

Abstract
Until recently, denitrification was thought to be the only significant pathway for N2 formation and, in turn, the removal of nitrogen in aquatic sediments. The discovery of anaerobic ammonium oxidation in the laboratory suggested that alternative metabolisms might be present in the environment. By using a combination of 15N-labeled NH4+, NO3−, and NO2− (and 14N analogues), production of 29N2 and 30N2 was measured in anaerobic sediment slurries from six sites along the Thames estuary. The production of 29N2 in the presence of 15NH4+ and either 14NO3− or 14NO2− confirmed the presence of anaerobic ammonium oxidation, with the stoichiometry of the reaction indicating that the oxidation was coupled to the reduction of NO2−. Anaerobic ammonium oxidation proceeded at equal rates via either the direct reduction of NO2− or indirect reduction, following the initial reduction of NO3−. Whether NO2− was directly present at 800 μM or it accumulated at 3 to 20 μM (from the reduction of NO3−), the rate of 29N2 formation was not affected, which suggested that anaerobic ammonium oxidation was saturated at low concentrations of NO2−. We observed a shift in the significance of anaerobic ammonium oxidation to N2 formation relative to denitrification, from 8% near the head of the estuary to less than 1% at the coast. The relative importance of anaerobic ammonium oxidation was positively correlated (P < 0.05) with sediment organic content. This report of anaerobic ammonium oxidation in organically enriched estuarine sediments, though in contrast to a recent report on continental shelf sediments, confirms the presence of this novel metabolism in another aquatic sediment system.
Since the 1970s, substantial research has focused on the ability of estuarine sediments to attenuate riverine nitrogen (N) loads before they affect coastal seas (4, 18, 19, 31, 33). Estuarine sediments are essentially anaerobic below a few surface millimeters, and the mineralization of organic matter proceeds via alternate electron acceptors such as NO3− and SO42− (20, 27). In turn, the reduction of NO3− removes NO3− from the overlying waters. Until recently, it was largely thought that NO3− could be either reduced to N2 gas via denitrification (a facultative metabolism mediated by a variety of bacteria) and lost from the system or reduced to ammonium (NH4+) by fermentative metabolisms and hence conserved within the sediments (dissimilatory nitrate reduction to ammonium [DNRA]) (8, 23). It had been demonstrated that changes in sediment organic loadings and estuarine NO3− concentrations may affect the partitioning between these two end products of NO3− reduction (11, 12). The discovery within the laboratory (17) of anaerobic ammonium oxidation revealed a novel metabolism that could short circuit the N cycle, bypassing what was previously thought to be a critical aerobic nitrification phase and potentially providing an alternative pathway for N2 gas formation in the environment (Fig. 1).
FIG. 1.

Simplified N cycle showing how anaerobic ammonium oxidation (Anammox) (broken arrows) bypasses the classic coupling of aerobic nitrification to denitrification, with the oxidation of NH4+ and N2 formation being driven by the reduction of NO2−. Note the process of DNRA and that NO2− is an intermediate of both DNRA and nitrification.
Originally, it was thought (17) that anaerobic ammonium oxidation coupled the oxidation of NH4+ to the reduction of NO3−:
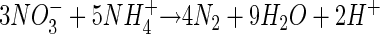 |
(1) |
Further work, however, showed that the oxidation of ammonium was actually coupled to the reduction of nitrite rather than nitrate (34, 35):
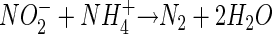 |
(2) |
The application of this process to the treatment of nitrogenous waste has received a great deal of attention (9, 10), and more recently, the organism responsible has been classified as a new autotrophic planctomycete (28).
Anaerobic ammonium oxidation was recently reported to account for as much as 24 and 60% of N2 formation in continental shelf sediments in relatively deep water (380 and 695 m, respectively) but less than 2% of N2 formation in eutrophic shallow coastal bay sediments (30). The drop in the significance of anaerobic ammonium oxidation for N2 formation relative to denitrification was attributed to changes in organic matter availability and hence sediment reactivity. If this trend continues inshore, where organic content and reactivity further increase, anaerobic ammonium oxidation might be assumed to be insignificant in organically enriched estuarine sediments (33).
The purpose of this research was to look for direct evidence of environmental anaerobic ammonium oxidation in estuarine sediments. Initial trials were designed to simply assay sediment at one site in the Thames estuary. Having established that anaerobic ammonium oxidation was detectable in estuarine sediments, the trials were extended downstream to reflect the changing sediment characteristics of both organic content and reactivity.
MATERIALS AND METHODS
Sampling sites.
The Thames estuary is a major macrotidal estuary on the southeastern coast of England which flows into the North Sea at Southend-on-Sea (Fig. 2). Oxygen saturation in the Thames estuary generally drops to 20% in the summer at site 1 and does not return to full saturation until some 50 km seaward at Southend-on-Sea (33). In contrast, the waters are highly enriched with NO3− and NO2−, and their concentrations are on average for the year ∼550 and 9 μM, respectively, at 0 salinity. At the outset of this study, no other studies of anaerobic ammonium oxidation in the environment had been carried out, and until techniques were proven to be reliable, experiments were restricted to the most-NO3−-enriched and -oxygen-depleted sediments near the major sewage discharge point at Crossness (site 1). Sediments from along the estuary were collected from sites close to those described in detail earlier (33). At sites 1 and 2, the sediments have a high organic and silt-clay content, both of which then decrease seaward along the estuary from sites 3 to 6 (Table 1 and see below).
FIG. 2.

Map of the Thames estuary on the southeastern coast of England showing locations of the six sample sites (filled circles) and the two major STWs (open circles).
TABLE 1.
Sediment characteristics and representative salinity of the overlying water at the six sites in the Thames estuary
| Site | Particle sizea (0-5 cm [≥50%, wt/wt]) | Organic carbon (% of dry wt) | Porosity | Specific gravity (g cm−3) | Salinity (%) |
|---|---|---|---|---|---|
| 1 Crossness | Silts and clays | 2.91 | 0.76 | 1.21 | 2 |
| 2 Grays | Silts and clays | 3.59 | 0.76 | 1.24 | 10 |
| 3 Gravesend | Very fine sands | 2.70 | 0.67 | 1.45 | 10 |
| 4 Allhallows | Very fine sands | 1.18 | 0.65 | 1.46 | 26 |
| 5 Southend | Fine sands | 0.45 | 0.50 | 1.68 | 30 |
| 6 Grain | Fine sands | 0.26 | 0.47 | 1.74 | 30 |
For detailed descriptions, see reference 33.
Sediment preparation.
Sediment samples (oxic and suboxic layers, 0 to 2 cm) were collected from intertidal flats along the Thames estuary at low tide (Fig. 2), stored in plastic bags, and returned to the laboratory within 1 h. Sediment slurries (30 ml; 50%, vol/vol) were prepared in serum bottles (37 ml) with low-nutrient seawater, adjusted to in situ salinity with distilled water, sealed, degassed (oxygen-free nitrogen, 20 min), and preincubated on rollers in the dark at 15°C for 6 h in a constant-temperature room. All subsequent experiments were carried out in a constant-temperature room at 15°C. Anoxia was checked by sampling the headspace in each serum bottle and measuring the O2 with a gas chromatograph fitted with an electron capture detector. Preliminary trials showed that ambient NO3− and NO2− were reduced to less than 1 μM during the preincubations.
Measuring anaerobic ammonium oxidation.
Slurries from site 1 were enriched to approximately 10% above the ambient level with concentrated stocks of labeled 15NH4+ (18 mM 15NH4Cl [99.3 15N atom%]; Sigma-Aldrich, Poole, United Kingdom) and either 14NO2− or 14NO3− (or both) at either 800 μM or a range up to 3,200 μM and incubated as described above. Some additional trials were carried out with autoclaved sediment in serum bottles to confirm that the N transformations were biologically mediated. Initial trials at 200 μM NO2− or NO3− had demonstrated a very rapid turnover (1 to 2 h) in these sediments, and concentrations were increased to enable production of 29N2 to be measured with time. Under anaerobic conditions and the above-described isotopic labeling, any anaerobic ammonium oxidation coupled to the reduction of NO2− or NO3− would yield 15N-labeled gas according to the following formulas:
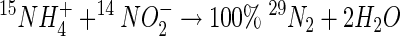 |
(3) |
 |
(4) |
The sole production of 29N2 would confirm anaerobic ammonium oxidation coupled to the reduction of NO2− rather than NO3− in these sediments. Assuming that the pool of 14NH4+ turned over at the same rate as 15NH4+, the total rate of anaerobic ammonium oxidation could then be calculated with the equation (29)
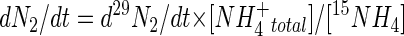 |
(5) |
(where dN2 is total N2 production by anaerobic ammonium oxidation per unit of time [dt], d29N2 is the measurement of 29N2 per unit of time, NH4+total is the total soluble NH4+ pool, and [15NH4+] is the concentration of 15NH4+ determined by difference from the nonenriched reference samples) and expressed as a proportion of the total NO2− or NO3− reduction in the slurries. Separate slurries were enriched with their respective analogues, e.g., 14NH4+, 15NO2−, and 15NO3− (>99% of the ambient level, 93 mM stock Na15NO3−/NO2− [99.2 15N atom%]; Sigma-Aldrich), which gives a measure of total 15N gas production (29N2 and 30N2) from either 15NO2− or 15NO3−. With 15NO2− or 15NO3−, the production of 29N2 can be due to both anaerobic ammonium oxidation (A29) and denitrification (D29) (30). Essentially, the proportion due to each respective process can be calculated from the equation A29 = P29 − D29, where A29 equals the production of 29N2 gas due to anaerobic ammonium oxidation, D29 equals the production of 29N2 gas due to denitrification, and P29 equals the total production of 29N2 measured by isotope ratio mass spectrometry (see below). In contrast, the production of 30N2 (P30) is assumed to be due solely to denitrification of 15NO2−/NO3−; hence, P30 equals D30 and, from this, D29 can be calculated, assuming random pairing of 14N and 15N from the labeled NO2− or NO3− pools, which, with only trace amounts of 14NO2−/NO3− and enrichment with 99.2% atom 15NO2−/NO3−, would usually be less than 1.5% (6). Overall, the calculations (30) give an estimate of total anaerobic ammonium oxidation and denitrification in the sediment slurries. These estimates can then be compared to 29N2 production in the presence of 15NH4+ after taking into account the proportionate labeling of the 14N- and 15N-NH4+ pools. Trials were extended along the estuary by using labeling with 15NO3−, as this gives a measure of total denitrification and anaerobic ammonium oxidation in the same serum bottle.
During the incubations, independent slurries were sacrificed with time, and a gas sample (4 ml) was collected from the slightly pressurized headspace with a gastight syringe (SGE gastight Luer lock syringe; Alltech Associates Ltd., Carnforth, Lancashire, United Kingdom), with allowance for equilibration to atmospheric pressure, and transferred through a septum to an inverted water-filled vial (12-ml Exetainer vial; Labco Ltd., High Wycombe, United Kingdom), with venting of water to atmosphere through a small-bore needle. Microbial activity was then inhibited by injecting ZnCl2 (250 μl; 50%, wt/vol) through the septum. Pore waters were recovered by centrifugation of the slurry, filtered (0.2-μm-pore-size Minisart Plus filter; Sartorius UK Ltd.), and frozen (−20°C) until analysis. Exchangeable ammonium was recovered by using a KCl extraction technique, which recovered >95% of adsorbed NH4+ by a double extraction with 2 M KCl (16).
Analytical procedures.
All nutrient analyses (NO3−, NO2−, and NH4+) were carried out with a continuous-flow autoanalyzer (SAN++; Skalar, De-Breda, The Netherlands) and standard colorimetric techniques (13). Salinity was measured with a handheld refractometer. Water content, specific gravity, and porosity were determined from the dry weights and wet weights of known volumes of sediment. Acidified dried sediment samples (with allowance for hydroscopic adsorption [see reference 7]), were analyzed for organic carbon with an elemental analyzer coupled to a continuous-flow isotope ratio mass spectrometer calibrated with known quantities of urea (Delta Matt Plus; Thermo Finnigan, Bremen, Germany).
Mass/charge ratios for m/z 28, m/z 29, and m/z 30 nitrogen (28N2, 29N2, and 30N2) were measured by the Natural Environment Research Council (NERC) 15N stable isotope facility (Centre for Ecology and Hydrology-Merlewood, Cumbria, United Kingdom). The headspace of each respective vial was sampled (20 μl) with a gastight precision syringe and directly injected into a N2 preparative interface coupled to an Isoprime isotope ratio mass spectrometer (Micromass U.K., Wythenshawe, United Kingdom) via an open split. Instrument stability checks were performed prior to each analysis by running a series of 10 reference pulses of N2 until a standard deviation of better than 10−6 was achieved.
RESULTS
Anaerobic ammonium oxidation at site 1.
Production of 29N2 from the oxidation of 15NH4+ occurred at equal rates in the presence of either 14NO3−, 14NO2−, or both (Fig. 3) but could not be measured in control slurries enriched with only 15NH4+ (data not shown). Production of 29N2 was linear with time until the pools of either NO2− or NO3− were depleted and, hence, was independent of the decreasing concentration of either acceptor. Measurable production of 29N2 ceased after 8 h, when either electron acceptor had been depleted, reconfirming the production measured in the controls. When the slurries were enriched with NO3−, little, if any, transient accumulation of NO2− could be detected as the NO3− diminished. When spiked together with 14NO2− and 14NO3−, production of 29N2 occurred at the same rate as in the independent trials, but NO3− was consumed preferentially to NO2− by the NO3−-reducing community (Fig. 3c). In addition, the production of 29N2 continued to the end of the trial (12 h), being sustained by the NO2− pool, which remained largely untouched until after 8 h, when the concentration of NO3− had been reduced to approximately 200 μM. The rates of turnover were consistently equal for NO3− and NO2−, being equivalent to 0.11 and 0.12 μmol ml of wet sediment−1 h−1, respectively, at a starting concentration of 800 μM. Anaerobic ammonium oxidation produced 0.16 and 0.15 nmol of 29N2 ml of wet sediment−1 h−1 for NO3− and NO2−, respectively. With 10.3% labeling of the NH4+ pool, this gives an average total anaerobic ammonium oxidation for both acceptors of 1.5 nmol of N2 ml of wet sediment−1 h−1, equivalent to 1.3% of the total NO3− or NO2− reduction.
FIG. 3.

Production of 29N2 from labeled 15NH4+ oxidation in the direct presence of 14NO3− (a), 14NO2− (b), or both (c). Values are means ± 1 standard error of the mean (SEM) (n = 4). The same symbols are used for each panel. Data were collected July 2002.
The sediments at site 1 reached an average maximum NO3− or NO2− reduction rate of 383 μM h−1 at 3,200 μM (Fig. 4a). Because the pool of either substrate from 200 to 800 μM was completely turned over during the incubation, the data presented in Fig. 4 are for the whole 4-h incubation period, and rates are estimated from data points above 800 μM. The yield of 29N2 from anaerobic ammonium oxidation increased with the turnover of substrate, up to a maximum of 0.8 or 1 nmol of 29N2 ml of wet sediment−1 for approximately 1.5 μmol of NO3− or NO2− ml of wet sediment−1 (Fig. 4b). Though NO2− accumulated in the NO3−-enriched slurries at a constant 20 μM between 800 and 3,200 μM NO3−, this had no effect on the yield of 29N2. Although total NO3− or NO2− reduction increased above 1.5 μmol ml of wet sediment−1, production of 29N2 did not change significantly, and in turn, the yield of 29N2 as a proportion of total NO3− or NO2− reduction decreased (Fig. 4b and c).
FIG. 4.

(a) Total rate of turnover for either NO3− or NO2− as a function of the initial enrichment concentration; (b) production of 29N2 from labeled 15NH4+ oxidation; (c) ratio of 29N2 per mole of NO3− or NO2− reduced as a function of total NO3− or NO2− reduction. Values are means ± 1 SEM (four replicates were performed). The same symbols were used for each panel. Data were collected August 2002.
The addition of ∼9 μmol of 15NH4+ to the slurries increased the soluble NH4+ concentration at site 1 by 150 μM, i.e., 10% (or half that predicted), which indicated that half of the label was exchanged and bound to the sediments. Throughout all the slurry experiments, the total NH4+ pool remained essentially constant, suggesting that NH4+ oxidation (and/or assimilation) was balanced by ammonification (data not shown). The 15NH4+ pool was diluted during these incubations, but as 29N2 production remained linear, this dilution was not significant and did not affect our measure of anaerobic ammonium oxidation. There was no significant production of 30N2 in the 15NH4+ incubations and, in addition, no 15N gases were produced in slurries that had previously been autoclaved, confirming the theory that anaerobic ammonium oxidation is a biological process.
Anaerobic ammonium oxidation along the estuary.
Slurries spiked with 14NH4+ and 15NO3− produced >90% 30N2 (Table 2). With >98 atom% 15N labeling of the NO3− pool, the predicted proportion of 29N2 was <2%; but under these conditions, 29N2 contributed up to 9.42% of the total 15N gas production at site 2, and anaerobic ammonium oxidation could account for 7.82% of total 15N gas formation. Overall, there was good agreement between the rates of anaerobic ammonium oxidation measured with 15N labeling of either the NH4+ or the NO3− pool (Table 2) (P = 0.497).
TABLE 2.
Anaerobic ammonium oxidation measured by production of 29N2 with 15N labelling of either the NO3− or NH4+ pools
| Site | Actual 30N2 (% 15N gas) | Predicted 29N2 (% 15N gas)a | Actual 29N2 (% 15N gas)b | A29 (% 15N gas)c | Probability of 15N from 15NH4+d | AAOe from:
|
|
|---|---|---|---|---|---|---|---|
| 15NO3− (μmol) | 15NH4+ (μmol) | ||||||
| 2 Grays | 90.58 | 1.86 | 9.42 | 7.82 | 0.49 | 0.261 (0.019) | 0.296 (0.0289) |
| 3 Gravesend | 93.77 | 1.99 | 6.23 | 4.37 | 0.56 | 0.120 (0.0228) | 0.160 (0.0164) |
| 4 Allhallows | 95.44 | 1.93 | 4.56 | 2.71 | 0.27 | 0.040 (0.0045) | 0.026 (0.0033) |
| 6 Grain | 97.60 | 1.85 | 2.40 | 0.57 | 0.54 | 0.004 (0.0006) | 0.003 (0.0005) |
From >98 atom % 15N-NO3− and random pairing of 14N and 15N.
As a proportion of total measured 15N gases.
A29 = P29 − D29 (see the text and reference 30).
[15NH4+]/[total NH4+].
Anaerobic ammonium oxidation (AAO) with 15N-labeled NO3− (n = 4) or NH4+ (n = 3). Figures in parentheses are SEM.
Maximum rates of anaerobic ammonium oxidation were measured at site 2 in October and November 2002; the rates then decreased significantly between sites (by analysis of variance, P was <0.05) to a minimum at site 6 at the mouth of the estuary (Fig. 5a). Denitrification rates followed a similar pattern, though the peak was found further downstream at site 3, and were significantly positively correlated with rates of anaerobic ammonium oxidation (P < 0.05). In a similar pattern, maximum N2 formation via anaerobic ammonium oxidation was measured at site 2 (8%); the proportion then decreased steadily to <1% at site 6 at the mouth of the estuary (Fig. 5b). Overall, there was good positive correlation (P < 0.05) between sediment organic carbon content and the significance of anaerobic ammonium oxidation to N2 formation along the estuary, although site 1 was a distinct outlier in this relationship (Fig. 5c; Table 1). In November 2002, when all six sites were visited, there was a distinct decrease in the rate of NO3− reduction along the estuarine gradient and a distinct shift in the fate of reduced NO3− (Fig. 5d). At sites 1 and 2, 60 to 70% recovery of reduced NO3− as 15N gas (as in the initial trials at site 1 [data not shown]) suggested significant DNRA, while 100% recovery at sites 3 to 6 suggested the dominance of denitrification. Nitrite accumulated in the sediments to 3 μM during the incubations with 15NO3− (800 μM) at all sites (except site 1, as in the initial trials). A large accumulation of NO2− (150 μM) at site 5 explains the difference between NO3− reduction and 15N gas formation but does not necessarily suggest significant DNRA.
FIG. 5.

(a) Anaerobic ammonium oxidation (AAO) and denitrification (Den) at six sites along the Thames estuary in November 2002; (b and c) anaerobic ammonium oxidation as a proportion of total 15N gas formation in October and November 2002 (b) and as a scatter against sediment organic carbon content (c); (d) total NO3− reduction and 15N gas formation in November 2002. Values are means ± 1 SEM (n = 4). sed, sediment. Note that a large accumulation of NO2− at site 5 explains differences between reduction and gas formation (see the text).
DISCUSSION
Biogeography of anaerobic ammonium oxidation.
The results presented here confirm the presence of anaerobic ammonium oxidation in another aquatic system and extend the findings recently reported for marine sediments and anoxic marine basins (2, 15, 30). However, the pattern reported here is in stark contrast to that reported for marine sediments. For example, the significance of anaerobic ammonium oxidation to N2 production decreased along the estuarine gradient with decreasing organic content to <1% at the most seaward site (Fig. 5c; Table 1). Though a similar low proportion of anaerobic ammonium oxidation was reported at an inshore coastal site (30), an increase in the significance of anaerobic ammonium oxidation was measured with decreasing organic content and decreasing sediment reactivity in an offshore direction, i.e., in the opposite direction to that of our estuarine gradient. In addition, the maximum proportion of anaerobic ammonium oxidation to N2 production in the Thames sediments was 8%; therefore, denitrification still dominated N2 production overall, while up to 60% of N2 production via anaerobic ammonium oxidation was reported to occur in offshore marine sediments (30).
Anaerobic ammonium oxidation via nitrite reduction.
The sole production of 29N2 from sediments incubated with 15NH4+ and either 14NO3− or 14NO2− agreed with the 1:1 stoichiometry of equation 3. With the stoichiometry of equation 4, if any oxidation of ammonium was directly coupled to the reduction of NO3−, then at least 25% of the 15N-labeled gases should have been 30N2; as this was not the case, it was concluded that the anaerobic ammonium oxidation in these estuarine sediments was coupled to the reduction of NO2−, which agrees with the original hypotheses and that recently reported for marine sediments (3, 30, 34).
In the presence of both electron acceptors, the concentration of NO2− remained essentially the same (Fig. 3c), which can be explained only if the rate of NO2− reduction was balanced by the supply of NO2− from NO3− reduction. Given that anaerobic ammonium oxidation could account for only 1% of NO2− reduction at site 1, this change remained unnoticeable. Even if obligate NO2− respirers, as well as anaerobic ammonium oxidizers, were present (5), their impact on the NO2− pool while in the presence of NO3− was minimal. After 8 h, NO3− became limiting, and the community that reduced NO3−changed to a community that directly reduced NO2−. Despite the increase in the rate of turnover, and therefore competition, for NO2−, the rate of anaerobic ammonium oxidation remained unaffected. Saturation of anaerobic ammonium oxidation at low concentrations of NO2− would explain why there was no measurable difference between the rates measured with either trace or significant (∼20 μM) accumulation of NO2− from NO3− (or in the direct presence of NO2−) but also suggests that the availability of NO2− would seldom be limiting for anaerobic ammonium oxidation in estuarine sediments. Anaerobic ammonium-oxidizing bacteria are reliant on the in situ formation of NO2− via NO3− reduction, and as concentrations of NO2− are usually less than 5 μM in upper sediment layers (M. Trimmer, unpublished results), such an affinity is suited to scavenging NO2− at low concentrations. Whereas the NO3−- and NO2−-reducing community has a high capacity to reduce NO2− (up to 383 μM h−1), anaerobic ammonium oxidation appears to be saturated at a comparatively low NO2− concentration, which agrees with Km estimates (3 μM) for NO2− consumption via anaerobic ammonium oxidation in marine sediments (3).
In the only other published environmental study of anaerobic ammonium oxidation in aquatic sediments, the vast majority (>91%) of NO3− or NO2− reduced was recovered as N2 gas (30). This finding suggests that DNRA was insignificant and that anaerobic ammonium oxidation was reliant on NO2− formed intracellularly by the denitrifying community “leaking” into the sediment. In contrast, both the denitrification and DNRA pathways may support anaerobic ammonium oxidation at sites 1 and 2. In previous studies on the end products of NO3− reduction in organically enriched sediments, it was speculated that NO2− formed by DNRA may later be respired by denitrifying bacteria (11). Yet this may be an additional pathway for NO2− formation and, in turn, a pathway for NO2− coupling to anaerobic ammonium oxidation in estuarine sediments. Hence, N transformations in estuarine sediments may actually be more complex than originally thought, with potential interplay between DNRA, denitrification, and anaerobic ammonium oxidation. At the remaining sites, however, anaerobic ammonium oxidation must have been coupled to the in situ formation of NO2− via the denitrifying community.
The quantification of anaerobic ammonium oxidation using 15NO3− is based on the assumption that the process produces only 28N2 and 29N2 (30). This assumption may be violated in the presence of DNRA, which, via the reduction of 15NO3− to 15NH4+, may in turn generate 30N2 by anaerobic ammonium oxidation. At site 2, where the contribution of anaerobic ammonium oxidation to N2 formation was greatest, DNRA may account for approximately 30% of the reduced 15NO3−. The starting concentration for 15NO3− was 800 μM, and with a 50% turnover and 30% DNRA, these conditions would produce 120 μM 15NH4+. As only approximately 50% of 15NH4+ remains in the soluble pool, 60 μM 15NH4+ was left, which represents about 5% of the total soluble NH4+ pool of ∼1,200 μM. At the start, the probability of 29N2 production with 15NO3− and 14NH4+ was 100%, but with a 50% turnover, this estimate dropped to 95%, and in turn, our estimate of anaerobic ammonium oxidation from our measured production of 29N2 is underestimated by ∼5%. The magnitude of this violation via DNRA depends upon its contribution to NO3− reduction and the relative size of the nonlabeled NH4+ pool.
Anaerobic ammonium oxidation along the estuary.
The distribution of anaerobic ammonium oxidation along the Thames estuary may simply reflect changes in the abundance of bacteria per unit of wet sediment. At site 1, despite having the highest concentrations of NO2− in the water column throughout the estuary, the sediments are highly reduced (visible observations) and concentrations of NO2− are permanently low (<1 μM), which may in part be due to denitrification, substantial DNRA, and the reoxidation of sulfides. Such a low NO2− supply may be capable of sustaining only a relatively small population of anaerobic ammonium-oxidizing bacteria; hence, anaerobic ammonium oxidation accounted for only ∼1% of N2 formation. Though anaerobic ammonium oxidation was saturated at low concentrations of NO2−, it accumulated at all sites except for site 1, which was a clear outlier in the overall relationship between organic content and anaerobic ammonium oxidation (Fig. 5c). At site 2, conditions must have been favorable for both anaerobic ammonium-oxidizing bacteria and denitrifying bacteria, e.g., sustained though low supplies of NO2− and organic substrates and NO3−, respectively. Despite the saturation of anaerobic ammonium oxidation at a low NO2− concentration, a sustained population would still be reliant on an in situ supply of NO2−, hence the correlation between sediment organic C and the significance of anaerobic ammonium oxidation. Higher organic loadings, i.e., the loadings at site 2, stimulated NO3− reduction (26) and, in turn, the in situ availability of NO2−, which sustains a greater population of anaerobic ammonium-oxidizing bacteria. The overall positive correlation between rates of denitrification and anaerobic ammonium oxidation supports this hypothesis, though DNRA may help support the maximum rates of anaerobic ammonium oxidation at site 2. The marked decrease in anaerobic ammonium oxidation and denitrification seaward of site 2 suggested a decline in both populations as organic C, NO3−, and NO2− decreased along the estuary.
The organism responsible for anaerobic ammonium oxidation in the original bioreactor studies has been identified as a novel planctomycete (17, 28), but the organism(s) that is responsible for anaerobic ammonium oxidation in the Thames estuary, as well as what conditions regulate its significance relative to denitrification, remains unknown. Given that the anaerobic ammonium oxidation reaction was first discovered in a sewage bioreactor (17) and that two very large sewage treatment works (STWs) (among many others) discharge into the Thames at site 1 (combined output, 1.3 × 106 m3 day−1), the sediments along the estuary may have been seeded by these STWs, yet the activity was most significant some 13 km downstream from the main discharge area. In addition, the well-studied classic nitrifier Nitrosomonas eutropha can perform anaerobic ammonium oxidation under anaerobic or O2-limiting conditions and, as such, is distinct from the anaerobic ammonium-oxidizing bacteria (1). Indeed, anaerobic ammonium oxidation may in fact be due to a combination of both of these metabolisms (24).
Although it is possible to measure the proportion of N2 production from either anaerobic ammonium oxidation or denitrification in anaerobic slurries, regulation of either process maybe more complex under in situ conditions, and such measurements should be taken as estimates of potential rates. For example, anaerobic ammonium oxidation is an obligate anaerobic process, while denitrification is facultative, and because of this, denitrification is likely to occur along the gradient of decreasing oxygen concentrations and below the oxic-anoxic interface within sediments (14). Oxygen penetration (typically 0.2 to 2 mm) into intact estuarine sediment cores will directly affect the demand for NO3− from the overlying water and, in turn, the in situ rates of NO2− formation via nitrification and reduction (21, 22, 23). Oxygen penetration is likely to be more significant where the dominant source of NO2− and NO3− within sediments is that of nitrification, as in unpolluted estuaries or coastal seas (25, 32). While total anaerobic ammonium oxidation is unlikely to be limited by the availability of NO2−, oxygen penetration will affect the source of NO2− and overall rates of denitrification.
The organically enriched sediments of the Thames estuary, although representing only 18% of the total sediment area, are responsible for 34% of the total mineralization of organic material (33). While the maximum contribution from anaerobic ammonium oxidation to N removal was only 8%, this maximum activity occurred within these organically enriched sediments, which again highlights the significance of the depositional zone (i.e., around the turbidity maxima) to N processing—including novel pathways—in estuarine sediments.
Acknowledgments
We acknowledge the staff of the Environment Agency (Thames Region) and the Port of London Authority for site access. Special thanks go to Andrew Stott from the NERC 15N stable isotope facility at CEH-Merlewood. Jakob Petersen is gratefully acknowledged for his technical and field assistance.
This research was funded by a research grant (NER/A/S/1999/00037) to M.T. from the NERC. Isotope analyses were provided by a grant-in-kind from the NERC Isotope Life Sciences Steering Committee and Science Programmes Group (Swindon, United Kingdom).
REFERENCES
- 1.Bock, E., I. Schmidt, R. Stüven, and D. Zart. 1995. Nitrogen loss caused by denitrifying Nitrosomonas cells using ammonium or hydrogen as electron donors and nitrite as electron acceptor. Arch. Microbiol. 163:16-20. [Google Scholar]
- 2.Dalsgaard, T., D. E. Canfield, J. Petersen, B. Thamdrup, and J. Acuna-González. 2003. N2 production by the anammox reaction in the anoxic water column of Golfo Dulce, Costa Rica. Nature 422:606-608. [DOI] [PubMed] [Google Scholar]
- 3.Dalsgaard, T., and B. Thamdrup. 2002. Factors controlling anaerobic ammonium oxidation with nitrite in marine sediments. Appl. Environ. Microbiol. 68:3802-3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong, L. F., D. C. O. Thornton, D. B. Nedwell, and G. J. C. Underwood. 2000. Denitrification in sediments of the River Colne estuary, England. Mar. Ecol. Prog. Ser. 203:109-122. [Google Scholar]
- 5.Dong, L. F., D. B. Nedwell, G. J. C. Underwood, D. C. O. Thornton, and I. Rusmana. 2002. Nitrous oxide formation in the Colne estuary, England: the central role of nitrite. Appl. Environ. Microbiol. 68:1240-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hauck, R. D., S. W. Melsted, and P. E. Yankwich. 1958. Use of N-isotope distribution in nitrogen gas in the study of denitrification. Soil Sci. 86:287-291. [Google Scholar]
- 7.Hedges, J. I., and J. H. Stern. 1984. Carbon and nitrogen determinations of carbonate-containing solids. Limnol. Oceanogr. 29:657-663. [Google Scholar]
- 8.Herbert, R. A., and D. B. Nedwell. 1990. Role of environmental factors in regulating nitrate respiration in intertidal sediments, p. 77-91. In N. P. Revsbech and J. Søorensen (ed.), Denitrification in soil and sediment. Plenum Press, New York, N.Y.
- 9.Jetten, M. S. M., S. J. Horn, and M. C. M. van Loosdrecht. 1997. Towards a more sustainable municipal wastewater treatment system. Water Sci. Technol. 35(9):171-180. [Google Scholar]
- 10.Jetten, M. S. M. 2001. New pathways for ammonia conversion in soil and aquatic systems. Plant Soil 230:9-19. [Google Scholar]
- 11.King, D. H., and D. B. Nedwell. 1985. The influence of nitrate concentration upon the end-products of nitrate dissimilation by bacteria in anaerobic saltmarsh sediment. FEMS Microbiol. Ecol. 31:23-28. [Google Scholar]
- 12.King, D. H., and D. B. Nedwell. 1987. The adaptation of nitrate-reducing bacterial communities in estuarine sediments in response to overlying nitrate load. FEMS Microbiol. Ecol. 45:15-20. [Google Scholar]
- 13.Kirkwood, D. S. 1996. Nutrients: practical notes on their determination in seawater. Techniques in marine environmental science no. 17. ICES, Copenhagen, Denmark.
- 14.Körner, H., and W. G. Zumft. 1989. Expression of denitrification enzymes in response to the dissolved oxygen level and respiratory substrate in continuous culture of Pseudomonas stutzeri. Appl. Environ. Microbiol. 55:1670-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuypers, M. M. M., A. O. Sliekers, G. Lavik, M. Schmid, B. B. Jørgensen, J. G. Kuenen, J. S. Sinninghe Damsté, M. Strous, and M. S. M. Jetten. 2002. Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. Nature 422:608-611. [DOI] [PubMed] [Google Scholar]
- 16.Laima, M. C. J. 1992. Extraction and seasonal variation of NH4+ pools in different types of coastal marine sediments. Mar. Ecol. Prog. Ser. 82:75-84. [Google Scholar]
- 17.Mulder, A., A. A. van de Graaf, L. A. Robertson, and J. G. Kuenen. 1995. Anaerobic ammonium oxidation discovered in a denitrifying fluidized bed reactor. FEMS Microbiol. Ecol. 16:177-183. [Google Scholar]
- 18.Nedwell, D. B. 1975. Inorganic nitrogen metabolism in a eutrophicated tropical mangrove estuary. Water Res. 9:221-231. [Google Scholar]
- 19.Nielsen, K., L. P. Nielsen, and P. Rasmussen. 1995. Estuarine nitrogen retention independently estimated by the denitrification rate and mass balance methods: a study of Norsminde Fjord, Denmark. Mar. Ecol. Prog. Ser. 119:275-283. [Google Scholar]
- 20.Revsbech, N. P., and B. B. Jørgensen. 1986. Microelectrodes: their use in microbial ecology. Adv. Microb. Ecol. 9:293-356. [Google Scholar]
- 21.Risgaard-Petersen, N., S. Rysgaard, L. P. Nielsen, and N. P. Revsbech. 1994. Diurnal variation of denitrification and nitrification in sediments colonised by benthic microphytes. Limnol. Oceanogr. 39:573-579. [Google Scholar]
- 22.Rysgaard, S., P. B. Christensen, and L. P. Nielsen. 1995. Seasonal variation in nitrification and denitrification in estuarine sediment colonised by benthic microalgae and bioturbating infauna. Mar. Ecol. Prog. Ser. 126:111-121. [Google Scholar]
- 23.Rysgaard, S., N. Risgaard-Petersen, and N. P. Sloth. 1996. Nitrification, denitrification, and nitrate ammonification in sediments of two coastal lagoons in southern France. Hydrobiology 329:133-141. [Google Scholar]
- 24.Schmidt, I., C. Hermelink, K. van de Pas-Schoonen, M. Strous, H. J. op den Camp, J. G. Kuenen, and M. S. M. Jetten. 2002. Anaerobic ammonia oxidation in the presence of nitrogen oxides (NOx) by two different lithotrophs. Appl. Environ. Microbiol. 68:5351-5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seitzinger, S. P. 1987. Nitrogen biogeochemistry in an unpolluted estuary: the importance of benthic denitrification. Mar. Ecol. Prog. Ser. 41:177-186. [Google Scholar]
- 26.Sloth, N. P., T. H. Blackburn, L. S. Hansen, N. Risgarrd-Petersen, and B. A. Lomstein. 1995. Nitrogen cycling in sediments with different organic loading. Mar. Ecol. Prog. Ser. 116:163-170. [Google Scholar]
- 27.Sørensen, J., B. B. Jørgensen, and N. P. Revsbech. 1979. A comparison of oxygen, nitrate and sulphate respiration in coastal marine sediments. Microb. Ecol. 5:105-115. [DOI] [PubMed] [Google Scholar]
- 28.Strous, M., J. A. Fuerst, E. H. M. Kramer, S. Logemann, G. Muyzer, K. T. van de Pas-Schoonen, R. Webb, G. J. Kuenen, and M. S. M. Jetten. 1999. Missing lithotroph identified as new planctomycete. Nature 400:446-449. [DOI] [PubMed] [Google Scholar]
- 29.Thamdrup, B., and T. Dalsgaard. 2000. The fate of ammonium in anoxic manganese oxide-rich marine sediment. Geochim. Cosmochim. Acta 64:4157-4164. [Google Scholar]
- 30.Thamdrup, B., and T. Dalsgaard. 2002. Production of N2 through anaerobic ammonium oxidation coupled to nitrate reduction in marine sediments. Appl. Environ. Microbiol. 68:1312-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trimmer, M., D. B. Nedwell, D. Sivyer, and S. J. Malcolm. 1998. Nitrogen fluxes through the lower estuary of the Great Ouse, England: the role of the bottom sediments. Mar. Ecol. Prog. Ser. 163:109-124. [Google Scholar]
- 32.Trimmer, M., R. J. Gowen, B. M. Stewart, and D. B. Nedwell. 1999. The spring bloom and its impact on benthic mineralisation rates in western Irish Sea sediments. Mar. Ecol. Prog. Ser. 185:37-46. [Google Scholar]
- 33.Trimmer, M., D. B. Nedwell, D. B. Sivyer, and S. J. Malcolm. 2000. Seasonal benthic organic matter mineralisation measured by oxygen uptake and denitrification along a transect of the inner and outer River Thames estuary, UK. Mar. Ecol. Prog. Ser. 197:103-119. [Google Scholar]
- 34.van de Graaf, A. A., P. De Bruijn, L. A. Robertson, M. S. M. Jetten, and J. G. Kuenen. 1996. Autotrophic growth of anaerobic ammonium-oxidising microorganisms in a fluidized bed reactor. Microbiology 142:2187-2196. [Google Scholar]
- 35.van de Graaf, A. A., A. Mulder, P. De Bruijn, M. S. M. Jetten, L. A. Robertson, and J. G. Kuenen. 1995. Anaerobic oxidation of ammonium is a biologically mediated process. Appl. Environ. Microbiol. 61:1246-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]