

Abstract
The genome of the Kaposi sarcoma-associated herpesvirus (KSHV or HHV8) was mapped with cosmid and phage genomic libraries from the BC-1 cell line. Its nucleotide sequence was determined except for a 3-kb region at the right end of the genome that was refractory to cloning. The BC-1 KSHV genome consists of a 140.5-kb-long unique coding region flanked by multiple G+C-rich 801-bp terminal repeat sequences. A genomic duplication that apparently arose in the parental tumor is present in this cell culture-derived strain. At least 81 ORFs, including 66 with homology to herpesvirus saimiri ORFs, and 5 internal repeat regions are present in the long unique region. The virus encodes homologs to complement-binding proteins, three cytokines (two macrophage inflammatory proteins and interleukin 6), dihydrofolate reductase, bcl-2, interferon regulatory factors, interleukin 8 receptor, neural cell adhesion molecule-like adhesin, and a D-type cyclin, as well as viral structural and metabolic proteins. Terminal repeat analysis of virus DNA from a KS lesion suggests a monoclonal expansion of KSHV in the KS tumor.
Kaposi sarcoma (KS) is a vascular tumor of mixed cellular composition (1). Its histology and relatively benign course in persons without severe immunosuppression has led to suggestions that KS tumor cell proliferation is cytokine-induced (2). Epidemiologic studies indicate the tumor is under strict immunologic control and is likely to be caused by a sexually transmitted infectious agent other than HIV (3). KS-associated herpesvirus (KSHV) was discovered in an AIDS–KS lesion by representational difference analysis and shown to be present in almost all AIDS–KS lesions (4). These findings have been confirmed and extended to nearly all KS lesions examined from the various epidemiologic classes of KS (5–9). KSHV is the eighth presumed human herpesvirus (HHV8) identified to date.
The virus was initially identified from two herpesvirus DNA fragments, KS330Bam and KS631Bam (4). Subsequent sequencing of a 21-kb AIDS–KS genomic library fragment (KS5) hybridizing to KS330Bam demonstrated that KSHV is a gamma-herpesvirus related to herpesvirus saimiri (HVS) belonging to the genus Rhadinovirus (10). Colinear homology (synteny) of genes in this region is maintained between KSHV and HVS, as well as Epstein–Barr virus (EBV) and equine herpesvirus 2. A 12-kb region (bacteriophage clones L54 and SGL-1) containing the KS631Bam sequence includes cyclin D and interleukin 8 receptor type a (IL-8Ra) homologs unique to rhadinoviruses (11).
KSHV is not readily transmitted to uninfected cell lines (10), but it is present in a rare B-cell primary effusion (body cavity-based) lymphoma (PEL) frequently associated with KS (12). BC-1 is a PEL cell line containing a high KSHV genome copy number and is coinfected with EBV (13). The KSHV genome form in BC-1 and its parental tumor comigrates with 270-kb linear markers on pulsed field gel electrophoresis (10). However, the genome size based on encapsidated DNA from an EBV-negative cell line (14) is estimated to be 165 kb (15). Estimates from KS lesions indicate a genome size larger than that of EBV (172 kb) (16).
To determine the genomic sequence of KSHV and identify novel virus genes, we mapped contiguous, overlapping virus DNA inserts from BC-1 genomic libraries. With the exception of a small, unclonable repeat region at its right end, the genome was sequenced to high redundancy allowing definition of the viral genome structure and identification of genes that may play a role in KSHV-related pathogenesis.
MATERIALS AND METHODS
Library Generation and Screening.
BC-1, HBL-6, and BCP-1 cells were maintained in RPMI 1640 medium with 20% fetal calf serum (10, 13, 17). DNA (18) from BC-1 cells was commercially cloned into either Lambda FIXII or S-Cos1 vectors (Stratagene). Phage and cosmid libraries were screened by standard methods (19, 20).
Initial library screening was performed using the KS330Bam and KS631Bam representational difference analysis fragments (4). Overlapping clones were sequentially identified using probes synthesized from the ends of previously identified clones (see Fig. 1) (21, 22). The map was considered circularly permuted by the presence of multiple, identical terminal repeat (TR) units in cosmids Z2 and Z6. Each candidate phage or cosmid was confirmed by tertiary screening.
Figure 1.
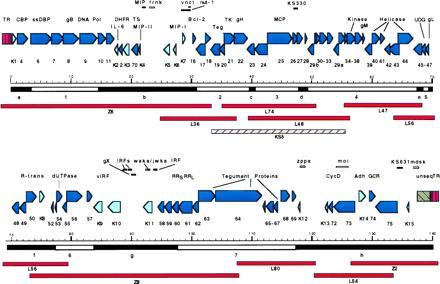
Annotated LUR and TR of the KSHV genome. The orientation of identified ORFs in the LUR are denoted by the direction of arrows, with HVS homologous ORFs in dark blue and nonhomologous ORFs in light blue. Seven blocks (numbered) of conserved herpesvirus genes with nonconserved interblock (IB) regions (lettered) are shown under the kilobase marker; the block numbering scheme differs from the original description by Chee et al. (30). The overlapping cosmid (Z prefix) and lambda (L prefix) clones used to map the KSHV genome are compared with the KS5 lambda phage clone from a KS lesion and shown below. Features and putative coding regions not specifically designated are shown above the ORF map. Repeat regions are shown as white lines (frnk, vnct, waka/jwka, zppa, moi, mdsk). Putative coding regions and other features (see text) not designated as ORFs are shown as solid lines.
Shotgun Sequencing and Sequence Verification.
Lambda and cosmid DNA were purified by standard methods (18). Shotgun sequencing (23, 24) was performed on sonicated DNA. A 1–4 kb fraction was subcloned into M13 mp19 (New England Biolabs) and propagated in XL1-Blue cells (Stratagene) (18). M13 phages were positively screened by using insert DNA from the phage or cosmid, and negatively screened with vector arm DNA or adjacent genome inserts.
Automated dideoxy cycle sequencing was performed with M13 (−21) CS+ or FS dye primer kits (Perkin–Elmer) on Applied Biosystems models 373A or 377 sequenators. Approximately 300 M13 sequences were typically required to achieve initial coverage for each 10 kb of insert sequence. Minimum sequence fidelity standards were defined as complete bidirectional coverage with at least four overlapping sequences at any given site. For regions with sequence gaps, ambiguities, and frameshifts, or that did not meet these criteria, primer walking was done with custom primers (Perkin–Elmer) and dye terminator chemistry (FS or Ready Reaction kits; Perkin–Elmer). An unsequenced 3-kb region adjacent to the right end TR sequence in the Z2 cosmid insert could not be cloned into M13 or Bluescript despite repeated efforts.
Sequence Assembly and Open Reading Frame Analysis.
Sequence data were edited using factura (Applied Biosystems) and assembled into contiguous sequences using electropherograms with AutoAssembler (Applied Biosystems) and into larger assemblies with assemblylign (IBI–Kodak). Base positions not clearly resolved by multiple sequencing attempts (less than 10 bases in total) were assigned the majority base pair designation. The entire sequence (in 1- to 5-kb fragments) and all predicted ORFs were analyzed by using blastx, blastp, and blastn (25). The sequence was further analyzed using motifs (10), repeat, and bestfit (Genetics Computer Group, Madison, WI), and macvector (IBI).
ORF Assignment and Nomenclature.
All ORFs with homologies to HVS were identified. These and other potential ORFs having >100 amino acids were found using macvector. ORFs not homologous to HVS ORFs were included in the map (see Fig. 1) based on homology to known genes, optimum initiation codon context (26), size, and position. Conservative selections were made to minimize spurious assignments; this underestimates the number of true reading frames. KSHV ORF nomenclature is based on HVS homologies; KSHV ORFs not homologous to HVS genes are numbered in consecutive order with a K prefix. ORFs with sequence but not positional homology to HVS ORFs were assigned the HVS ORF number (e.g., ORF2). As new ORFs are identified, we suggest they be designated by decimal notation. The standard map orientation (see Fig. 1) of the KSHV genome is the same as for HVS (27) and equine herpesvirus 2 (28), and reversed relative to the EBV standard map (29).
RESULTS
Genomic Mapping and Sequence Characteristics.
Complete genome mapping was achieved with seven lambda and three cosmid clones (Fig. 1). The structure of the BC-1 KSHV genome is similar to HVS in having a long unique region (LUR) flanked by TR units. The ≈140.5-kb LUR sequence has 53.5% G+C content and includes all identified KSHV ORFs. TR regions consist of multiple 801-bp direct repeat units having 84.5% G+C content (Fig. 2A) with potential packaging and cleavage sites. Minor sequence variations are present among repeat units. The first TR unit at the left (Z6) TR junction (205 bp) is deleted and truncated in BC-1 compared with the prototypical TR unit.
Figure 2.
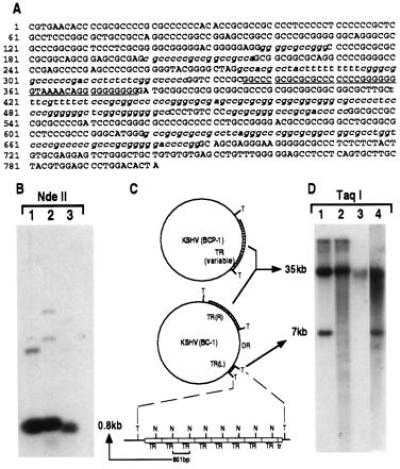
(A) Sequence of TR unit demonstrating its high G+C content. Sequences highly homologous to conserved herpesvirus pac1 sites are underlined, with lesser homology sites to specific pac1 and pac2 sequences italicized. (B) Southern blot of DNA from BC-1 (lane 1), BCP-1 (lane 2) and a KS lesion (lane 3) digested with NdeII which cuts once in the TR sequence and probed with a plasmid containing the TR sequence. The intense hybridization band at 0.8 kb represents multiple copies of the NdeII-digested single unit TR (C). A schematic representation (C) of genome structures of KSHV in BCP-1 and BC-1 cell lines consistent with the data presented in B and D. Taq I (T) sites flank the TR regions and NdeII (N) sites are within the TRs. Lowercase tr refers to the deleted truncated TR unit at the left end of the unique region. DR represents the duplicated region of the LUR buried within the TR. (D) Southern blot hybridization with TR probe of DNA from BC-1 (lane 1), BCP-1 (lane 2), a KS lesion (lane 3), and HBL-6 (lane 4) digested with Taq I, which does not cut in the TR. Taq I-digested DNA from both BC-1 (lane 1) and HBL-6 (lane 4) show similar TR hybridization patterns, suggesting identical insertion of a unique sequence into the TR region, which sequencing studies demonstrate is a duplicated portion of the LUR (see text). BCP-1 TR hybridization (lane 2) shows laddering consistent with a virus population having variable TR region lengths within this cell line due to lytic replication. The absence of TR laddering in KS lesion DNA (lane 3) suggests that a clonal virus population is present in the tumor.
The genome sequence abutting the right terminal repeat region is incomplete due to a 3-kb region in the Z2 cosmid insert that could not be cloned into sequencing vectors. Partial sequence information from primer walking indicates that this region contains stretches of 16-bp A+G-rich imperfect direct repeats interspersed with at least one stretch of 16-bp C+T-rich imperfect direct repeats. These may form a larger inverted repeat that could have contributed to our difficulty in subcloning this region. Greater than 12-fold average sequence redundancy was achieved for the entire LUR with complete bidirectional coverage by at least four overlapping reads except in the unclonable region.
The BC-1 TR region was examined by Southern blot analysis because sequencing of the entire region is not possible due to its repeat structure. BC-1, BCP-1 (an EBV-negative, KSHV infected cell line), and KS lesion DNAs have an intense ≈800-bp signal consistent with the unit length repeat sequence when digested with enzymes that cut once in the TR and hybridized to a TR probe (Fig. 2 B and C). Digestion with enzymes that do not cut in the TR indicates that the BC-1 strain contains a unique region buried in the TR, flanked by ≈7-kb and ≈35-kb TR sequences (Fig. 2 C and D). An identical pattern occurs in HBL-6, a cell line independently derived from the same tumor as BC-1, suggesting that this duplication was present in the parental tumor (Fig. 2 C and D). The restriction pattern with NotI, which also cuts only once within the TR but rarely within the LUR, suggests that the buried region is at least 33 kb (data not shown). Partial sequencing of this region demonstrates that it is a precise genomic duplication of the region beginning at ORF K8 (data not shown). The LUR is 140.5 kb including the right end unsequenced gap (<3 kb). The estimated KSHV genomic size in BC-1 and HBL-6 (including the duplicated region) is ≈210 kb.
Based on the EBV replication model used in clonality studies (31), the polymorphic BCP-1 laddering pattern may reflect lytic virus replication and superinfection (Fig. 2C). The EBV laddering pattern occurs when TR units are deleted or duplicated during lytic replication and is a stochastic process for each infected cell (31). No laddering is present for BC-1 which is under tight latent KSHV replication control (10). KS lesion DNA also shows a single hybridizing band suggesting that virus in KS tumor cells may be of monoclonal origin. A monoclonal or oligoclonal banding pattern is present in most KS lesions examined (data not shown).
Features and Coding Regions of the KSHV LUR.
The KSHV genome shares the seven block organization [blocks (B) 1–7; Fig. 1] of other herpesviruses (30), with subfamily-specific or unique ORFs present between blocks [interblock regions (IB) a–h; Fig. 1]. ORF analysis indicates that only 79% of the sequenced 137.5-kb LUR encodes 81 identifiable ORFs, which is likely to be due to our conservative assignment of ORF positions. Additional potential coding regions are included as features in the map until experimental verification is available. The overall LUR CpG dinucleotide observed/expected (O/E) ratio is 0.75 consistent with a moderate loss of methylated cytosines, but there is marked regional variation. The lowest CpG O/E ratios (<0.67) occur in IBa (bp 1–3200), in B5 (68,602–69,405), and IBh (117,352–137,507). The highest O/E ratios (>0.88) extend from B2 to B3 (30,701–47,849), in IBe (67,301–68,600), and in B6 (77,251–83,600). Comparison to the KS5 sequence (10) shows a high sequence conservation between these two strains with only 21 point mutations over the comparable 20.7-kb region (0.1%). A frameshift within BC-1 ORF28 (position 49,004) compared with KS5 ORF28 was not resolvable despite repeated sequencing of KS5 and PCR products amplified from BC-1. Two additional frameshifts in noncoding regions (bp 47,862 and 49,338) are also present compared with the KS5 sequence.
Several repeat regions are present in the LUR (Fig. 1). A 143-bp sequence is repeated within ORF K11 at positions 92,678–92,820 and 92,852–92,994 (waka/jwka). Complex repeats are present in other regions of the genome: 20- and 30-bp repeats in the region from 24,285–24,902 (frnk), a 13-bp repeat between bases 29,775 and 29,942 (vnct), two separate 23-bp repeat stretches between bases 118,123 and 118,697 (zppa), and 15 different 11- to 16-bp repeats throughout the region from 124,527 to 126,276 (moi). A complex A+G-rich repeat region (mdsk) begins at 137,099 and extends into the unsequenced gap.
Conserved ORFs with homologs found in other herpesviruses are listed in Table 1, along with their polarity, map positions, sizes, relatedness to HVS and EBV homologs, and putative functions. Conserved ORFs coding for presumed viral structural proteins and enzymes include genes involved in viral DNA replication [e.g., DNA polymerase (ORF9)], nucleotide synthesis [e.g., dihydrofolate reductase (DHFR, ORF2), thymidylate synthase (TS, ORF70)], regulators of gene expression [R transactivator (ORF50)], and five conserved herpesvirus structural capsid and five glycoprotein genes.
Table 1.
KSHV ORFs with homology to genes in other herpesviruses
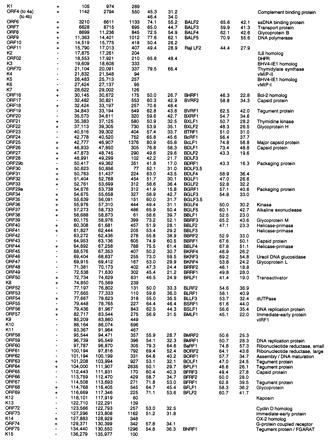
| Name | Pol. | Start | Stop | Size aa | KSHV vs. HVS
|
KSHV vs. EBV
|
|||
|---|---|---|---|---|---|---|---|---|---|
| % Sim. | % Id. | EBV name | % Sim. | % Id. | Putative function | ||||
% Sim., percent similar; % Id., percent identical; ssDNA, single-stranded DNA; DHFR, dihydrofolate reductase; FGARAT, N-formylglycinamide ribotide amidotransforase.
Several genes with homologs to HVS ORFs have unique features. ORF45 has sequence similarity to nuclear and transcription factors (chicken nucleolin and yeast SIR3) and has an extended acidic domain typical for transactivator proteins between amino acids 90 and 115. ORF73 also has an extended acidic domain separated into two regions by a glutamine-rich sequence encoded by the moi repeat. The first region consists almost exclusively of aspartic and glutamic acid residue repeats while the second glutamic acid-rich region has a repeated leucine heptad motif suggestive of a leucine zipper structure. ORF75, a putative tegument protein, has a high level of homology to the purine biosynthetic enzyme of Escherichia coli and Drosophila melanogaster N-formylglycinamide ribotide amidotransferase (FGARAT).
ORFs K3 and K5 are not homologous to HVS genes but are similar to the major immediate early bovine herpesvirus type 4 (BHV-4) gene IE1 (12 and 13% identity, respectively) (32). While BHV-4 IE1 is spliced, it is not known if splicing occurs in either KSHV transcript. These genes have no significant homology to the herpes simplex virus 1 (HSV-1) α0 (which is homologous to BHV-4 IE1), but encode proteins sharing with the HSV-1 ICP0 protein a cysteine-rich region which may form a zinc finger motif (32). The hypothetical protein encoded by ORF K5 has a region homologous to the nuclear localization site present in the late form of the BHV-4 protein. ORF K8 has a purine binding motif (GLLVTGKS) in the C terminus of the protein that is similar to a motif present in the KSHV thymidine kinase (ORF21) (10).
No homologs to HVS ORF1, -3, -5, -12, -13, -14, -15, -51, and -71 were identified in the KSHV LUR sequence. HVS ORF1 codes for a transforming protein, responsible for HVS-induced in vitro lymphocyte transformation (33) and has poor sequence conservation among HVS strains (34, 35). Functional homologs to this gene may be present but were not identifiable by sequence homology. Similarly, no homologs to EBV latency and transformation-associated proteins (EBNA-1, EBNA-2, EBNA-LP, LMP-1, LMP-2, or gp350/220) were found despite some homology to repeat sequences present in these genes. KSHV also does not have a sequence homolog to the BZLF1 EBV transactivator gene.
Several sequences were not given ORF assignments although they have characteristics of expressed genes. The sequence between bp 90,173 and 90,643 shares homology to the precursor of secreted glycoprotein X (gX), encoded by a number of alpha-herpesviruses (pseudorabies, EHV1), and which does not form part of the virion structure. Like its homolog in EHV1, the KSHV form lacks the highly acidic carboxy terminus of the pseudorabies gene.
Two polyadenylylated transcripts expressed at high copy number in BCBL-1 (15) are present at positions 28,661–29,741 (T1.1) in IBb and 118,130–117,436 (T0.7) in IBh. T0.7 encodes a 60-aa peptide (ORF K12) called Kaposin, and T1.1, also referred to as nut-1 (D. Ganem, personal communication), has been speculated to be a U RNA-like transcript (15).
Cell Cycle Regulation and Cell Signaling Protein Homolog Genes.
A number of ORFs that are either unique to KSHV or shared only with other gamma-herpesviruses encode homologs to oncoproteins and cell signaling proteins. ORF16, homologous to EBV BHRF-1 and HVS ORF16, encodes a functional Bcl-2-like protein that can inhibit Bax-mediated apoptosis (R. Sarid, T. Sato, and Y.C., unpublished data). ORF72 (11) encodes a functional cyclin D homolog, also found in HVS (37), that can substitute for human cyclin D in phosphorylating the retinoblastoma tumor suppressor protein (38).
KSHV also encodes a functionally active IL-6 (ORF K2) and two macrophage inflammatory proteins (MIPs) (ORFs K4 and K6), which are not found in other human herpesviruses (39). The viral (v)IL-6 has 62% amino acid similarity to the human IL-6 and can substitute for human IL-6 in preventing mouse myeloma cell apoptosis (39). Both MIP-like proteins have conserved C-C dimer signatures characteristic of β-chemokines and near sequence identity to human MIP-1α in their N-terminus regions. MIP-I (ORF K6) can inhibit CCR-5-dependent HIV-1 replication (39). A sequence at bp 22,529–22,185 has low conservation with MIP-1β (blastx poisson, P = 0.0015) but retains the C-C dimer motif. ORF K9 (vIRF) encodes a hypothetical 449 aa protein with homology to the family of interferon regulatory factors (IRFs) (40). It has 13.4% amino acid identity to human interferon consensus sequence binding protein and partial conservation of the IRF DNA binding domain (39). Three additional nondesignated sequences at bp 88,910–88,410, 90,541–89,600 and 94,127–93,636 also have low homology to IRF-like proteins (P ≥ 0.35). No conserved interferon consensus sequences were found in this region of the genome.
Other potential signal transduction homologs, which are also found in other herpesviruses, include a complement-binding protein homolog (v-CBP, ORF4), a neural cell adhesion molecule (NCAM)-like protein (v-adh, ORF K14), and an IL-8 receptor homolog (ORF74). Homologs to ORF4 and -74 are present in other rhadinoviruses and ORF4 is homologous to variola B19L and D12L proteins. ORF K14 (v-adh) is homologous to the rat and human OX-2 membrane antigens, various NCAMs and the poliovirus receptor-related protein PRR1. OX-2 is in turn homologous to ORF U85 of human herpesviruses 6 and 7, but there is no significant homology between the KSHV and beta-herpesvirus OX-2/NCAM homologs. Like other immunoglobulin family adhesion proteins, v-adh has V-like, C-like, transmembrane and cytoplasmic domains, and a potential RGB binding site at residues 268–270. The vIL-8R has a seven transmembrane spanning domain structure characteristic of G-protein coupled chemoattractant receptors (11) which includes the EBV-induced EBI1 protein (41).
DISCUSSION
The full-length sequence of the KSHV genome in BC-1 cells provides the opportunity to investigate molecular mechanisms of KSHV-associated pathogenesis. The KSHV genome has standard features of rhadinovirus genomes including a single unique coding region flanked by high G+C terminal repeat regions which are the presumed sites for genome circularization. In addition to having 66 conserved herpesvirus genes involved in herpesvirus replication and structure, KSHV is unique in encoding a number of proteins mimicking cell cycle regulatory and signaling proteins.
Our estimated size of the BC-1 derived genome (210 kb including the duplicated portion) is consistent with that found using encapsidated virion DNA (15). Genomic rearrangements are common in cultured herpesviruses (29, 42). However, the genomic duplication present in the BC-1 KSHV probably did not arise during tissue culture passage. TR hybridization studies indicate that this insertion of a duplicated LUR fragment into the BC-1 TR is also present in KSHV from the independently derived HBL-6 cell line (43). It is not known whether this duplication accounts for the inability of KSHV in BC-1 cells to be induced into full lytic replication.
Despite this genomic rearrangement, the KSHV genome is well conserved within coding regions. There is less than 0.1% base pair variation between the BC-1 and the 21-kb KS5 fragment isolated from a KS lesion. Higher levels of variation may be present in strains from other geographic regions or other disease conditions. Within the LUR, synteny to HVS is lost at ORF2 and -70, but there is concordance in all other regions conserved with HVS. Several conserved genes, such as thymidine kinase (10), thymidylate synthase and dihydrofolate reductase [which is present in HVS (27) but not human herpesviruses], may encode proteins that are appropriate targets for existing drugs.
Molecular mimicry by KSHV of cell cycle regulatory and signaling proteins is a prominent feature of the virus. The KSHV genome has genes encoding homologs to cellular complement-binding proteins (ORF4), cytokines (ORFs K2, K4 and K6), a Bcl-2 protein (ORF16), a cytokine transduction pathway protein (K9), an IL-8R-like protein (ORF74), and a D-type cyclin (ORF72). Additional regions potentially coding for proteins with low homology to MIP and IRF-like proteins are also present in the KSHV genome. There is a striking parallel between the cellular homologs encoded by KSHV and cellular genes known to be induced by EBV infection. Cellular cyclin D, CD21/CR2, Bcl-2, an IL-8R-like protein (EBI1), IL-6 and adhesion molecules are upregulated by EBV infection (41, 44–47). This suggests that KSHV modifies the same signaling and regulation pathways as EBV modifies after infection, but does so by introducing exogenous genes from its own genome.
Cellular defense against virus infection commonly involves cell cycle shutdown, apoptosis (for review, see ref. 48) and elaboration of cell-mediated immunity (CMI). The KSHV-encoded v-Bcl-2, v-cyclin, and vIL-6 are active in preventing either apoptosis or cell cycle shutdown (refs. 38 and 39 and unpublished observation). Functional studies for other gene homologs, such as v-IRF, v-CBP, and ORF74 are in progress. These may be either agonists or antagonists of their cellular counterparts. At least one of the β-chemokine homologs, v-MIP-I, prevents CCR5-mediated HIV infection of V-MIP-I transfected cells (39). β-Chemokines are not known to be required for successful EBV infection of cells although EBV-infected B cells express higher levels of MIP-1α than normal tonsillar lymphocytes (49). The autocrine dependence of EBV-infected B cells on small and uncharacterized protein factors in addition to IL-6 (36) leads to speculation that β-chemokines may also play a role in the EBV life cycle.
KSHV has not formally been shown to be a transforming virus and homologs to the major transforming genes of HVS and EBV are not present in the BC-1 strain KSHV. Nonetheless, dysregulation of cell proliferation control caused by the identified KSHV-encoded protooncogenes and cytokines may contribute to neoplastic expansion of virus-infected cells. Preliminary studies suggest that subgenomic KSHV fragments can transform NIH 3T3 cells (R. Sarid, unpublished data). If KSHV replication, like that of EBV, involves recombination of TR units (30), a monomorphic TR hybridization pattern present in a KS lesion would indicate a clonal virus population in the tumor. This is consistent with KS being a true neoplastic proliferation arising from single transformed, KS-infected cell rather than KSHV being a “passenger virus.” Identification of KSHV homologs to known oncoproteins and cell proliferation factors in the current study provides evidence that KSHV is likely to be a transforming virus.
Acknowledgments
We would like to acknowledge Gifty Asamani and Zhou Ju-Yen for excellent technical assistance, Ai-En Thlick for assistance with the figures, and Xiao-Lu Ye and Peisen Zhang for help with DNA analysis. We also thank Dr. Phil Pellett for critical reading of the manuscript. This work was supported by central funds of the Columbia Genome Center and National Institutes of Health Grant CA67391 awarded to Y.C. and P.S.M.
Footnotes
Abbreviations: KS, Kaposi sarcoma; KSHV, KS-associated herpesvirus; HVS, herpesvirus saimiri; EBV, Epstein–Barr virus; LUR, long unique region; TR, terminal repeat; ORF, open reading frame; MIP, macrophage inflammatory peptide; IRF, interferon regulatory factor, IL, interleukin; IB, interblock region; R, receptor; V, viral.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base [accession nos. U75698U75698 (long unique unit) and U75699U75699 and U75700U75700 (terminal repeat unit)].
References
- 1.Tappero J W, Conant M A, Wolfe S F, Berger T G. J Am Acad Dermatol. 1993;28:371–395. doi: 10.1016/0190-9622(93)70057-z. [DOI] [PubMed] [Google Scholar]
- 2.Ensoli B, Barillari G, Gallo R C. Immunol Rev. 1992;127:147–155. doi: 10.1111/j.1600-065x.1992.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 3.Peterman T A, Jaffe H W, Beral V. AIDS. 1993;7:605–611. doi: 10.1097/00002030-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Chang Y, Cesarman E, Pessin M S, Lee F, Culpepper J, Knowles D M, Moore P S. Science. 1994;265:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 5.Boshoff C, Whitby D, Hatziionnou T, Fisher C, van der Walt J, Hatzakis A, Weiss R, Schulz T. Lancet. 1995;345:1043–1044. doi: 10.1016/s0140-6736(95)90780-7. [DOI] [PubMed] [Google Scholar]
- 6.Dupin N, Grandadam M, Calvez V, Gorin I, Aubin J T, Harvard S, Lamy F, Leibowitch M, Huraux J M, Escande J P, Agut H. Lancet. 1995;345:761–762. doi: 10.1016/s0140-6736(95)90642-8. [DOI] [PubMed] [Google Scholar]
- 7.Moore P S, Chang Y. N Engl J Med. 1995;332:1181–1185. doi: 10.1056/NEJM199505043321801. [DOI] [PubMed] [Google Scholar]
- 8.Schalling M, Ekman M, Kaaya E E, Linde A, Biberfeld P. Nat Med. 1995;1:707–708. doi: 10.1038/nm0795-707. [DOI] [PubMed] [Google Scholar]
- 9.Chang Y, Ziegler J L, Wabinga H, Katongole-Mbidde E, Boshoff C, Whitby D, Schulz T, Weiss R A, Jaffe H A, Moore P S. Arch Intern Med (Moscow) 1996;156:202–204. [PubMed] [Google Scholar]
- 10.Moore P S, Gao S-J, Dominguez G, Cesarman E, Lungu O, Knowles D M, Garber R, McGeoch D J, Pellett P, Chang Y. J Virol. 1996;70:549–558. doi: 10.1128/jvi.70.1.549-558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cesarman E, Nador R G, Bai F, Russo J, Moore P S, Chang Y, Knowles D M. J Virol. 1996;70:8218–8223. doi: 10.1128/jvi.70.11.8218-8223.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cesarman E, Chang Y, Moore P S, Said J W, Knowles D M. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 13.Cesarman E, Moore P S, Rao P H, Inghirami G, Knowles D M, Chang Y. Blood. 1995;86:2708–2714. [PubMed] [Google Scholar]
- 14.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Ganem D. Nat Med. 1996;2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 15.Zhong W, Wang H, Herndier B, Ganem D. Proc Natl Acad Sci USA. 1996;93:6641–6646. doi: 10.1073/pnas.93.13.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Decker L L, Shankar P, Khan G, Freeman R B, Dezube B J, Lieberman J, Thorley-Lawson D A. J Exp Med. 1996;184:283–288. doi: 10.1084/jem.184.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao S-J, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, Newton R, Rinaldo C R, Saah A, Phair J, Detels R, Chang Y, Moore P S. Nat Med. 1996;2:925–928. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- 18.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 19.Benton W D, Davis R W. Science. 1977;196:180–182. doi: 10.1126/science.322279. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D, Meselson M. Methods Enzymol. 1983;100:333–342. doi: 10.1016/0076-6879(83)00066-x. [DOI] [PubMed] [Google Scholar]
- 21.Feinberg A P, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 22.Melton D A, Krieg P A, Rebagliati M R, Maniatis T, Zinn K, Green M R. Nucleic Acids Res. 1984;12:7035–7056. doi: 10.1093/nar/12.18.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deininger P L. Anal Biochem. 1983;129:216–223. doi: 10.1016/0003-2697(83)90072-6. [DOI] [PubMed] [Google Scholar]
- 24.Bankier A T, Weston K M, Barrell B G. Methods Enzymol. 1987;155:51–93. doi: 10.1016/0076-6879(87)55009-1. [DOI] [PubMed] [Google Scholar]
- 25.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Kozak M. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albrecht J-C, Nicholas J, Biller D, Cameron K R, Biesinger B, Newman C, Wittmann S, Craxton M A, Coleman H, Fleckenstein B, Honess R W. J Virol. 1992;66:5047–5058. doi: 10.1128/jvi.66.8.5047-5058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Telford E A, Watson M S, Aird H C, Perry J, Davison A J. J Mol Biol. 1995;249:520–528. doi: 10.1006/jmbi.1995.0314. [DOI] [PubMed] [Google Scholar]
- 29.Baer R, Bankier A T, Biggin P L, Deininger P L, Farrell P J, Gibson T J, Hatfull G, Hudson G S, Satchwell S C, Séguin C, Tuffnell P S, Barrell B G. Nature (London) 1984;310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- 30.Chee M S, Bankier S B, Bohni C M, Brown R C, Horsnell T, Hutchison C A, Kouzarides T, Martignetti J A, Preddie E, Satchwell S C, Tomlinson P, Weston K M, Barrell B G. Curr Top Microbiol Immunol. 1990;154:125–169. doi: 10.1007/978-3-642-74980-3_6. [DOI] [PubMed] [Google Scholar]
- 31.Raab-Traub N, Flynn K. Cell. 1986;47:883–889. doi: 10.1016/0092-8674(86)90803-2. [DOI] [PubMed] [Google Scholar]
- 32.van Santen V. J Virol. 1991;65:5211–5224. doi: 10.1128/jvi.65.10.5211-5224.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akari H, Mori K, Terao K, Otani I, Fukasawa M, Mukai R, Yoshikawa Y. Virology. 1996;218:382–388. doi: 10.1006/viro.1996.0207. [DOI] [PubMed] [Google Scholar]
- 34.Jung J U, Desrosiers R C. J Virol. 1991;65:6953–6960. doi: 10.1128/jvi.65.12.6953-6960.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung J U, Desrosiers R C. Mol Cell Biol. 1995;15:6506–6512. doi: 10.1128/mcb.15.12.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tosato G, Tanner G, Jones K D, Revel M, Pike S E. J Virol. 1990;64:3033–3041. doi: 10.1128/jvi.64.6.3033-3041.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholas J, Cameron K R, Honess R W. Nature (London) 1992;355:362–365. doi: 10.1038/355362a0. [DOI] [PubMed] [Google Scholar]
- 38.Chang Y, Moore P S, Talbot S J, Boshoff C H, Zarkowska R, Godden-Kent D, Peterson H, Weiss R A, Mittnacht S. Nature (London) 1996;382:410. doi: 10.1038/382410a0. [DOI] [PubMed] [Google Scholar]
- 39.Moore, P. S., Boshoff, C., Weiss, R. A. & Chang, Y. (1996) Science, in press. [DOI] [PubMed]
- 40.David M. Pharmacol Ther. 1995;65:149–161. doi: 10.1016/0163-7258(94)00050-d. [DOI] [PubMed] [Google Scholar]
- 41.Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, Kieff E. J Virol. 1993;67:2209–2220. doi: 10.1128/jvi.67.4.2209-2220.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cha T-A, Tom E, Kemble G W, Duke G M, Mocarski E S, Spaete R R. J Virol. 1996;70:78–83. doi: 10.1128/jvi.70.1.78-83.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaidano G, Cechova K, Chang Y, Moore P S, Knowles D M, Dalla-Favera R. Leukemia. 1996;10:1237–1240. [PubMed] [Google Scholar]
- 44.Palmero I, Holder A, Sinclair A J, Dickson C, Peters G. Oncogene. 1993;8:1049–1054. [PubMed] [Google Scholar]
- 45.Finke J, Fritzen R, Ternes P, Trivedi P, Bross K J, Lange W, Mertelsmann R, Dolken G. Blood. 1992;80:459–469. [PubMed] [Google Scholar]
- 46.Finke J, Lange W, Mertelsmann R, Dolken G. Leuk Lymphoma. 1994;12:413–419. doi: 10.3109/10428199409073782. [DOI] [PubMed] [Google Scholar]
- 47.Jones K, Rivera C, Sgadari C, Franklin J, Max E E, Bhatia K, Tosato G. J Exp Med. 1995;182:1213–1221. doi: 10.1084/jem.182.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen Y, Shenk T E. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 49.Harris P E, Maffei A, Liu Z, Colovai I, Reed E F, Inghirami G, Suciu-Foca N. J Immunol. 1993;151:5975–5983. [PubMed] [Google Scholar]