion of the HeLa-CGM1a cells. As shown in Fig. 5, pGEM (Opa−) could not adhere to or enter the HeLa-CGM1a. In contrast, HeLa-CGM1a bound and strongly engulfed pEXI (OpaI+). Almost 30% of HeLa cell-associated bacteria were gentamicin resistant. pEXA (OpaA+) adhered to HeLa-CGM1a (Fig. 5A), but were not internalized (Fig. 5B). When coinfected with pEXI, pEXA still was unable to invade the HeLa-CGM1a cells (data not shown). This indicates that the bacteria need to bind to a specific receptor to activate the internalization system and this is distinctly different from the macropinocytosis reported for Salmonella subspecies (28). The invasion by pEXI was confirmed by electron microscopy. Both surface adherent bacteria (Fig. 6A) as well as bacteria deep in the cells in a membrane bounded compartment were seen (Fig. 6B).
Figure 5.
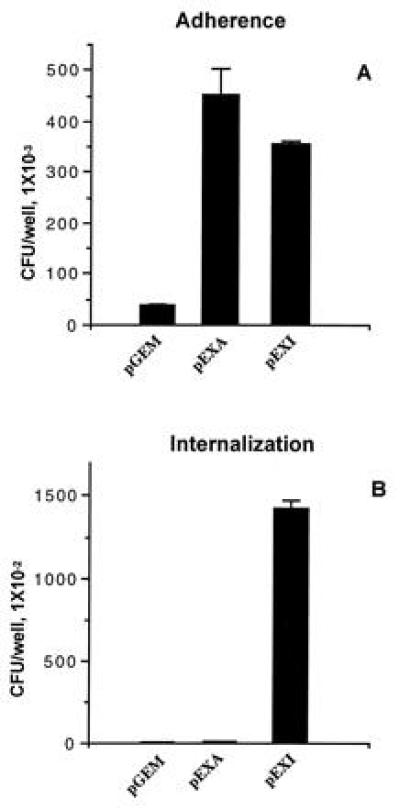
The internalization of pEXI by HeLa-CGM1a cells. pGEM, pEXA, and pEXI at OD540 = 0.04 were incubated with HeLa-CGM1a cells in RPMI 1640 medium for 6 h. Unbound cells were removed by replacing the supernatants with fresh RPMI medium 1640 (1 ml per well per wash) at 3-h intervals. The adherent and intracellular E. coli were distinguished by incubation with gentamicin. Only pEXI were recovered in large numbers following gentamicin treatment, although both pEXA and pEXI adhered to the HeLa-CGM1a.
Figure 6.
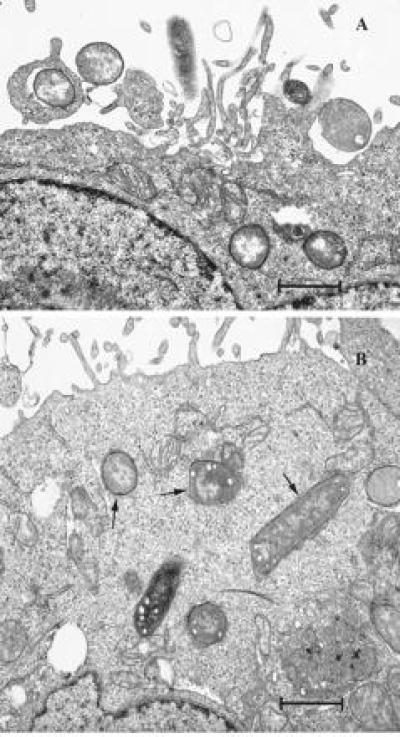
Transmission electron micrograph of internalization of pEXI by HeLa-CGM1a cells. (A) OpaI+ E. coli are shown in the early stage of interaction with HeLa-CGM1a cells. (B) The internalized E. coli are clearly enclosed within vesicles as indicated with arrows. (Bar = 1 μm.)
DISCUSSION
Clinical GC isolated either from the male urethra or from the cervix of an infected female (except at the time of menses), are most often Opa+ (29–31). Male volunteers inoculated intraurethrally with Opa− strains of GC, when they became infected, shed primarily Opa+ variants of most Opa protein types (7, 8). Furthermore, a recent study showed high level of Opa-specific antibody and protection against gonococcal salpingitis (32). Taken together, these data suggest strongly that in vivo expression of Opa proteins plays an important role in gonococcal pathogenesis. Recent work from two independent laboratories demonstrated that the OpaA protein of MS11 utilized heparan sulfate on syndecan glycoproteins as receptor molecules to interact with epithelial cells (14, 15). This interaction results in the internalization of OpaA GC into epithelial cells (15). In contrast, PMN lack receptors for OpaA, but have receptors for other Opa proteins (10, 13) and these seem to be stored within secondary granules (22).
The current study demonstrates that human CGM1a appears to be a receptor for the interaction of Opa+ GC with PMN. Attention was directed to the role of the CGM1a antigen by the observation that OpaI+ E. coli bound specifically a ≈30-kDa protein band, which correlated well with the predicted molecular weight of CGM1a. The CGM1 antigens are only expressed in neutrophils, not in human monocytes, lymphocytes and HL-60 cells (24). Transfected HeLa cells expressing CGM1a were able to avidly bind and internalize OpaI+ E. coli, but not E. coli containing only the pGEM plasmid vector. This interaction did not occur with HeLa cells transfected with the neomycin-resistance gene only. The interaction was not inhibited by addition of heparin, but was strongly inhibited by a mAb specific for CGM1.
CGM1 antigens of neutrophils belongs to the family of CEA. Studies trying to identify tumor-specific antigens led to the discovery of CEA, a 180-kDa tumor-associated cell-surface glycoprotein on colon cancer cells (33, 34). Subsequently, it was found that antibodies to CEA also reacted with normal cells and identified a number of closely related, crossreacting antigens, initialy termed NCAs (35–37). The CEA gene, located in human chromosome 19 (38), is a member of a family of >17 expressible closely related genes (39) that belong to the Ig gene superfamily (40). The human CEA family, recognized by CD66 and CD67 antibodies, consists of two major subfamilies, one termed the CEA subgroup, containing CEA, NCA, biliary glycoprotein (41), CGM1 (CGM1a, CGM1b, and CGM1c) (24, 42), CGM2, CGM6, and CGM7 (CGM = CEA gene family member). The second subfamily consists of the pregnancy-specific glycoproteins (43–45).
Our studies have not addressed the role of any other member of the CEA family as Opa protein receptors. Virji et al.* recently reported that COS cells transfected with CD66a antigen—i.e., binary glycoprotein—caused opacity protein specific adherence of N. meningitidis. This activity was dependent on the immunoglobulin-like N-terminal domain This IgV-like portion of the molecule is strongly conserved among different members of the CEA family and CGM1 consists solely of this domain. Taken together with our results presented for CGM1a neutrophil antigen, it would appear that several CEA family members may serve as receptors. CD66a antigen is expressed in many tissues including urogenital epithelial cells (46). It remains to be determined whether the opa-mediated interaction recognizes the CEA protein structure or a glycosylation pattern shared by this family of proteins. It has been previously reported that the type I fimbriae of E. coli attach to purified CEA, binary glycoprotein, and NCA (47), and that mannose-inhibitable adherence to neutrophils was dependent on the presence of NCA-50 (48). It has been demonstrated that this reactivity is due to high mannose type oligosaccharides found on the three glycosylation sites in the N-terminal domain of this antigen (49).
Internalization of microorganisms into either professional or nonprofessional cells by phagocytosis require the reorganization of the actin-based cytoskeleton. This actin assembly is initiated by signals arising from the interaction of phagocytosis-promoting receptors on the cell surface with ligands on the surface of the microorganisms such as invasin on Yersinia pseudotuberculosis (50, 51). Several receptors have been identified to mediate binding and ingestion of phagocytic particles. The best-characterized receptors are the opsonin-recognizing receptors including the various types of receptors for the Fc portion of IgG (FcγRs) (52). After the opsonized bacteria bind the Fcγ RIII, the tyrosine-activation motif on the cytoplasmic domain of Fcγ RIII is activated by phosphorylation. These phosphorylated tyrosine residues within tyrosine-activation motifs, can recruit another tyrosine kinase, Syk, whereupon it becomes activated. Activation of Syk as a result of clustering of receptors by antigen, in the case of T and B cells, or by immune complexes, in the case of Fcγ and Fcɛ receptors, leads to phagocytosis or actin polymerization (53). In fact, CGM1a has a cytoplasmic domain where tyrosines lie within a sequence context (YX2LX7YX2M) (24), which is similar to the consensus sequence (“YLYL” motif: YX2LX7YX2L/I) found in the cytoplasmic domain of molecules of multichain immune recognition receptors (54). Since the cytoplasmic domains of CGM1a contains a tyrosine-activation motif-like motif, it is possible that in the HeLa transfectants CGM1a may have acted not only as receptor for the adherence of OpaI+ E. coli, but also as a signal transducing molecule initiating the internalization of the bacteria. The role of CGM1a in PMN physiology remains to be explored.
The biological role of the GC Opa proteins is now becoming clearer as the eukaryotic ligands for these proteins are being elucidated. It has been established that a subset of opa proteins binds to syndecans (14, 15). Syndecans serve as receptors or coreceptors for growth factors, for cell-to-cell interactions and for cell interactions with extracellular matrix components (reviewed in ref. 55). The finding that MS11 OpaI as well as OpaB and OpaC bind to neutrophil CGM1a defines a new molecular basis for the adherence of GC to host surfaces. The extent that this specificity applies to other members of the CEA family of proteins and the role that these interactions may have in signaling the internalization of GC by both neutrophils and epithelial cells will prove a fertile area for investigation.
Acknowledgments
We wish to thank Drs. Fritz Grunert and Wolfgang Zimmermann for generously providing the HeLa-CGM1a cells. We are indebted to Dr. Kathleen A. Haines for insightful scientific and technical advice. We also thank Drs. Asesh Banerjee and Vijay Pancholi for useful suggestions and editorial comments on the manuscript. This work was supported by U.S. Public Health Service Grant AI 10615.
Footnotes
Abbreviations: GC, Neisseria gonorrhoeae; PMN, polymorphonuclear leukocytes; Opa protein, phase-variable opacity protein; CEA, carcinoembryonic antigen; NCA, nonspecific crossreacting antigen.
Virji, M., Watt, S. M., Barker, S. & Makepeace, K., Tenth International Pathogenic Neisseria Conference, September 8–13, 1996, Baltimore.
References
- 1.Evans B A. J Infect Dis. 1977;136:248–255. doi: 10.1093/infdis/136.2.248. [DOI] [PubMed] [Google Scholar]
- 2.Ward M E, Watt P J. J Infect Dis. 1972;126:601–605. doi: 10.1093/infdis/126.6.601. [DOI] [PubMed] [Google Scholar]
- 3.Ward M E, Watt P J, Robertson J N. J Infect Dis. 1974;129:650–659. doi: 10.1093/infdis/129.6.650. [DOI] [PubMed] [Google Scholar]
- 4.McGee Z A, Robinson E N., Jr . In: The Pathogenesis of Bacterial Infections. Jackson G G, Thomas H, editors. Berlin: Springer; 1985. pp. 8–16. [Google Scholar]
- 5.McGee Z A, Johnson A P, Taylor-Robinson D. J Infect Dis. 1981;143:413–421. doi: 10.1093/infdis/143.3.413. [DOI] [PubMed] [Google Scholar]
- 6.Swanson J L, Robbins K, Barrera O, Corwin D, Boslego J, Ciak J, Blake M S, Koomey J M. J Exp Med. 1987;165:1344–1357. doi: 10.1084/jem.165.5.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swanson J, Barrera O, Sola J, Boslego J. J Exp Med. 1988;168:2121–2129. doi: 10.1084/jem.168.6.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jerse A E, Cohen M S, Drown P M, Whicker L G, Isbey S F, Seifert H S. J Exp Med. 1994;179:911–920. doi: 10.1084/jem.179.3.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhat K S, Gibbs C P, Barrera O, Morrison S G, Jähnig F, Stern A, Kupsch E M, Meyer T F, Swanson J. Mol Microbiol. 1991;5:1889–1901. doi: 10.1111/j.1365-2958.1991.tb00813.x. [DOI] [PubMed] [Google Scholar]
- 10.Belland R J, Chen T, Swanson J, Fischer S H. Mol Microbiol. 1992;6:1729–1737. doi: 10.1111/j.1365-2958.1992.tb01345.x. [DOI] [PubMed] [Google Scholar]
- 11.Weel J F L, Hopman C T P, van Putten J P M. J Exp Med. 1991;173:1395–1405. doi: 10.1084/jem.173.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makino S, van Putten J P M, Meyer T F. EMBO J. 1991;10:1307–1315. doi: 10.1002/j.1460-2075.1991.tb07649.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kupsch E -M, Knepper B, Kuroki T, Heuer I, Meyer T F. EMBO J. 1993;12:641–650. doi: 10.1002/j.1460-2075.1993.tb05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen T, Belland R J, Wilson J, Swanson J. J Exp Med. 1995;182:511–517. doi: 10.1084/jem.182.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Putten J P M, Paul S M. EMBO J. 1995;14:2144–2154. doi: 10.1002/j.1460-2075.1995.tb07208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon D, Rest R F. Proc Natl Acad Sci USA. 1992;89:5512–5516. doi: 10.1073/pnas.89.12.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson J, King G, Zeligs B. Infect Immun. 1975;11:65–68. doi: 10.1128/iai.11.1.65-68.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swanson J L, Sparks E, Young D, King G. Infect Immun. 1975;11:1352–1361. doi: 10.1128/iai.11.6.1352-1361.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Virji M, Everson J S. Infect Immun. 1981;31:965–970. doi: 10.1128/iai.31.3.965-970.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Virji M, Heckels J E. J Gen Microbiol. 1986;132:503–512. doi: 10.1099/00221287-132-2-503. [DOI] [PubMed] [Google Scholar]
- 21.Fischer S H, Rest R F. Infect Immun. 1988;56:1574–1579. doi: 10.1128/iai.56.6.1574-1579.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farrell C F, Rest R F. Infect Immun. 1990;58:2777–2784. doi: 10.1128/iai.58.9.2777-2784.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farrell C F, Naids F L, Rest R F. In: Neisseriae 1990: Proceeding of the Seventh International Pathogenic Neisseria Conference. Achtman M, Kohl P, Marchal C, Morelli G, Seiler A, Thiesen B, editors. Berlin: de Gruyter; 1991. pp. 579–584. [Google Scholar]
- 24.Nagel G, Grunert F, Kuijpers T W, Watt S M, Thompson J, Zimmermann W. Eur J Biochem. 1993;214:27–35. doi: 10.1111/j.1432-1033.1993.tb17892.x. [DOI] [PubMed] [Google Scholar]
- 25.Towbin H, Staehlin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fall R R. Methods Enzymol. 1979;62:390–398. doi: 10.1016/0076-6879(79)62246-2. [DOI] [PubMed] [Google Scholar]
- 27.Skubitz K M, Micklem K, van der Schoot E. In: Leukocyte Typing V. Schlossman S F, editor. Oxford: Oxford Univ. Press; 1995. pp. 889–899. [Google Scholar]
- 28.Francis C L, Ryan T A, Jones B D, Smith S J, Falkow S. Nature (London) 1993;364:639–642. doi: 10.1038/364639a0. [DOI] [PubMed] [Google Scholar]
- 29.James J F, Swanson J L. Infect Immun. 1978;19:332–340. doi: 10.1128/iai.19.1.332-340.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James J F, Swanson J. In: Immunobiology of Neisseria gonorrhoeae. Brooks G F Jr, Gotschlich E C, Holmes K K, Sawyer W D, Young F E, editors. Washington, DC: Am. Soc. Microbiol.; 1978. pp. 338–343. [Google Scholar]
- 31.Ison C A, Brooks G F. In: Neisseriae 1990: Proceeding of the Seventh International Pathogenic Neisseria Conference. Achtman M, Kohl P, Marchal C, Morelli G, Seiler A, Thiesen B, editors. Berlin: de Gruyter; 1991. pp. 597–602. [Google Scholar]
- 32.Plummer F A, Chubb H, Simonsen J N, Bosire M, Slaney L, Nagelkerke N J, Ndinya-Achola J O, Waiyaki P, Brunham R C. J Clin Invest. 1994;93:1748–1755. doi: 10.1172/JCI117159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gold P, Freedman S O. J Exp Med. 1965;121:439–462. doi: 10.1084/jem.121.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barnett T, Zimmermann W. Tumour Biol. 1990;11:59–63. doi: 10.1159/000217643. [DOI] [PubMed] [Google Scholar]
- 35.Von Kleist S, Chavanel G, Burtin P. Proc Natl Acad Sci USA. 1972;69:2492–2494. doi: 10.1073/pnas.69.9.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mach J P, Pusztaszeri G. Immunochemistry. 1972;9:1031–1034. doi: 10.1016/0019-2791(72)90113-9. [DOI] [PubMed] [Google Scholar]
- 37.Bordes M, Knobel S, Martin F. Eur J Cancer. 1975;11:783–786. doi: 10.1016/0014-2964(75)90171-1. [DOI] [PubMed] [Google Scholar]
- 38.Tynan K, Olsen A, Trask B, de Jong P, Thompson J, Zimmermann W, Mohrenweiser H. Nucleic Acids Res. 1992;20:1629–1636. doi: 10.1093/nar/20.7.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson J, Zimmermann W, Osthus-Bugat P, Schleussner C, Eades-Perner A M, Barnert S, Von Kleist S, Willcocks T, Craig I, Tynan K. Genomics. 1992;12:761–772. doi: 10.1016/0888-7543(92)90307-e. [DOI] [PubMed] [Google Scholar]
- 40.Paxton R J, Mooser G, Pande H, Lee T D, Shively J E. Proc Natl Acad Sci USA. 1987;84:920–924. doi: 10.1073/pnas.84.4.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watt S M, Fawcett J, Murdoch S J, Teixeira A M, Gschmeissner S E, Simmons D L. Blood. 1994;84:200–210. [PubMed] [Google Scholar]
- 42.Oikawa S, Inuzuka C, Kuroki M, Arakawa F, Matsuoka Y, Kosaki G. J Biol Chem. 1991;266:7995–8001. [PubMed] [Google Scholar]
- 43.Tatarinov Y S, Masyukevich V N. Bull Exp Biol Med. 1970;69:66–68. [Google Scholar]
- 44.Oikawa S, Inuzuka C, Kosaki G, Nakazato H. Biochem Biophys Res Commun. 1988;156:68–77. doi: 10.1016/s0006-291x(88)80806-4. [DOI] [PubMed] [Google Scholar]
- 45.Oikawa S, Inuzuka C, Kuroki M, Matsuoka Y, Kosaki G, Nakazato H. Biochem Biophys Res Commun. 1989;163:1021–1031. doi: 10.1016/0006-291x(89)92324-3. [DOI] [PubMed] [Google Scholar]
- 46.Prall F, Nollau P, Neumaier M, Haubeck H D, Drzeniek Z, Helmchen U, Wagener C. J Histochem Cytochem. 1996;44:35–41. doi: 10.1177/44.1.8543780. [DOI] [PubMed] [Google Scholar]
- 47.Leusch H-G, Drzeniek Z, Markos-Pusztai Z, Wagener C. Infect Immun. 1991;59:2051–2057. doi: 10.1128/iai.59.6.2051-2057.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sauter S L, Rutherfurd S M, Wagener C, Shively J E, Hefta S A. Infect Immun. 1991;59:2485–2493. doi: 10.1128/iai.59.7.2485-2493.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sauter S L, Rutherfurd S M, Wagener C, Shively J E, Hefta S A. J Biol Chem. 1993;268:15510–15516. [PubMed] [Google Scholar]
- 50.Isberg R R, Voorhis D L, Falkow S. Cell. 1987;50:769–778. doi: 10.1016/0092-8674(87)90335-7. [DOI] [PubMed] [Google Scholar]
- 51.Isberg R R, Leong J M. Cell. 1990;60:861–871. doi: 10.1016/0092-8674(90)90099-z. [DOI] [PubMed] [Google Scholar]
- 52.Ravetch J V. Cell. 1994;78:553–560. doi: 10.1016/0092-8674(94)90521-5. [DOI] [PubMed] [Google Scholar]
- 53.Greenberg S. Trends Cell Biol. 1995;5:93–99. doi: 10.1016/s0962-8924(00)88957-6. [DOI] [PubMed] [Google Scholar]
- 54.Keegan A D, Paul W E. Immunol Today. 1992;13:63–68. doi: 10.1016/0167-5699(92)90136-U. [DOI] [PubMed] [Google Scholar]
- 55.Couchman J R, Woods A. J Cell Biochem. 1996;61:578–584. doi: 10.1002/(sici)1097-4644(19960616)61:4<578::aid-jcb11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]