

Abstract
Background:
Flow cytometric analysis of human P2X7 pore activity segregates variant from common P2RX7 genotypes and may serve as a biomarker for cancer, pain, inflammation and immune responses to infection. Standardization is needed to accommodate variable sample age and instrumentation differences in a multi-center clinical trial.
Methods:
CD14-PE stained whole blood samples were treated with YO-PRO-1 combined with a P2X7 agonist (BzATP) or control, followed by the addition of PI after closure of the P2X7 pore. Recalled instrument settings from previous publications were used to adapt a standardized fluorescent particle-adjusted set up method. Experiments were performed to compare the two methods while evaluating components of systematic variability and facilitating reliable processing of samples with varied ages.
Results:
The median YO-PRO-1 fluorescence of BzATP treated samples had less variability when collected by the bead-adjusted method, and was less influenced by the compensation strategy employed. The average day-to-day coefficient of variance for assessments of P2X7 pore activity by this method was 0.11 ± 0.04, and the exclusion of non-viable cells was found to accommodate samples aged up to 4 days after phlebotomy. The bead-adjusted set up method produced measurements differing by only 2.0 ± 1.5% on two analog cytometers, and within similar decades when comparing analog to digital instruments.
Conclusions:
These results provide a standardized method for quantitative flow cytometric analysis of P2X7 receptor phenotypes in blood monocytes with minimal intra-laboratory variation and potential for inter-laboratory comparisons that can greatly facilitate multi-center functional genomic clinical studies.
Keywords: flow cytometry, standardization, whole blood, functional genomic assay, P2X7
The purinergic nucleotide receptor P2X7 is increasingly associated with diverse pathological conditions including cancer, pain, inflammation and aberrant responses to infection (1). This ligand-gated cation channel is encoded by P2RX7, a 55 kB gene with 13 exons on human chromosome 12q24 (2,3), and is expressed by all classes of leukocytes studied to date, epithelial cells and select neuronal populations (1,4,5). Current bioinformatics data suggest there is a recombination hotspot in the middle of intron 5, associated with over 380 single nucleotide polymorphisms in the region (www.hapmap.org, accessed 5/23/07). Consistent with its role in amplification of innate immune responses (6-10), genetic association studies link variants of this receptor to the control of Mycobacterium tuberculosis in different populations on three continents (11-13). To facilitate additional association studies, our laboratory developed a genomically validated functional assay to rapidly identify and bridge disparate P2RX7 genetic, phenotypic and clinical results, while also increasing statistical power in the face of sample size constraints (10,14-16).
Our assay segregates variant from common P2X7 receptor genotypes/phenotypes by taking advantage of a unique feature of the P2X7 receptor. This feature is the reversible expansion of its selectivity filter following continuous cellular exposure to extracellular nucleotides that results in increased cation (Ca2+, K+, and Na+) permeability and the passage of larger molecules (≤ 900 Da), a phenomenon referred to as pore activity (2,17). Using a kinetic functional assay with features expressly designed to segregate samples containing loss-of-function alleles, we identified a threshold of pore activity below which these genotypes are greatly enriched. Specifically, logistic regression analysis with the full range of pore activity data as a continuous variable and a combined binomial variable derived from three well validated loss of function genotypes had a receiver-operator area under the curve of 0.927 (p < 0.001). The optimal threshold of low pore activity was thereby established as 22-fold of P2X7 agonist (BzATP)-induced YO-PRO-1 uptake over that stimulated by the saline control, where the fold-uptake was defined as the ratio of fluorescence measurements in BzATP- and buffer-treated samples (14). The performance characteristics at this threshold for identifying samples with 1513 CC, 1729 TA or 946 GA genotypes were as follows: sensitivity 85%, specificity 91%, positive predictive value 59% and negative predictive value of 98%. For example, a blood sample with normal pore activity (i.e. > 22-fold of BzATP stimulated YO-PRO-1 uptake) has a 98% chance of not harboring the three aforementioned loss-of-function genotypes. Utilizing this cellular assay, our laboratory identified that 19% of individuals residing in the upper Midwest of North America have attenuated monocyte P2X7-regulated pore function, and that discordant low-pore samples not containing the 1513 CC, 1729 TA or 946 GA genotypes were enriched for novel loss-of-function alleles (14). Moreover, we demonstrated an association between loss-of-function P2X7 receptor genotypes to attenuated nucleotide-stimulated P2X7 pore function and an anti-inflammatory cytokine profile by monocytes following in vitro challenge with LPS and nucleotides (10). Thus, the precise method we have shown previously for our pore assay is tightly linked to the identification of samples with P2XR7 genetic variants, and attenuation in P2X7 receptor pore function may serve as a biomarker for infectious/inflammatory diseases and disorders.
We presently have the opportunity to further validate this assay in an ongoing multi-center clinical trial with overnight shipping of samples to our facility. In preparation for this trial, one of our goals was to refine our set up method to increase the ease of use on any cytometer, while also accommodating aged samples. Recent examples from the clinical literature demonstrate that differences in instrument settings can contribute to discrepant results (18,19). This functional assay also presents difficulties in defining standard setup since it is performed in whole blood. Moreover, we have noticed a trend in unpublished data that sample age increases the day-to-day variability. Specifically, when all samples are processed on the day of phlebotomy with our prior method, the coefficient of variance is 0.10 ± 0.07, whereas if any of the samples have delayed processing the day-to-day CV in 0.21 ± 0.13 (p = 0.09). Considering previous work on standardization (20-29), we have revised our method to further homogenize data collection using readily available fluorescent particles and the viability marker propidium iodide to identify responding cells. In addition, the following variables inherent to multi-center clinical assessments were examined: sample age, different instrumentation, and biological variability. We report that our revised bead-adjusted setup method produces less intra-laboratory variability than our former method with recalled settings, particularly when the raw median YO-PRO-1 fluorescence data are presented from BzATP-treated samples. Moreover, the bead-adjusted setup accommodates processing of samples with varying age by excluding nonviable cells, and produces comparable results when transferred between instruments, suggesting that it may also reduce inter-laboratory differences.
METHODS
Human Subject Participation and Sample Collection
Investigations were carried out with approval of the University of Wisconsin Institutional Review Board, and ancillary studies to two protocols (single center and multi-center) involving patients with asthma were performed after obtaining informed consent. For all experiments, five to ten mL of whole blood were obtained by routine phlebotomy using plastic vacutainers containing citrate for anticoagulation. All samples were stored exclusively at room temperature with or without overnight shipping until the time of processing. Eight participants were recruited for 2 additional phlebotomy visits within a 3-month span. All samples were processed and analyzed at the Flow Cytometry Facility of the University of Wisconsin Paul P. Carbone Comprehensive Cancer Center.
Sample Preparation
Although all leukocytes studied to date express P2X7, monocytes were selected as the cell population to screen because of the greater sensitivity of pore function noted between individuals participating in a small study with 45 healthy subjects (5,30). All reagents have been optimized and are prepared in large batches with aliquots sufficient for at least 200 subjects, using previously described staining procedures (10,14,30). Briefly, aliquots of whole blood (500 μL/aliquot) were washed twice in HEPES-buffered saline (HBS; 130 mM NaCl, 5 mM KCl, 20 mM HEPES pH 7.4, 0.1% bovine serum albumin, 10 mM glucose; components purchased at Sigma, St. Louis, MO) and resuspended in the original 500 μL volume. A 10 μL aliquot of anti-human CD14 antibody conjugated to phycoerythrin (CD14-PE, 50 μg/mL; BD Biosciences, San Diego, CA) was added to each sample and incubated at room temperature for 20 min. The cells were washed twice in a potassium glutamate buffer (130 mM potassium glutamate, 5 mM KCl, 20 mM HEPES pH 7.4, 0.1% bovine serum albumin, 10 mM glucose; components from Sigma) to maximize the differences between high and low pore activities (17). In the absence of NaCl, cells were stimulated for 20 min with 0 or 250 μM 2′-3′-O-(4-Benzoylbenzoyl) adenosine 5′-triphosphate (BzATP; Sigma) in the presence of 1 μM YO-PRO-1 (Molecular Probes, Eugene, OR). Samples were adjusted to 10 mM magnesium chloride, washed in HEPES-buffered saline and diluted to a volume of 2.5 mL in HBS. These latter steps have been previously shown to close the pore rapidly, allowing for kinetic precision in a large clinical study (10,30). For our previous studies in healthy subjects and the data from the Natural Cold in Asthma protocol (refs (10,30) and Table 2), the vast majority of samples were processed on the same day as the phlebotomy without inclusion of propidium iodide. For the methods described here, including all the samples from the MIA trial (Table 2), PI was added to each sample (5 μg/mL) and incubated for 15 min prior to sample acquisition using flow cytometry as described below.
Table 2.
Descriptive statistics for measurements of P2X7 pore activity in clinical samples from asthma patients. Whole blood samples were obtained locally as a part of the Natural Cold in Asthma study or through an ongoing multi-center clinical trial (Macrolides In Asthma; ACRN-MIA) via the Asthma Clinical Research Network. Data represent the BzATP-induced fold-stimulation of YO-PRO-1 uptake fluorescence. Seventeen of the thirty-one Natural Cold subjects had samples obtained both during the acute and baseline phases of a naturally occurring upper respiratory viral infection, in which case the mean value was compiled for the group descriptors. The remainder of the Natural Cold, and all of the ACRN-MIA, patients had samples measured from one visit only. Samples greater than one day old were excluded from analysis in the Natural Cold protocol due to the absence of dead cell exclusion. CD14-PEpos/PIpos cells were uniformly excluded in the ACRN-MIA study and samples up to four days old were processed with the data presented. ACRN-MIA subjects with samples older than 4 days due to shipping delays were redrawn at another visit if possible.
| Natural Cold Data (Recalled Settings) N = 31 |
ACRN-MIA Data (Bead-adjusted Settings) N = 99 |
|
|---|---|---|
| Minimum | 13.8 | 2.3 |
| 1st Quartile | 359 | 274 |
| Median | 669.5 | 598 |
| 3rd Quartile | 992 | 893.5 |
| Maximum | 1676 | 1666 |
Instrument Settings Determination and Calculation of Spectral Overlap
All instruments in the facility are cleaned daily and assessed for laser alignment, photomultiplier tube (PMT) performance, linearity and noise. All fluorochromes were excited with a 488 nm laser and the following filter sets were used in all flow cytometers: a BP 530/30 for YO-PRO-1, a BP 585/42 for PE and a LP 670 for propidium iodide. Experiments were performed on FACScan, FACSCalibur, and LSR II flow cytometers (Becton Dickinson, San Jose, CA) in conjunction with the CellQuest, CellQuestPro, FACSDiva acquisition and analysis software programs (v. 3.3, v. 4.0, and v. 5.0.1, respectively; Becton Dickinson).
Previously, pore activity was measured on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) using recalled instrument settings: YO-PRO-1 at 410 mV, PE at 412 mV, acquisition threshold for PE at 324 mV, and PE – 32.6% YO-PRO-1 compensation (10). To facilitate comparisons across instrument platforms and among laboratories, we used these recalled settings as a template to adapt desired PMT voltages for SpheroTM Rainbow fluorescent particles (mid-range 3.0-3.4 μm; Sperotech, Lake Forest IL) shown in Supplementary Figure 1. Rainbow particles have a very stable fluorescence and can be stored and used over a long period of time, allowing the intensities of these particles in each fluorescent detector to serve as target values for determining the voltages applied to each PMT for all of the cytometers used. The same target values were used for the analog instruments, FACScan and FACSCalibur (BD Biosciences, San Jose, CA) because these instruments process fluorescence signals in the same way and the intensities determined were very close (data not shown). The LSR II (BD Biosciences, San Jose, CA) uses digital processing and fluorescence is collected and sampled differently so a second set of target values using these same samples was determined for each color detector on this cytometer. Specifically, the target value ranges of 558 ± 20 (YO-PRO-1), 511 ± 20 (PE) and 676 ± 20 (PI) were used on the LSR II.
To calculate spectral overlap, BD Calibrite™ fluorescent particles were used as a substitute for single stained control samples because the detection of monocytes using scatter properties is very problematic in whole blood. Samples of the FITC- and PE-labeled beads from the Calibrite bead kit (BD Biosciences, San Jose, CA) were prepared individually. The fluorescence contribution of YO-PRO-1 to the PE detector signal and that of PE to the YO-PRO-1 detector and the PI detector signals were calculated (Supplementary Figure 2). The contribution of positively stained beads was subtracted from detectors until the median fluorescence was equal to that of unstained beads in detectors in which they overlapped (Supplementary Figure 2).
PBMC isolation
PBMCs were isolated for select experiments to verify that the Calibrite method discussed above was providing appropriate compensation for PE and YO-PRO-1. Six mLs of citrate-treated blood was underlayed with three mLs of Lymphoprep (Axis-Sheild, Oslo, Norway) and centrifuged at 800 g for 30 minutes at room temperature. The cellular interface was collected and washed twice with sterile phosphate-buffered saline (PBS). Cells were counted and assayed for pore activity as described below.
Flow Cytometry Data and Statistical Analyses
For each sample, 10,000 CD14-PEpos/PIneg events were acquired. Flow cytometry data was analyzed using FlowJo data analysis software (version 8.4. TreeStar, Inc., Ashland, OR). Analysis was restricted to either CD14pos events or CD14pos/PIneg events. The median fluorescence values for YO-PRO-1 are reported after saline-treatement or BzATP-stimulation. In some cases, data are expressed as a BzATP-induced fold of YO-PRO-1 uptake, i.e. the ratio of these two measurements, to be consistent with our prior work (10,30). One-way ANOVA and students' t-tests were performed using the JMP 6.0 software package (SAS Institute, Cary NC).
RESULTS
Contribution of non-viable cells to analysis of pore activity
We previously demonstrated that the use of our original method on a single instrument provides a rapid, high-throughput, genomically validated, functional screening assay for variant P2RX7 alleles with minimal intra-laboratory variability (10,14). In order to facilitate the analysis of P2X7 pore function as a continuous variable in larger epidemiological studies, we needed to refine the method to accommodate samples of varying age in a way that also maximizes platform flexibility and reduces potential for inter-laboratory variability. Figure 1A shows the decline in monocyte viability in whole blood samples stored at room temperature for up to four days post phlebotomy. Although the cell impermeant DNA intercalating cyanine dye YO-PRO-1 gains access to nuclear material after BzATP-stimulation of the P2X7 pore, it may also stain cells no longer possessing an intact plasma membrane. In aged samples, a second YO-PRO-1 positive population of BzATP-stimulated cells appears, which is minimally present in whole blood processed within a few hours of phlebotomy (Figure 1B). To restrict the analysis to viable monocytes, the inclusion of a second vital dye after P2X7 pore closure is thereby required. Flow cytometric analysis of YO-PRO-1 fluorescence detected in basal and BzATP-stimulated CD14-PEpos monocytes was conducted with or without PI addition at the end of the staining procedure. Because our previous measure of P2X7 pore activity (BzATP stimulated fold of YO-PRO-1 uptake) is a ratio of agonist-induced and control measurements, it is not surprising that the exclusion of non-viable cells increases the apparent fold-uptake (viable = 76.5 ± 20.5 vs. total = 32.5 ± 6.2 expressed as mean ± SEM, N = 12; p = 0.04). Thus, to minimize the effects of variable sample age, exclusion of non-viable cells is required to measure true YO-PRO-1 fluorescence in monocytes following BzATP challenge as a measure of P2X7 pore function.
Figure 1.
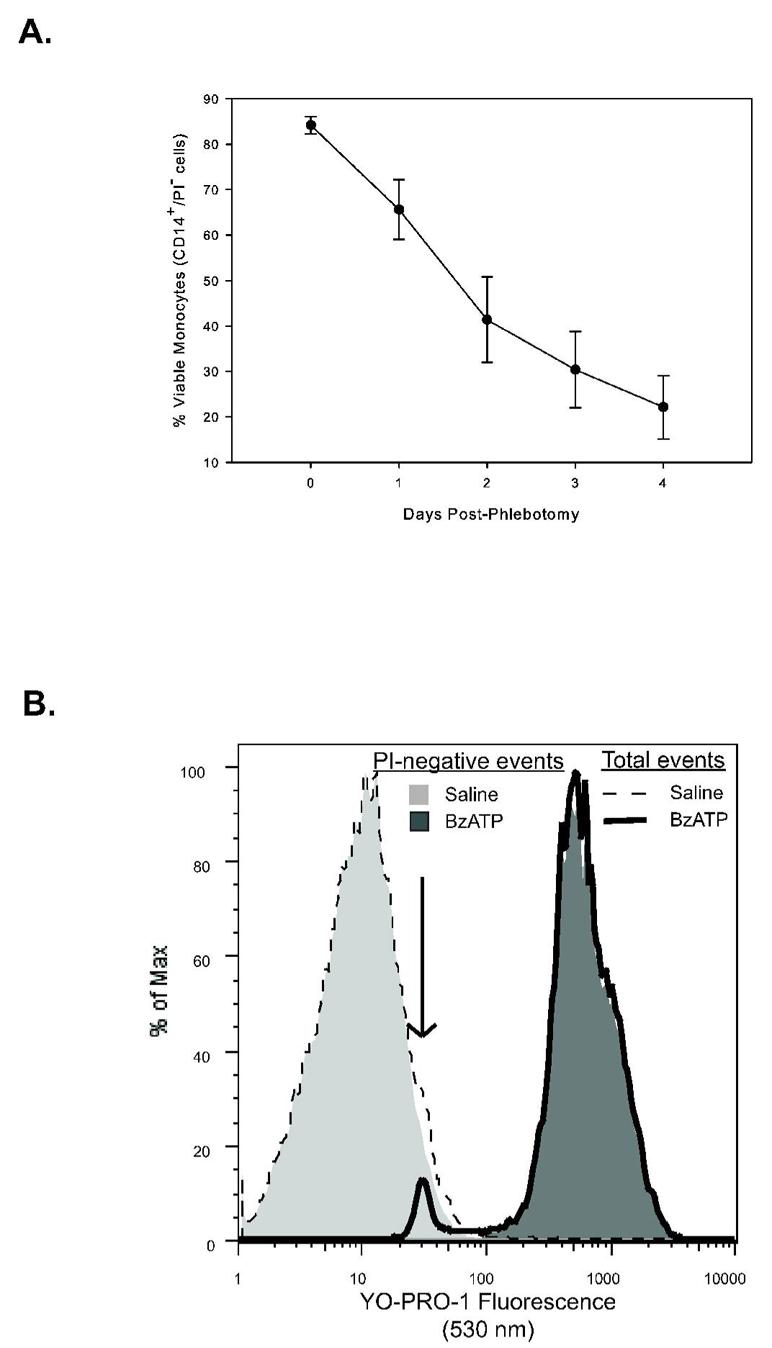
Effect of monocyte viability on BzATP-stimulated P2X7 pore function. A. Peripheral blood was obtained from eight laboratory volunteers and processed immediately or stored at room temperature for up four days to simulate the conditions of shipping blood from centers around the country. On the day of processing, aliquots of the blood samples were stained and data was acquired using bead-adjusted settings on a FACSCalibur flow cytometer. Data is expressed as % CD14pos/PIneg events in the sample (mean ± SEM; n =8). B. Anti-CD14-PE labeled human periphral blood from a representative subject was stimulated with control vehicle or 250 mM BzATP for 20 minutes in the presence of 1 mM YO-PRO-1 dye as described in Materials and Methods and beads were used to set the cytometer. Overlayed histogram analysis of YO-PRO-1 flurescence detected in live (dashed or solid outline) and total (light or dark shading) monocyte populations with or without stimulation with BzATP as denoted by the inset symbol key. The arrow indicates the presence of non-viable monocytes in both the saline and the BzATP samples when analyzing total events, that are also separate populations on PE vs YO-PRO-1 plots.
Standardization of instrument setup for measurements of P2X7 pore function in viable monocytes
To minimize operator- and platform-dependent differences in settings adjustment, the present study adapted methods of establishing fluorescent particle targeted PMT sensitivities and calculation of spectral overlap (22-27) using our recalled settings as a guide. After establishing appropriate compensation for PI (discussed below), Table 1 illustrates the effect of bead-adjusted versus recalled flow cytometer instrument settings on the day-to-day variability of control and BzATP-stimulated YO-PRO-1 fluorescence associated with viable (CD14-PEpos/PIneg) monocytes. Despite daily assessment of instrument performance and confirmation that our window of analysis is within the linear range for the PMTs (not shown), bead-adjusted voltage settings reduced day-to-day variability of these measurements in comparison to those obtained with recalled settings (Table 1).
Table 1.
Comparison of the methods of determining instrument settings on the day-to-day variability of the pore assay. Three separate blood samples drawn from the same two donors were drawn on three sequential days and assayed as described. All samples were assayed on a FACScan flow cytometer using either bead-adjusted settings as described or by recalling same instrument settings used in previous assays. Means are the average of three separate measurements of the median fluorescence intensity (MFI) of YO-PRO-1 in live monocytes, stimulated or unstimulated with BzATP.
| Donor |
YO-PRO-1 Measurement |
PMT Method |
Mean |
Standard Deviation |
Coefficient of Variance |
|---|---|---|---|---|---|
| Low Responder |
Unstimulated | Recalled | 4.6 | 1.6 | 0.35 |
| Bead Adjusted |
4.0 | 0.8 | 0.20 | ||
| Normal Responder |
Unstimulated | Recalled | 2.2 | 0.6 | 0.26 |
| Bead Adjusted |
2.8 | 0.3 | 0.12 | ||
| Low Responder |
BzATP Stimulated |
Recalled | 30.0 | 6.3 | 0.21 |
| Bead Adjusted |
30.1 | 2.5 | 0.08 | ||
| Normal Responder |
BzATP Stimulated |
Recalled | 983.0 | 154.9 | 0.16 |
| Bead Adjusted |
1019.0 | 118.7 | 0.12 | ||
| Low Responder |
Fold Uptake | Recalled | 6.8 | 1.4 | 0.20 |
| Bead Adjusted |
7.7 | 2.0 | 0.26 | ||
| Normal Responder |
Fold Uptake | Recalled | 469.7 | 170.9 | 0.36 |
| Bead Adjusted |
365.4 | 63.7 | 0.17 |
Compensation settings are also a major source of intra- and inter-laboratory variability, and ideally should be calculated using single color controls. Our analysis plan was to gate out PIpos events, which simplifies the major compensation requirements to eliminate the PE signal in the YO-PRO-1 channel and vice versa. Figures 2A and 2B demonstrate that the identification of monocytes by forward and side scatter properties is confounded in whole blood relative to PBMCs, likely due to the vast excess of RBCs and platelets. The use of a CD14-PE threshold enriches data collection of this population in whole blood and PBMCs (Figures 2C and 2D). Accordingly, the requirement of a fluorescent CD14-PE tag to identify monocytes precludes single color compensation controls in whole blood samples, but is similar to the CD45-tagged TruCount method used by clinical labs for rapid complete blood counts with leukocyte differential assessment and other purposes (for examples, refer to citations (31,32)). The commercially available Calibrite bead set includes PE-beads, and FITC-labeled beads are used as a surrogate for YO-PRO-1. After setting compensation values by the Calibrite bead method, we validated the lack of spectral overlap using PBMCs, where appropriate single color controls can be generated (not shown). Figures 2E and 2F show PI vs. PE dot plots of whole blood and PBMCs with representative acquisition gates to identify a viable PE-CD14pos population, thereby allowing for appropriate data restriction when collecting YO-PRO-1 fluorescence measurements. Finally, Figures 2G and 2H show CD14-PE vs. YO-PRO-1 plots with overlays of control and BzATP-treated samples. These figures demonstrate the absence of events compressed against the PE vs YO-PRO-1 line of identity in whole blood and PBMCs, with the former plots providing a daily verification that the Calibrite method is performing appropriately for whole blood samples.
Figure 2.
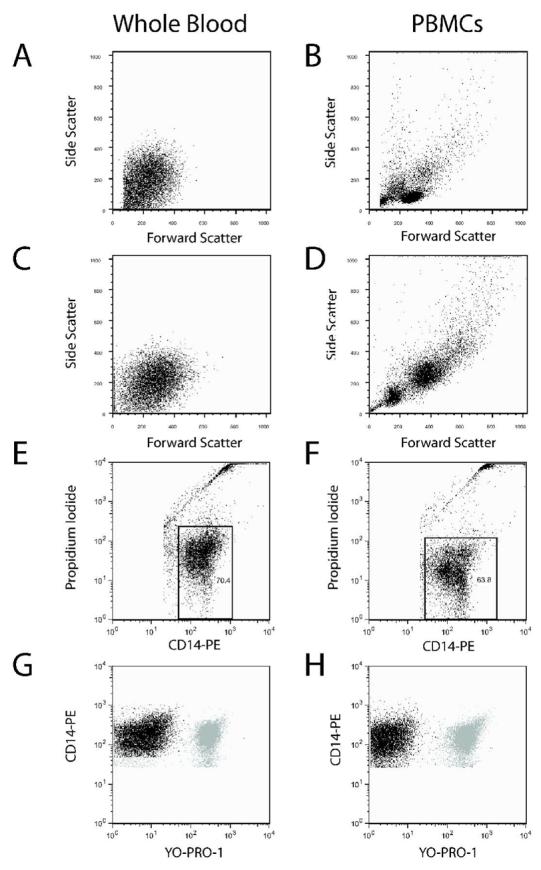
Identification of monocytes in whole blood samples requires CD14-PE thresholding and precludes single color compensation controls. Side scatter vs. forward scatter plots are show for whole blood and PBMC samples with thresholding on forward scatter (Panels A and B) or CD14-PE fluorescence (Panels C and D). Because monocyte data enrichment in whole blood samples cannot be done with SSC vs. FSC plots (Panel A), the Calibrite bead compensation method is used as a surrogate for YO-PRO-1 compensation controls in whole blood samples already containing CD14-PE. Using the Calibrite bead adjusted setup method, Panels E and F show that data restriction on viable CD14-PEpos/PIneg events is just as easy in whole blood as it is in PBMC samples. Finally, Panels G and H show the effects of bead-determined compensation on PE versus YO-PRO-1 analysis when restricted to live events in whole blood and PBMC samples respectively. The unstimulated YO-PRO-1 labeled monocytes (black) are overlayed with stimulated monocytes (gray).
Signal to noise determinations, such as the BzATP-stimulated fold uptake of YO-PRO-1, have intrinsic appeal but are complicated when both signals involve logarithmic amplification of the fluorescence signal. To begin to assess which measurement is the most reliable, we measured YO-PRO-1 MFI in samples that varied widely in terms of preparation or compensation strategy (Supplementary Table 1). Purification of PBMCs from the same donor markedly influences untreated MFI measurements, thereby influencing the fold stimulation. Similarly, intentional under- and over-compensation of PE in the YO-PRO-1 channel alters the baseline measurements and, thus, the fold calculations. By contrast, the BzATP-stimulated median YO-PRO-1 fluorescence appears reasonably independent of wide variation in sample preparation or compensation strategy (Supplementary Table 1). Subtraction of the unstimulated MFI has been suggested to be a more robust measure of signal to noise estimates (29), and appears relatively stable under these select conditions (Supplementary Table 1). Collectively, these data demonstrate that the bead-adjusted setup method for both PMT voltages and compensation calculation further reduce intra-laboratory variability when viable monocyte associated YO-PRO-1 fluorescence is measured in BzATP-stimulated whole blood samples.
Combined Estimates of Biological and Systematic Variability
Because P2X7 mRNA and protein expression is enhanced by inflammatory cytokines such as IFN-γ, subtle changes in systemic inflammation from any one donor could vary from day to day (33). Therefore, peripheral blood was obtained from eight individuals without any apparent cold symptoms on three different days throughout a 3-month period. Figure 3 shows the means and standard deviations of the BzATP-induced YO-PRO-1 fluorescence in viable monocytes for these 8 subjects measured on three independent days. The average day-to-day coefficient of variance is 0.11 ± 0.04. Of note, more day-today variation in the BzATP-induced fold of dye uptake occurred in normal responders (average CV 0.25 ± 0.28), likely due in part to the compensation differences affecting the basal values in this ratio as discussed above. These data strongly suggest that measurements of P2X7 pore activity by viable monocytes in blood obtained on different days is reliable, particularly when the raw BzATP-induced YO-PRO-1 fluorescence data are reported. These results provide a standardized method for quantitative flow cytometric analysis of P2X7 receptor phenotypes in blood monocytes with minimal intra-laboratory variation.
Figure 3.

Effect of day-to-day phlebotomy on measured P2X7 pore function in live monocytes. Staining and standardized calibration of FACSCalibur settings was conducted (see Methods) and 10000 CD14-PEpos/PIneg events were collected. Data indicate the fold stimulation in BzATP-induced YO-PRO-1 uptake by live (CD14-PEpos/PIneg) monocytes acquired on 3 different days within a 3-month span from 8 different donors.
Instrument- and operator-dependent differences have been shown to contribute to inter-laboratory variation in quantitative flow cytometry for clinical studies, particularly in the absence of a standardized set up protocol (34,35). Employing similar methods to those of this study, standardization of instrument settings with fluorescent particles, can minimize both platform and day-to-day variation and permits both intra-and inter-laboratory comparisons (34). Previously, the whole blood P2X7 pore assay was performed exclusively on a FACScan (10,30). To measure the differences between instruments, the same samples from eleven volunteers were analyzed on each cytometer in the facility that includes two analog instruments (FACScan and FACSCalibur) and a digital cytometer (LSR II). Figure 4 shows representative histograms of the BzATP-stimulated YO-PRO-1 fluorescence for a low responder and a normal responder processed on the same day on three different flow cytometers (FACScan, FACSCalibur, and LSR II). In general, the cytometers that use logarithmic amplifiers have very comparable measurements, while the LSR II has different values likely due to digital signal processing without logarithmic amplification. Specifically, when comparing data obtained from all eleven volunteers, the absolute difference between intra-subject measurements on the two analog instruments (FACScan and FACSCalibur) was 2.0 ± 1.5 percent of the average BzATP-stimulated YO-PRO-1 fluorescence. As shown in Figure 4, data collected with the bead-adjusted method on the LSR II produces histogram results in a decade similar to those obtained on analogue machines, when these values are considered in proportion to the total window of analysis for each instrument. Although the fold stimulation in BzATP-induced YO-PRO-1 uptake by variable monocytes (vs. the saline control) was similar across all three platforms (ANOVA, p = 0.78), the raw values for BzATP-induced YO-PRO-1 fluorescence obtained on the LSR II were 9.0 ± 0.8 times higher than the average values measured on the FACScan and the FACSCalibur. Collectively, these data indicate that the bead-adjusted setup method reduces systematic variability associated with raw fluorescence measurements and can be used to obtain comparable results between two analogue and one digital instruments.
Figure 4.
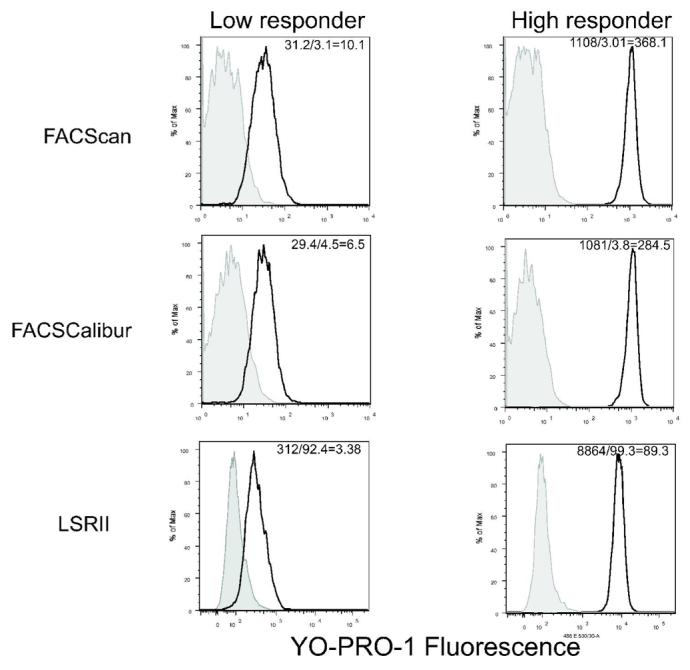
Comparative histograms of the YO-PRO-1 fluorescence of monocytes stimulated with BzATP in two donors: one low responder and one high responder. Data was acquired on the same sample on three different flow cytometers: FACScan, FACSCalibur, and the LSRII. The instruments were set using two sets of target values as described. The FACScan and FACSCalibur used the same values while the LSRII used a different set of values. Median intensitites of live monocytes are given for each instrument, with the values for the BzATP stimulated and the unstimulated samples as well as the MFI ratio presented in the insets.
Assay Compatibility with Aged Samples in an Ongoing Multi-Center Asthma Clinical Trial
The evaluation of P2X7 pore function as a predictor of symptoms and other clinical endpoints in the Asthma Clinical Research Network Macrolides In Asthma trial involves overnight shipping of room-temperature blood samples to our facility. To begin assessing the impact of declining monocyte viability (Figure 1A) on the performance of our revised assay, time course experiments with samples from laboratory volunteers were performed with serial processing on multiple days after sample collection (Figure 5). Samples from four of the eight subjects appear to have a progressive rise in BzATP-stimulated YO-PRO-1 fluorescence, whereas the other four specimens do not have an appreciable change. Analysis of variance suggests there is more variability between subjects in this regard than there is with respect to time (p = 0.814). When the data are expressed as a percent of each subject's Day 0 measurement, there may be a trend towards rising values that could reach significance if more subjects were studied. Specifically, Day 1 is 108.2 ± 34.4%, Day 2 is 126.5 ± 55.1%, Day 3 is 156.6 ± 43.3% and Day 4 is 162.4 ±56.5% of the values obtained on Day 0 (ANOVA p = 0.104). Due to overnight shipping to our center of samples obtained from a multi-center clinical trial, the median sample age is 1 day with an inter-quartile range of 1 to 2 days. Table 2 shows descriptive statistics from clinical samples using our past (fixed settings, total CD14pos cells) and current (bead-adjusted, nonviable-excluded) methods from a local and multi-center asthma protocol, respectively. The distributions of these datasets from reasonably similar patient populations appear unchanged (Table 2, p = 0.846), suggesting that the revised method has not influenced the data while improving intra-laboratory reproducibility. Thus the exclusion of non-viable monocytes with our bead-adjusted setup method can accommodate samples of varying age and allows for the development of an age-related confidence interval around the measurement, while not appreciably altering the data distribution using our prior method at a single center.
Figure 5.

Median fluorescence intensities of YO-PRO-1 in live monocytes stimulated with BzATP. Blood was drawn from 8 different donors and aliquots of each blood was assayed daily for 5 days. All data was acquired on a FACSCalibur flow cytometer using bead-adjusted settings.
DISCUSSION
Flow cytometric analysis of P2X7 receptor pore activity in human blood monocytes segregates variant from common P2X7 receptor genotypes/phenotypes and may serve as a biomarker for infectious/inflammatory diseases and disorders. To facilitate association studies, our laboratory developed a genomically validated functional assay capable of bridging disparate P2RX7 genetic, phenotypic and clinical results, while also increasing statistical power in the face of sample size constraints (10,14). The present study set out to adapt bead files from our prior recalled settings to standardize flow cytometer instrument set up across platforms and further minimize intra-laboratory variability. Despite daily cleaning and fluorescent particle calibration of the cytometers in our facility, the bead-adjusted method was associated with less variability than measurements collected with recalled settings (Table 1). This may be due to fluctuations in cytometer performance as the laser ages, which are better accommodated by an assay specific PMT-adjustment method. Additionally, the reduced variability was more pronounced when the data are presented as the median YO-PRO-1 fluorescence associated with the samples stimulated with BzATP (Table 1). For analog instruments, which amplify fluorescence measurements logarithmically, ratio determinations such as our previously defined fold-uptake become susceptible to the variability associated with spectral overlap calculations. In this regard, the bead adjusted method we present offers another advantage in that the median YO-PRO-1 fluorescence in BzATP-stimulated samples is relatively unaffected by compensation strategy (Supplementary Table 1). Hence, the day-to-day variability is quite low (Figure 3) and likely, at least in part, reflects biological fluctuations in systemic cytokine levels that influence P2RX7 expression. This latter observation has also been made by others using PBMCs and a flow cytometry protocol that does not include daily standards for PMT voltages or compensation calculations (36). Collectively, the present bead-adjusted set up method is relatively simple, minimizes intra-laboratory variability, and facilitates future inter-laboratory comparisons.
Because declining monocyte viability in aged samples influences assessment of P2X7 pore function (Figure 1), we needed to revise our assay to exclude non-viable cells. Specifically, non-viable cells within the total monocyte population affect the measurement of true P2X7 pore function due to unregulated YO-PRO-1 uptake associated with loss of plasma membrane integrity influencing the basal rate as well as attenuated responsiveness to BzATP-stimulation. Closure of the P2X7 pore by divalent cations (17) allows inclusion of a second vital dye to assess plasma membrane permeability. In this regard, eliminating PIpos events accommodates samples aged up to 4 days after phlebotomy (Figure 5). Therefore, exclusion of non-viable cells in the analysis of P2X7 pore activity in monocytes is warranted for all future studies in order to measure true P2X7 pore function.
An additional feature of our revised method is that it facilitates comparisons between different types of cytometers. We found that our recalled settings from the FACScan were not transferable to another analog cytometer (not shown). In contrast, the bead adjusted set up method produced median YO-PRO-1 fluorescence measurements in BzATP-treated samples that differed by only 2.0 ± 1.5 percent (also see Figure 4) when processed simultaneously on FACScan and FACSCalibur cytometers. The digital LSR II produced higher numerical fluorescence values than the analog instruments but the results were visually comparable in terms of the decades relative to the scale for the individual machine (Figure 4). PMT targets established by this method require that they fall within the dynamic range for the instrument used. Thus, inclusion of other instruments and manufacturers for use in this assay will require validation of the PMT targets, however, the methods we describe in the present study lay the ground-work for maximal inter-laboratory consistency.
We are currently using this assay in an ongoing multi-center clinical trial within the Asthma Clinical Research Network Trial-Macrolides in Asthma (ACRN-MIA; see http://www.clinicaltrials.gov, identifier NCT00318708) to test whether a continuous measure of P2X7 pore function is associated with clinical symptoms or endpoints in this population. Although P2RX7 has not previously been referred to as an asthma gene, its chromosomal location is thought to contain numerous such candidates based on linkage in multiple populations to measurements of lung function (37-40). Additionally, P2RX7 is thought to control the immune response to infection with Chlamydia species (41,42), an intracellular pathogen thought to contribute to asthma pathogenesis which is also one of the therapeutic targets for the ACRN-MIA clinical trial (43). It also gives us another opportunity to validate the assay at the genetic level. Although our prior method was sufficient for identifying samples with validated and novel loss of function alleles with an area under the curve of 0.927 (14), the presently described correction for sample age and the exclusion of nonviable cells will require refinement of the receiver-operator curve analysis using results obtained by our present method in order to accurately segregate variant from common P2X7 receptor genotypes/phenotypes in future studies. In general, this approach of genomically validating results from the P2X7 pore assay could serve as an additional control as other laboratories begin to adopt this method.
In conclusion, this study provides a standardized method for quantitative detection of P2X7 pore function by monocytes in human whole blood examined within 4 days post-phlebotomy that will likely facilitate future cohort studies by minimizing intra- and inter-laboratory variation in sample acquisition. Because individuals with reduced capacity for P2X7 pore formation are suggested to be predisposed to an anti-inflammatory cytokine profile in the setting of immune system perturbation (10), utilization of our novel standardized calibration method will facilitate the segregation of variant from common P2X7 receptor genotypes/phenotypes and potentially identify variation in P2X7 receptor pore function as a biomarker for infectious/inflammatory diseases and disorders.
Supplementary Material
ACKNOWLEGEMENTS
This work was supported by the National Institutes of Health grants NCRR K12 RR01761401, AADC PO1 AI5050001 and NHLBI U10 HL074212. The principal investigators for the Natural Cold in Asthma study at the University of Wisconsin are Drs. William W. Busse and James E. Gern. The Asthma Clinical Research Network-Macrolides In Asthma clinical trial principal investigators are Drs. Richard Martin and E. Rand Sutherland (National Jewish Respiratory Center, Denver), Dr. Robert F. Lemanske, Jr. (University of Wisconsin, Madison), Dr. Elliot Israel (Brigham & Womens' Hospital, Boston), Dr. Homer Boushey (University of California, San Francisco), Dr. Vern Chinchilli (Pennsylvania State University, Hershey), Dr. Stephen Peters (Wake Forest University, Winston-Salem), Dr. Mario Castro (Washington University, St. Louis), Dr. Stephen Wasserman (University of California, San Diego), Dr. William Calhoun (University of Texas, Galveston), Dr. Emily DiMango (Columbia University, New York) and Dr. Monica Kraft (Duke University, Durham). This work would not be possible without the concerted effort of the dozens of Clinical Study Coordinators in Madison and throughout the ACRN, as well as the personnel of the ACRN Data Coordinating Center.
REFERENCES
- 1.Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442(7102):527–32. doi: 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- 2.Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997;272(9):5482–6. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- 3.Buell GN, Talabot F, Gos A, Lorenz J, Lai E, Morris MA, Antonarakis SE. Gene structure and chromosomal localization of the human P2X7 receptor. Receptors Channels. 1998;5(6):347–54. [PubMed] [Google Scholar]
- 4.Di Virgilio F, Chiozzi P, Ferrari D, Falzoni S, Sanz JM, Morelli A, Torboli M, Bolognesi G, Baricordi OR. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood. 2001;97(3):587–600. doi: 10.1182/blood.v97.3.587. [DOI] [PubMed] [Google Scholar]
- 5.Gu BJ, Zhang WY, Bendall LJ, Chessell IP, Buell GN, Wiley JS. Expression of P2X(7) purinoceptors on human lymphocytes and monocytes: evidence for nonfunctional P2X(7) receptors. Am J Physiol Cell Physiol. 2000;279(4):C1189–97. doi: 10.1152/ajpcell.2000.279.4.C1189. [DOI] [PubMed] [Google Scholar]
- 6.Mehta VB, Hart J, Wewers MD. ATP-stimulated release of interleukin (IL)-1beta and IL-18 requires priming by lipopolysaccharide and is independent of caspase-1 cleavage. J Biol Chem. 2001;276(6):3820–6. doi: 10.1074/jbc.M006814200. [DOI] [PubMed] [Google Scholar]
- 7.Tonetti M, Sturla L, Giovine M, Benatti U, De Flora A. Extracellular ATP enhances mRNA levels of nitric oxide synthase and TNF-alpha in lipopolysaccharide-treated RAW 264.7 murine macrophages. Biochem Biophys Res Commun. 1995;214(1):125–30. doi: 10.1006/bbrc.1995.2265. [DOI] [PubMed] [Google Scholar]
- 8.Perregaux DG, McNiff P, Laliberte R, Conklyn M, Gabel CA. ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J Immunol. 2000;165(8):4615–23. doi: 10.4049/jimmunol.165.8.4615. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Fisette PL, Denlinger LC, Guadarrama AG, Sommer JA, Proctor RA, Bertics PJ. Purinergic receptor modulation of lipopolysaccharide signaling and inducible nitric-oxide synthase expression in RAW 264.7 macrophages. J Biol Chem. 1998;273(42):27170–5. doi: 10.1074/jbc.273.42.27170. [DOI] [PubMed] [Google Scholar]
- 10.Denlinger LC, Angelini G, Schell K, Green DN, Guadarrama AG, Prabhu U, Coursin DB, Bertics PJ, Hogan K. Detection of human P2X7 nucleotide receptor polymorphisms by a novel monocyte pore assay predictive of alterations in lipopolysaccharide-induced cytokine production. J Immunol. 2005;174(7):4424–31. doi: 10.4049/jimmunol.174.7.4424. [DOI] [PubMed] [Google Scholar]
- 11.Li CM, Campbell SJ, Kumararatne DS, Bellamy R, Ruwende C, McAdam KP, Hill AV, Lammas DA. Association of a polymorphism in the P2X7 gene with tuberculosis in a Gambian population. J Infect Dis. 2002;186(10):1458–62. doi: 10.1086/344351. [DOI] [PubMed] [Google Scholar]
- 12.Fernando SL, Saunders BM, Sluyter R, Skarratt KK, Goldberg H, Marks GB, Wiley JS, Britton WJ. A polymorphism in the P2X7 gene increases susceptibility to extrapulmonary tuberculosis. American Journal of Respiratory and Critical Care Medicine. 2006;175:360–366. doi: 10.1164/rccm.200607-970OC. [DOI] [PubMed] [Google Scholar]
- 13.Nino-Moreno P, Portales-Perez D, Hernandez-Castro B, Portales-Cervantes L, Flores-Meraz V, Baranda L, Gomez-Gomez A, Acuna-Alonzo V, Granados J, Gonzalez-Amaro R. P2X7 and NRAMP1/SLC11 A1 gene polymorphisms in Mexican mestizo patients with pulmonary tuberculosis. Clin Exp Immunol. 2007;148(3):469–77. doi: 10.1111/j.1365-2249.2007.03359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denlinger LC, Coursin DB, Schell K, Angelini G, Green DN, Guadarrama AG, Halsey J, Prabhu U, Hogan KJ, Bertics PJ. Human P2X7 pore function predicts allele linkage disequilibrium. Clin Chem. 2006;52(6):995–1004. doi: 10.1373/clinchem.2005.065425. [DOI] [PubMed] [Google Scholar]
- 15.Gordon D, Finch SJ. Factors affecting statistical power in the detection of genetic association. J Clin Invest. 2005;115(6):1408–18. doi: 10.1172/JCI24756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–83. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 17.Gudipaty L, Humphreys BD, Buell G, Dubyak GR. Regulation of P2X(7) nucleotide receptor function in human monocytes by extracellular ions and receptor density. Am J Physiol Cell Physiol. 2001;280(4):C943–53. doi: 10.1152/ajpcell.2001.280.4.C943. [DOI] [PubMed] [Google Scholar]
- 18.Akbari O, Faul JL, Hoyte EG, Berry GJ, Wahlstrom J, Kronenberg M, DeKruyff RH, Umetsu DT. CD4+ invariant T-cell-receptor+ natural killer T cells in bronchial asthma. N Engl J Med. 2006;354(11):1117–29. doi: 10.1056/NEJMoa053614. [DOI] [PubMed] [Google Scholar]
- 19.Vijayanand P, Seumois G, Pickard C, Powell RM, Angco G, Sammut D, Gadola SD, Friedmann PS, Djukanovic R. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med. 2007;356(14):1410–22. doi: 10.1056/NEJMoa064691. [DOI] [PubMed] [Google Scholar]
- 20.Bayer J, Grunwald D, Lambert C, Mayol JF, Maynadie M. Thematic workshop on fluorescence compensation settings in multicolor flow cytometry. Cytometry B Clin Cytom. 2007;72(1):8–13. doi: 10.1002/cyto.b.20153. [DOI] [PubMed] [Google Scholar]
- 21.Roederer M. Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry. 2001;45(3):194–205. doi: 10.1002/1097-0320(20011101)45:3<194::aid-cyto1163>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz A, Fernandez Repollet E, Vogt R, Gratama JW. Standardizing flow cytometry: construction of a standardized fluorescence calibration plot using matching spectral calibrators. Cytometry. 1996;26(1):22–31. doi: 10.1002/(SICI)1097-0320(19960315)26:1<22::AID-CYTO4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz A, Marti GE, Poon R, Gratama JW, Fernandez-Repollet E. Standardizing flow cytometry: a classification system of fluorescence standards used for flow cytometry. Cytometry. 1998;33(2):106–14. [PubMed] [Google Scholar]
- 24.Waxdal MJ, Monical MC, Palini AG. Inter-laboratory relative fluorescence intensity measurements using FlowCal 575 calibration beads: a baseline study. Cytometry. 1998;33(2):213–8. [PubMed] [Google Scholar]
- 25.Brown RD, Zarbo RJ, Linden MD, Torres FX, Nakhleh RE, Schultz D, Mackowiak PG. Two-color multiparametric method for flow cytometric DNA analysis. Standardization of spectral compensation. Am J Clin Pathol. 1994;101(5):630–7. doi: 10.1093/ajcp/101.5.630. [DOI] [PubMed] [Google Scholar]
- 26.Zhang YZ, Kemper C, Bakke A, Haugland RP. Novel flow cytometry compensation standards: internally stained fluorescent microspheres with matched emission spectra and long-term stability. Cytometry. 1998;33(2):244–8. doi: 10.1002/(sici)1097-0320(19981001)33:2<244::aid-cyto20>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 27.Fluorescence Calibration and Quantitative Measurement of Fluorescence Intensity; Approved Guideline Clinical and Laboratory Standards Institute. Report nr I/LA24-A. 24(26) [Google Scholar]
- 28.Schwartz A, Gaigalas AK, Wang L, Marti GE, Vogt RF, Fernandez-Repollet E. Formalization of the MESF unit of fluorescence intensity. Cytometry B Clin Cytom. 2004;57(1):1–6. doi: 10.1002/cyto.b.10066. [DOI] [PubMed] [Google Scholar]
- 29.Davis BH, Bigelow NC, Curnutte JT, Ornvold K. Neutrophil high affinit Fc receptor (CD 64) is a marker of in vivo activation and interferongamma therapy. Lab Hematol. 1995;1:3–12. [Google Scholar]
- 30.Denlinger LC, Schell K, Angelini G, Green D, Guadarrama A, Prabhu U, Coursin DB, Hogan K, Bertics PJ. A novel assay to detect nucleotide receptor P2X7 genetic polymorphisms influencing numerous innate immune functions. J Endotoxin Res. 2004;10(2):137–42. doi: 10.1179/096805104225004040. [DOI] [PubMed] [Google Scholar]
- 31.Nicholson JKA, Jones BM, Hubbard M. CD4 T-lymphocyte determinations on whole blood specimens using a single-tube three-color assay. Cytometry. 1993;14:685–89. doi: 10.1002/cyto.990140614. [DOI] [PubMed] [Google Scholar]
- 32.Hubl W, Iturraspe J, Martinez GA, Hutcheson CE, Roberts CG, Fisk DD, Sugrue MW, Wingard JR, Braylan RC. Measurement of absolute concentration and viability of CD34+ cells in cord blood and cord blood products using fluorescent beads and cyanine nucleic acid dyes. Cytometry. 1998;34(3):121–7. doi: 10.1002/(sici)1097-0320(19980615)34:3<121::aid-cyto2>3.3.co;2-u. [DOI] [PubMed] [Google Scholar]
- 33.Humphreys BD, Dubyak GR. Modulation of P2X7 nucleotide receptor expression by pro- and anti-inflammatory stimuli in THP-1 monocytes. J Leukoc Biol. 1998;64(2):265–73. doi: 10.1002/jlb.64.2.265. [DOI] [PubMed] [Google Scholar]
- 34.Zenger VE, Vogt R, Mandy F, Schwartz A, Marti GE. Quantitative flow cytometry: inter-laboratory variation. Cytometry. 1998;33(2):138–45. doi: 10.1002/(sici)1097-0320(19981001)33:2<138::aid-cyto8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 35.Levering WH, Preijers FW, van Wieringen WN, Kraan J, van Beers WA, Sintnicolaas K, van Rhenen DJ, Gratama JW. Flow cytometric CD34+ stem cell enumeration: lessons from nine years' external quality assessment within the Benelux countries. Cytometry B Clin Cytom. 2007;72(3):178–88. doi: 10.1002/cyto.b.20351. [DOI] [PubMed] [Google Scholar]
- 36.Jursik C, Sluyter R, Georgiou JG, Fuller SJ, Wiley JS, Gu BJ. A quantitative method for routine measurement of cell surface P2X7 receptor function in leucocyte subsets by two-colour time-resolved flow cytometry. J Immunol Meth. 2007;325:67–77. doi: 10.1016/j.jim.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Xu J, Postma DS, Howard TD, Koppelman GH, Zheng SL, Stine OC, Bleecker ER, Meyers DA. Major genes regulating total serum immunoglobulin E levels in families with asthma. Am J Hum Genet. 2000;67(5):1163–73. doi: 10.1086/321190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malerba G, Lauciello MC, Scherpbier T, Trabetti E, Galavotti R, Cusin V, Pescollderungg L, Zanoni G, Martinati LC, Boner AL. Linkage analysis of chromosome 12 markers in Italian families with atopic asthmatic children. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1587–90. doi: 10.1164/ajrccm.162.4.9909031. others. [DOI] [PubMed] [Google Scholar]
- 39.Raby BA, Silverman EK, Lazarus R, Lange C, Kwiatkowski DJ, Weiss ST. Chromosome 12q harbors multiple genetic loci related to asthma and asthma-related phenotypes. Hum Mol Genet. 2003;12(16):1973–9. doi: 10.1093/hmg/ddg208. [DOI] [PubMed] [Google Scholar]
- 40.Celedon JC, Soto-Quiros ME, Avila L, Lake SL, Liang C, Fournier E, Spesny M, Hersh CP, Sylvia JS, Hudson TJ. Significant linkage to airway responsiveness on chromosome 12q24 in families of children with asthma in Costa Rica. Hum Genet. 2007;120(5):691–699. doi: 10.1007/s00439-006-0255-5. others. [DOI] [PubMed] [Google Scholar]
- 41.Coutinho-Silva R, Perfettini JL, Persechini PM, Dautry-Varsat A, Ojcius DM. Modulation of P2Z/P2X(7) receptor activity in macrophages infected with Chlamydia psittaci. Am J Physiol Cell Physiol. 2001;280(1):C81–9. doi: 10.1152/ajpcell.2001.280.1.C81. [DOI] [PubMed] [Google Scholar]
- 42.Coutinho-Silva R, Stahl L, Raymond MN, Jungas T, Verbeke P, Burnstock G, Darville T, Ojcius DM. Inhibition of chlamydial infectious activity due to P2X7R-dependent phospholipase D activation. Immunity. 2003;19(3):403–12. doi: 10.1016/s1074-7613(03)00235-8. [DOI] [PubMed] [Google Scholar]
- 43.Johnston SL, Martin RJ. Chlamydophila pneumoniae and Mycoplasma pneumoniae: A Role in Asthma Pathogenesis? Am J Respir Crit Care Med. 2005;172(9):1078–89. doi: 10.1164/rccm.200412-1743PP. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.