

Abstract
The pore-forming α subunit of large conductance voltage- and Ca2+-sensitive K (MaxiK) channels is regulated by a β subunit that has two membrane-spanning regions separated by an extracellular loop. To investigate the structural determinants in the pore-forming α subunit necessary for β-subunit modulation, we made chimeric constructs between a human MaxiK channel and the Drosophila homologue, which we show is insensitive to β-subunit modulation, and analyzed the topology of the α subunit. A comparison of multiple sequence alignments with hydrophobicity plots revealed that MaxiK channel α subunits have a unique hydrophobic segment (S0) at the N terminus. This segment is in addition to the six putative transmembrane segments (S1–S6) usually found in voltage-dependent ion channels. The transmembrane nature of this unique S0 region was demonstrated by in vitro translation experiments. Moreover, normal functional expression of signal sequence fusions and in vitro N-linked glycosylation experiments indicate that S0 leads to an exoplasmic N terminus. Therefore, we propose a new model where MaxiK channels have a seventh transmembrane segment at the N terminus (S0). Chimeric exchange of 41 N-terminal amino acids, including S0, from the human MaxiK channel to the Drosophila homologue transfers β-subunit regulation to the otherwise unresponsive Drosophila channel. Both the unique S0 region and the exoplasmic N terminus are necessary for this gain of function.
Keywords: transmembrane topology
High-conductance voltage- and Ca2+-sensitive potassium channels are found virtually in all excitable and nonexcitable tissues, with the exception of heart. As sensors of both voltage and intracellular calcium, they are responsible for membrane hyperpolarization, associated with phenomena like repetitive firing, spike shaping, transmitter release, and regulation of vascular and visceral smooth muscle contractility (1–4). Cloning of high-conductance voltage-activated and Ca2+-sensitive K+ (MaxiK) channels revealed that they belong to the S4 superfamily of ion channels (5) but carry a unique C terminus containing four hydrophobic, possibly membrane-spanning regions (S7–S10) with a nonconserved linker between regions S8 and S9 (6–8). The C-terminal region after the nonconserved linker shows the highest sequence conservation between the Drosophila (Dslo) and mammalian clones and includes hydrophobic regions S9 and S10. This region can be expressed as a separate domain and has been proposed to determine the Ca2+ sensitivity of this channel (9). Alternative splicing rather than homologous genes seems to be responsible for the diversity of MaxiK channels (8, 10, 11).
The common features of voltage-dependent K+ channels and individual domains of Na+ and Ca2+ channels of the S4 superfamily are six putative transmembrane segments with a pore loop between transmembrane segments S5 and S6. The S4 region, which has been shown to move outward during depolarization and activation of these channels (12, 13), carries positive charges that are thought to interact with negative charges in regions S2 and S3 in Shaker K+ channels (14). By analyzing sequence alignments and hydrophobicity plots, we show that MaxiK channels may share these features, as initially proposed (7), but carry an additional hydrophobic region (S0) at the N terminus. Our data suggest that this hydrophobic region serves as a type I signal anchor directing the N terminus to the extracellular space.
MaxiK channels purified from smooth muscle are tightly associated with an accessory β subunit (15). Purification and cloning of this β subunit revealed that it has two putative membrane-spanning regions and a large extracellular loop with two glycosylation sites (16, 17). This β subunit dramatically increases the open probability of the pore-forming α subunit of mammalian MaxiK channels (18–21). We show herein that the Drosophila homologue (Dslo) is unaffected by the coexpression of this mammalian β subunit. We utilized this difference to map the region responsible for β-subunit regulation by constructing chimeras between the β-subunit responsive human MaxiK channel (Hslo) and the unresponsive Dslo. We show that 41 N-terminal amino acids, including S0, from Hslo are sufficient to confer β-subunit responsiveness to Dslo. Preliminary reports of these findings have been presented.‡
MATERIALS AND METHODS
Sequence Analysis.
We used the Genetics Computer Group software package (version 8.0) (22). Hydrophobicity analysis was done with the program pepplot; the program pileup was used to generate the multiple sequence alignments (in both cases using default settings). To obtain a reasonable alignment only the 400 N-terminal amino acids of Hslo and Dslo were used. The other K+-channel sequences were full-length. Accession numbers used are as follows: Hslo, U11058; Dslo, JH0697; Kv1.3, P22001; Shaker, X06742; Kv2.1 (drk1), P15387; Shab, P17970; Kv3.1, P15388; Shaw, P17972; Kv4, A39372; Shal, P17971.
In Vitro Translation.
H-S0 and D-S0 clones were made by introducing a stop codon after amino acid Arg-113 in Hslo and Arg-127 in Dslo. cRNA (0.5 μg in a 25-μl reaction) was translated in reticulocyte lysates in presence of microsomes (Promega) and [35S]methionine (NEN) by following the manufacturers instructions. Aliquots of 5–10 μl were diluted into 100 μl of 0.1 M Na2CO3 (pH 11) or phosphate-buffered saline (PBS, pH 7.4) [for N-glycosidase (PNGase) F digestions] and kept on ice for at least 30 min. Microsomes were collected by centrifugation (20,000 × g for 1 h). The pellets were rinsed two times with 100 μl of PBS. Soluble proteins (100 μl) were precipitated with acetone (200 μl). Equivalent amounts of pellet (P) and supernatant proteins (S) were loaded in each lane. To remove N-linked glycosylation, the microsomal pellet was resuspended in H2O and an aliquot was treated with 25 units of PNGase F (NEB) according to the manufacturers instructions. Control reactions were treated in the same way, but without adding enzyme. After SDS/PAGE, gels were fixed, stained, soaked in Amplify (Amersham), and dried before autoradiography.
Signal-Sequence Fusion Clones.
S-DCHT, S-Dslo, S-Hslo-M4, and S-ShH4-IR were made by ligating a PCR fragment coding for 33 N-terminal amino acids of the rat Na+ channel β1-subunit (23) into NcoI sites at the translational start. The correct orientation was determined by restriction analysis and confirmed by sequencing. These 33 amino acids contain a cleavable N-terminal signal sequence of 19 amino acids (24). Therefore, the mature proteins are predicted to contain 14 additional amino acids at the N terminus. Hslo-M4 was made by utilizing Met-10 in Hslo (20) as translational start site (see Fig. 5B). In ShH4-IR(S−) these 33 amino acids were removed from S-ShH4-IR.
Figure 5.
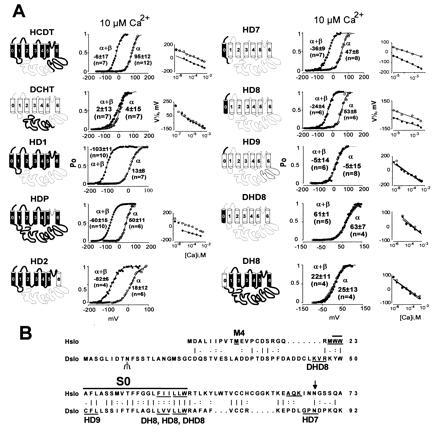
Transfer of β-subunit modulation from Hslo to Dslo. (A) Thick lines and solid membrane-spanning regions indicate sequences derived from Hslo; thin lines and open membrane regions are sequences derived from Dslo. Open probabilities in 10 μM Ca2+ for a representative experiment are plotted against voltage. Values are the mean half-activation potentials at 10 μM Ca2+ (±SD, n = number of experiments). V½ vs. Ca2+ are also shown. Oocytes were injected with 5 ng of α-subunit and 10 ng of β-subunit cRNA. Titration studies showed that even 10-fold less β-subunit cRNA is a saturating concentration (data not shown). (B) Sequence comparison of the N-terminal region of Hslo and Dslo. Hydrophobic region S0 is marked with a bar. Three amino acids at the junctions of chimeric constructs are underlined. M4 marks the beginning of clone Hslo-M4. The N-linked glycosylation site in Dslo is indicated with a Ψ and a consensus sequence for N-linked glycosylation in Hslo is indicated by an arrow. A vertical line, colon, and period indicate identical, highly conserved, and conserved amino acids according to the sequence comparison table of the GCG program.
Chimeric Constructs.
Chimeras were made by including appropriate restriction enzyme recognition sites into PCR primers or by overlap extension amplification (25). For all PCRs, high-fidelity Pfu polymerase (Stratagene) was used. A conserved SphI restriction site was utilized for generating the HCDT and DCHT chimeras. The amino acid sequences of the constructs (restriction sites used for cloning are given in brackets) are as follows: HCDT, Met-1 to Met-652 from Hslo and His-680 to Ser-1164 from Dslo (SphI); DCHT, Met-1 to Met-679 from Dslo and Arg-653 to Leu-1113 from Hslo (SphI); HD11, Tyr-318 to Asn-649 of Hslo replaced by Ser-332 to Thr-687 of Dslo (AccI, SphI); HD1, Met-1 to Ser-317 from Hslo and Ser-332 to Ser-1164 from Dslo (AccI); HD2, Met-1 to Glu-257 from Hslo and Asn-273 to Ser-1164 from Dslo (EcoRI); HDP, Asn-258 to Gly-300 of Hslo replaced by Asn-273 to Gly-314 from Dslo (KasI, EcoRI); HD7, Met-1 to Lys-65 from Hslo and Gly-85 to Ser-1164 from Dslo (ApaI); HD8, Met-1 to Ile-40 from Hslo and Leu-68 to Ser-1164 from Dslo (overlap extension); DH8, Met-1 to Val-67 from Dslo and Val-41 to Leu-1113 from Hslo (overlap extension); DHD8, Lys-48 to Val-67 of Dslo replaced by Met-21 to Ile-40 from Hslo (overlap extension); HD9, Met-1 to Trp-22 from Hslo and Trp-50 to Ser-1164 from Dslo (overlap extension); Hslo-M4, Met-10 to Leu-1113 from Hslo (NcoI); HΔN43, Met-Gly, Arg-44 to Leu-1113 from Hslo (NcoI introduced with PCR primer, EcoRI). All constructs were analyzed by restriction digestion. Sequences amplified by PCR and ligation connections were confirmed by sequencing. For cRNA synthesis, plasmids were linearized with appropriate restriction enzymes, and cRNA was transcribed using the mMESSAGE mMACHINE kit (Ambion, Austin, TX). The cRNA was precipitated with LiCl and dissolved in H2O; the concentration was measured in a spectrophotometer and confirmed by gel electrophoresis.
Electrophysiology.
Xenopus oocytes were injected with 5–10 ng of cRNA and measured after 2–5 days using inside-out patches. The pipette solution contained 105 mM potassium–methane sulfonate, 5 mM KCl, and 10 mM Hepes (pH 7.0). The bath solution had in addition 5 mM N-hydroxyethylethylenediaminetriacetic acid (HEDTA) and CaCl2, to obtain the desired free [Ca2+] and confirmed with a Ca2+ electrode (World Precision Instruments, Sarasota, FL). Data were analyzed as described (21). Each construct or their combinations were measured in at least four batches of oocytes.
RESULTS AND DISCUSSION
MaxiK Channels Contain an Additional Hydrophobic Region at the N Terminus.
Kyte–Doolittle hydrophobicity analysis (26) of the amino acid sequence of Hslo and Dslo channels revealed several hydrophobic regions at the N terminus (Fig. 1A). Hydrophobic regions S1–S6 were assigned based on multiple sequence alignments with other voltage-dependent potassium channels (Fig. 1B). Landmarks in this sequence alignment are conserved charged amino acids that are indicated with arrows in the sequence alignment and in the hydrophobicity plots (Fig. 1 A and B). Some of these residues in segments S2, S3, and S4 have been implicated in voltage-dependent gating (12, 13, 27, 28), and negatively charged amino acids in regions S2 and S3 are thought to interact with positive charges in the S4 region of Shaker K+ channels (boxed in Fig. 1B) (14).
Figure 1.
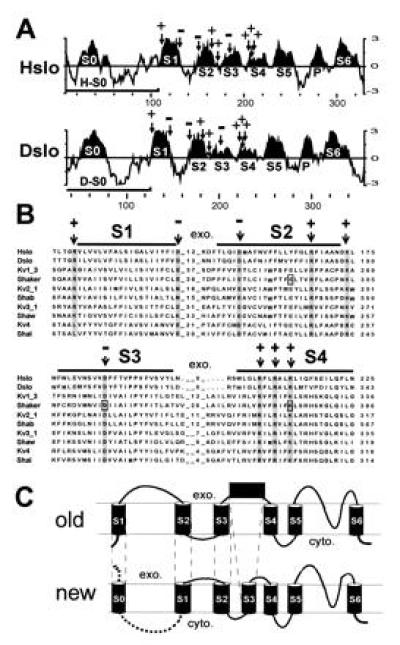
Unique hydrophobic region at the N terminus of MaxiK channels. (A) Kyte–Doolittle hydrophobicity plot (26) of Hslo and Dslo from the N terminus to the end of region S6. The curve represents the average of a residue-specific hydrophobicity index calculated from a window of 9 amino acids. Hydrophobic regions S1–S6 (and the pore region P) were designated according to the multiple sequence alignment (Fig. 2B) and are highlighted. Arrows mark the positions of conserved charged amino acids. Bars denote the coding region of clones H-S0 and D-S0. (B) Part of a multiple sequence alignment of Hslo and Dslo with voltage dependent potassium channels. The numbers between hydrophobic regions represent the number of amino acids of extracellular loops. The abundance of hydrophobic residues throughout the alignment and the hydrophobicity analysis of Hslo and Dslo were considered for designating transmembrane regions (marked with bars). Conserved amino acids are shaded and conserved charged amino acids are marked with an arrow. (C) Comparison of the previously suggested model (old) with the topology suggested to us by the sequence homology and hydrophobicity analysis (new). The exoplasmic (exo.) and cytoplasmic (cyto.) sides of the membrane are marked.
The sequence homology and the hydrophobicity pattern of regions S1–S6 of MaxiK channels, which closely resemble the pattern observed for other voltage-dependent ion channels, suggest that homology regions S1–S6 of MaxiK channels (excluding S0) have the same membrane topology and probably similar function as proposed for their counterparts in solely voltage-dependent ion channels. This notion is consistent with our finding that MaxiK channels become purely voltage-dependent at Ca2+ concentrations less than 100 nM (21). In addition, we also measure gating currents from Hslo channels (L.T., unpublished results), which are protein-bound charge movements characteristic of voltage-dependent ion channels.
From this analysis, it is obvious that both Hslo and Dslo have an additional hydrophobic segment (termed S zero, S0) at the N terminus. In previous reports (7, 10, 29), regions S1–S6 were assigned as shown in Fig. 1, but this additional hydrophobic region was not mentioned. Other authors (5, 6, 8, 11, 19, 30) have interpreted hydrophobic region S0 as transmembrane region S1. In the latter model, homology region S1 was shown as S2, S2 as S3, and region S3 was predicted as an extracellular loop as illustrated in Fig. 1C (old). In our revised model, the region previously thought to form a large extracellular loop between transmembrane segment S1 and S2 is intracellular and the previous transmembrane region S1 is now designated as S0 [Fig. 1C (new)].
Hydrophobic region S0 seems to be unique for MaxiK channels. In voltage-dependent K+ channels, the corresponding cytoplasmic regions are conserved, especially within subfamilies, and are responsible for subfamily-specific recognition and assembly (31, 32).
Hydrophobic Region S0 Is an Additional Transmembrane Segment at the N Terminus That Can Be Expressed as a Separable Domain.
Hydrophobic S0 region could be cytoplasmic and might interact with additional hydrophobic regions (S7–S10) at the C terminus. It may also be peripherally membrane associated or may be membrane spanning (Fig. 1C new). To distinguish among these possibilities, N termini (H-S0, D-S0; marked with bars in Fig. 1A) that contain only hydrophobic region S0 were in vitro-translated in presence of microsomes. After alkaline treatment, which opens the microsomal vesicles and releases peripheral membrane proteins (33, 34), membrane-spanning proteins were separated from soluble proteins by centrifugation. The majority of the in vitro-translated H-S0 and D-S0 fragments are found in the microsomal pellet (P) and are not released from the membrane by high pH treatment (Fig. 2A). This shows that H-S0 and D-S0 behave like integral membrane proteins and implies that hydrophobic region S0 is a membrane-spanning region. Both D-S0 and H-S0 contain a single consensus sequence for N-linked glycosylation (NXS/T), which in D-S0 is located before and in H-S0 after the hydrophobic segment S0 (Fig. 2C and see Fig. 5B). In vitro translation of D-S0 in the presence of microsomes gave an additional band, which is shifted to a higher molecular weight (Fig. 2A). To verify that this second band appeared because of N-linked glycosylation, we used N-glycosidase F treatment. This reduced the observed band shift in D-S0 but had no effect in the H-S0 protein (Fig. 2B). This result suggests that H-S0 and D-S0 insert into microsomal membranes as a type I signal anchor sequence (35) leading to an exoplasmic N terminus. This occurs in the absence of a cleavable N-terminal signal sequence, as found for most members of the superfamily of G-protein-coupled seven-helix receptors.
Figure 2.
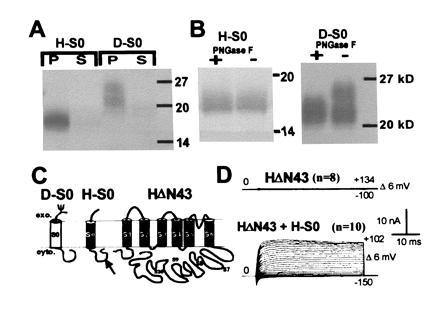
Hydrophobic region S0 is an additional transmembrane region. (A) Autoradiogram of H-S0 and D-S0 proteins after in vitro translations, high pH treatment, separation into microsomal pellet (P) and soluble fraction (S), and SDS/PAGE. The apparent molecular weights of H-S0 and D-S0 are higher than the calculated molecular weights (H-S0, 12.5 kDa; D-S0, 14 kDa). This may be due to a stable secondary structure, which might also explain the diffuse bands. (B) PNGase F treatment of microsomal pellets of H-S0 and D-S0; + and − indicates treatment with and without PNGase F. (C) Proposed membrane topology of H-S0, D-S0, and HΔN43. MaxiK channels possess additional hydrophobic regions (S7–S10) at the C terminus that are shown as intracellular. We prefer this topology because of the relatively low hydrophobicity of these potential transmembrane regions. (D) Functional rescue of the N-terminal deletion clone HΔN43 by coexpression of H-S0. Oocytes were injected with 10 ng of HΔN43 cRNA alone or along with 5 ng of H-S0 cRNA and currents were measured in inside-out patches in 10 μM free Ca2+.
To test whether Hslo can be expressed as a functional channel without the S0 region, we eliminated 43 N-terminal amino acids in Hslo (HΔN43). We anticipated that this truncated protein would fold into the membrane like normal voltage-dependent K+ channels with a cytoplasmic N terminus (Fig. 2C). Injection of HΔN43 cRNA alone did not produce any currents. However, coinjection of cRNA encoding H-S0 restored function (Fig. 2D) with half-activation potentials identical to those of the full-length wild-type channel. Moreover, coinjection of β-subunit cRNA induced the expected shift in the half-activation potentials in oocytes injected with HΔN43 and H-S0 cRNA (data not shown). These results suggest that indeed HΔN43 folds into the membrane in the correct functional orientation, although we cannot exclude the possibility that correct folding of HΔN43 may be dependent on the coexpressed H-S0 fragment.
As expected from the sequence homology and supported by experimental evidence (36) functional MaxiK channels are likely homotetramers. In the case of H-S0 being the region involved in functional tetramerization, as their corresponding counterparts in voltage-dependent K+ channels, we would not have expected to rescue function because this would induce multimerization of H-S0 but not the tetramerization of the pore-forming HΔN43 protein. Therefore, region S0 may be an essential part of the gating machinery of MaxiK channels rather than being involved in tetramerization.
Fusion of a Signal Sequence to the N Terminus Does Not Alter Functional Expression of MaxiK Channels.
To confirm the exoplasmic location of the N terminus in functional MaxiK channels expressed in oocytes, we fused 33 N-terminal amino acids containing a cleavable signal sequence from the rat Na+ channel β1-subunit (24) to the N termini of MaxiK channels and to the noninactivating Shaker K+ channel (ShH4-IR) (37). In the case of an already extracellular N terminus, the addition of a signal sequence would not affect the membrane orientation. If the N terminus is cytoplasmic, as in Shaker K+ channels, fusion of a signal sequence to the N terminus would be expected to result in a reverse orientation of hydrophobic region S1. This should either lead to loss of function or, although rather unlikely, to a reverse orientation§ of the channel in the membrane, which should be easily detected by electrophysiological measurements (Fig. 3D). In both Dslo and the chimeric construct DCHT (see Fig. 5A), the fusion of this signal peptide (clones S-Dslo and S-DCHT; S for signal sequence) to the N terminus resulted in normal functional expression of MaxiK channel activity in Xenopus oocytes (data not shown). Since Hslo and Dslo have more than one Kozak consensus sequence for initiation of translation (7, 20), we used the most downstream translation initiation codon (M4, see Fig. 5B) of Hslo as the fusion partner (S-Hslo-M4). This excludes the remote possibility of an internal ribosome entry (39) that could circumvent the translation of the signal peptide. As observed for S-Dslo and S-DCHT, functional expression of clone S-Hslo-M4 showed no obvious differences when compared with unmodified (wild type) channels in expression levels and electrophysiological properties (Fig. 3 A and B) including the sensitization caused by the β subunit (data not shown). In contrast, for Shaker K+ channels, the fusion of this signal peptide to the N terminus (S-ShH4-IR) resulted in loss of function, presumably due to a folding or trafficking defect (Fig. 3D). A reverse polarity pulse protocol was used to check for inverted channels in the membrane. Removal of the signal peptide from the same clone (ShH4-IR(S-) restored the normal function (Fig. 3E), showing that the loss of function was not due to cloning artifacts.
Figure 3.
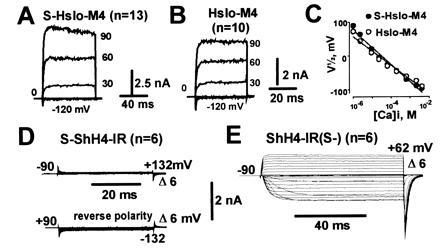
Addition of an N-terminal cleavable signal sequence does not alter functional expression of MaxiK channels. Representative inside-out macropatch currents from oocytes injected with S-Hslo-M4 (A) or Hslo-M4 (B) measured in symmetrical 110 mM K+ in presence of 10 μM Ca2+. (C) Half-activation potentials (V½) as a function of intracellular [Ca2+]. Signal sequence fusion on a Shaker potassium channel (S-ShH4-IR) (D) and after removal of the signal sequence from the same clone (ShH4-IR(S-) (E). Currents in D and E were measured in cell attached mode. Pulses to the indicated potentials were delivered to oocytes bathed in 105 mM potassium–methane sulfonate solution.
These experiments confirm that the extracellular orientation of the N terminus in both Hslo and Dslo, suggested from the in vitro translation experiments, is maintained in functional channels expressed in Xenopus oocytes.
Drosophila MaxiK Channels Are Not Regulated by the Human β Subunit.
Coexpression of Hslo α subunit with the human β subunit dramatically increases the channel open probability at Ca2+ concentrations higher than 100 nM (21). In marked contrast to the 100 mV shift of the half-activation potentials induced by coexpression of the β subunit with Hslo channels above 3 μM Ca2+ (Fig. 4 D and E), the Drosophila homologue [Dslo, splice variant A1/C2/EI/G3/I0 (10)] is unaffected by the coexpression of this mammalian β subunit over a wide range of Ca2+ concentrations (Fig. 4 B and C). Either such a β-subunit regulation is missing in Dslo channels or may require a Drosophila homologue of this β subunit for functional regulation. Such a Drosophila β-subunit homologue has not yet been isolated.
Figure 4.
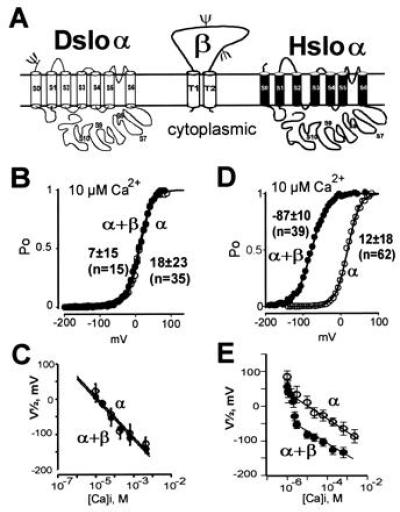
Dslo channels are not regulated by the human β subunit. (A) Proposed membrane topology of Dslo-α, Hslo-α, and the MaxiK channel β subunit. The proposed β-subunit topography (16) was confirmed by in vitro translation experiments showing an integral membrane protein with two N-linked glycosylation sites. (B and D) Open probability (Po) at steady-state current versus membrane potentials obtained in 10 μM intracellular Ca2+. Pulses were delivered from a holding potential of 0 mV in steps of 6 mV from −199 mV to >100 mV. (C and E) Mean V½ values with standard deviations plotted against the intracellular Ca2+ concentration in presence (•) and absence (○) of human β subunit for Dslo (C) and Hslo (E).
N-Terminal Region of Hslo Transfers β-Subunit Regulation to Dslo.
We anticipated that regions necessary for β-subunit regulation may be identified by exchanging regions between Hslo and Dslo. Many of the chimeric constructs (Fig. 5A) showed different apparent Ca2+ sensitivities in comparison with the wild-type channels despite the similar Ca2+ sensitivities of both Hslo and Dslo splice variants used in this study (Fig. 4). Therefore, the effect of the β subunit for each chimeric construct was evaluated by its ability to induce a shift of the voltage activation curve to the left. Because this effect is Ca2+-dependent (Fig. 4E), we performed experiments over a wide range of Ca2+ concentrations. This is especially important for constructs that are insensitive to β-subunit regulation to ensure that their unresponsiveness is not due to a shift in the Ca2+ dependence of the β-subunit effect.
Since MaxiK channels contain a unique C terminus with additional hydrophobic regions, we initially generated chimeras (HCDT, DCHT, and HD1), where we exchanged C-terminal regions. In chimeric constructs HCDT and DCHT, the highly conserved tail regions (containing S9 and S10), which have been proposed to transfer Ca2+ sensitivity (9), were exchanged. The results show (see Fig. 5, HCDT, DCHT, and HD1) that the N terminus (up to S6) but not the long C terminus from Hslo is required for β-subunit regulation.
It has been reported that charybdotoxin, a pore blocker of MaxiK channels, crosslinks to the β subunit (17), suggesting its proximity to the pore region. Therefore, we made a chimeric construct where we exchanged the pore region of Hslo with the homologous Dslo region (HDP) and a construct where the C terminus including S6 is from Dslo (HD2). The functional transfer of the pore region in chimeric construct HDP was confirmed by loss of charybdotoxin sensitivity (7) (data not shown). Surprisingly, both constructs were still modulated by the β subunit as evident from the shift of the voltage activation curve to more negative potentials. These findings also exclude the pore region and adjacent region S6 as being responsible for differences of β-subunit regulation between Hslo and Dslo.
To further circumscribe the region involved in β-subunit modulation, we replaced parts of the unresponsive Dslo with the corresponding Hslo sequences moving toward the N terminus. The sensitization induced by the β subunit was gained in clones containing the exoplasmic N terminus and S0 from Hslo (HD7, HD8) but was not established in chimeras where only the exoplasmic N terminus (HD9) or only S0 (DHD8) were from Hslo. As expected, a reverse chimera with the N terminus and S0 from Dslo and the rest of the protein from Hslo (DH8) was not affected by the β subunit. These results show that both the exoplasmic N terminus and S0 (41 amino acids) of Hslo, are required for transfer of β-subunit regulation from Hslo to Dslo. The amino acids required at the exoplamic N terminus can be limited further because a deletion of the first 10 amino acids in Hslo (Hslo-M4, Fig. 5B) is still regulated by the β subunit (data not shown). Therefore, we conclude that 31 amino acids at the N terminus of Hslo, which includes S0, are critical for β-subunit modulation in MaxiK channels.
Although our data do not show that this region is responsible for β-subunit binding, it is a plausible candidate. The exoplasmic N terminus and the transmembrane localization of S0 would provide sufficient surface for interaction with the β subunit. In the Kv1 family of voltage-dependent K+ channels, the corresponding cytoplasmic region was recently shown to bind the cytoplamic Kvβ1-subunit (40, 41).
From experiments expressing Hslo and Dslo “core” and “tail” regions as separate domains, it has been proposed that the highly conserved tail region of MaxiK channels exchanged the apparent Ca2+ sensitivity between a Dslo splice variant with low Ca2+ sensitivity and a mammalian highly sensitive clone (9). Although we used Hslo and Dslo splice variants with similar apparent Ca2+ sensitivities (similar half-activation potentials, V½, see Fig. 4), we found similar to Wei et al. (9) that the tail region of Dslo made Hslo less Ca2+ sensitive [V½ = 95 mV for HCDT vs. V½ = 12 mV in Hslo(α) in 10 μM Ca2+, see Figs. 5A and 4D]. These results indicate that the modification in Ca2+ sensitivities observed with the exchange of tail regions cannot be interpreted as a transfer of Ca2+ sensitivity. Additional evidence supporting this view is as follows: (i) the Dslo splice variant used in this study and the variant used by Wei et al. (9) are identical in the tail region; (ii) the reported differences in Ca2+ sensitivities of Dslo and Hslo are due to splice variations in the core region (10, 11); and (iii) most of our chimeric constructs where regions other than the tail regions were exchanged, differed in their apparent Ca2+ sensitivities when compared with the wild-type (see Figs. 5 and 4D). Thus, our findings stress the importance of the whole protein in determining the apparent Ca2+ sensitivity of MaxiK potassium channels.
Acknowledgments
We thank Dr. J. P. Adelman for kindly providing Dslo; Drs. E. Stefani, R. Latorre, and E. Wanker for helpful discussions; J. Yuguang for oocyte injections; and D. Grenet for oligonucleotide synthesis. L.T. is an Established Investigator of the American Heart Association. This work was supported by National Institutes of Health Grant HL54970 to L.T.
Footnotes
Abbreviations: MaxiK, high-conductance voltage-activated and Ca2+-sensitive K+ channel; Hslo, human MaxiK channel; Dslo, Drosophila MaxiK channel; PNGase F, N-glycosidase F.
P. Meera, M. Wallner, & L. Toro, 40th Annual Meeting of the Biophysical Society, Feb. 17–21, 1996, Baltimore, MD/A13.
A simple model for folding of polytopic eukaryotic membrane proteins suggested that the orientation might be determined only by the orientation of the first transmembrane region (38).
References
- 1.Nelson M T, Cheng M R, Santana L F, Bonev A D, Knot H J, Lederer W J. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 2.Gola M, Crest M. Neuron. 1993;10:689–699. doi: 10.1016/0896-6273(93)90170-v. [DOI] [PubMed] [Google Scholar]
- 3.Robitaille R, Garcia M L, Kaczorowski G J, Charleton M P. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- 4.Latorre R, Oberhauser A, Labarca P, Alvarez O. Annu Rev Physiol. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- 5.Jan L Y, Jan Y N. Cell. 1992;69:715–718. doi: 10.1016/0092-8674(92)90280-p. [DOI] [PubMed] [Google Scholar]
- 6.Atkinson N S, Robertson G A, Ganetzky B. Science. 1991;253:551–555. doi: 10.1126/science.1857984. [DOI] [PubMed] [Google Scholar]
- 7.Adelman J P, Shen K, Kavanaugh M P, Warren R A, Wu Y, Lagrutta A, Bond C T, North R A. Neuron. 1992;9:209–216. doi: 10.1016/0896-6273(92)90160-f. [DOI] [PubMed] [Google Scholar]
- 8.Butler A, Tsunoda S, McCobb D P, Wei A, Salkoff L. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- 9.Wei A, Solaro C, Lingle C, Salkoff L. Neuron. 1994;13:671–681. doi: 10.1016/0896-6273(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 10.Lagrutta A, Shen K, North R A, Adelman J P. J Biol Chem. 1994;269:20347–20351. [PubMed] [Google Scholar]
- 11.Tseng-Crank J, Foster C D, Krause J D, Mertz R, Godinot N, DiChiara T J, Reinhart P H. Neuron. 1994;13:1315–1330. doi: 10.1016/0896-6273(94)90418-9. [DOI] [PubMed] [Google Scholar]
- 12.Yang N, George A L, Horn R. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- 13.Mannuzzu L M, Moronne M M, Isacoff E Y. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- 14.Papazian D M, Shao X M, Seoh S A, Mock A F, Huang Y, Wainstock D H. Neuron. 1995;14:1239–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Calvo M, Knaus H G, McManus O G, Giangiacomo K M, Kaczorowski G J, Garcia M L. J Biol Chem. 1994;269:676–682. [PubMed] [Google Scholar]
- 16.Knaus H G, Folander K, Garcia-Calvo M, Garcia M L, Kaczorowski G J, Smith M, Swanson R. J Biol Chem. 1994;269:17274–17278. [PubMed] [Google Scholar]
- 17.Knaus H G, Eberhart A, Kaczorowski G J, Garcia M L. J Biol Chem. 1994;269:23336–23341. [PubMed] [Google Scholar]
- 18.McManus O B, Helms L M, Pallanck L, Ganetzky B, Swanson R, Leonard R J. Neuron. 1995;14:645–650. doi: 10.1016/0896-6273(95)90321-6. [DOI] [PubMed] [Google Scholar]
- 19.McCobb D P, Fowler N L, Featherstone T, Lingle C, Saito M, Krause J E, Salkoff L. Am J Physiol. 1995;269:H767–H777. doi: 10.1152/ajpheart.1995.269.3.H767. [DOI] [PubMed] [Google Scholar]
- 20.Wallner M, Meera P, Ottolia M, Kaczorowski G J, Latorre R, Garcia M L, Stefani E, Toro L. Receptors Channels. 1995;3:185–199. [PubMed] [Google Scholar]
- 21.Meera P, Wallner M, Jiang Z, Toro L. FEBS Lett. 1996;382:84–88. doi: 10.1016/0014-5793(96)00151-2. [DOI] [PubMed] [Google Scholar]
- 22.Devereux J, Haeberli P, Smithies O. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallner M, Weigl L, Meera P, Lotan I. FEBS Lett. 1993;336:535–539. doi: 10.1016/0014-5793(93)80871-q. [DOI] [PubMed] [Google Scholar]
- 24.Isom L L, DeJongh K S, Patton D E, Reber B F X, Offord J, Charbonneau H, Walsh K, Goldin A L, Catterall W A. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- 25.Horton R M, Hunt H D, Ho S N, Pullen J K, Pease L R. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 26.Kyte J, Doolittle R F. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 27.Planells-Cases R, Ferrer-Montiel A V, Patten C D, Montal M. Proc Natl Acad Sci USA. 1995;92:9422–9426. doi: 10.1073/pnas.92.20.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seoh S, Sigg D, Papazian D M, Bezanilla F. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 29.Pallanck L, Ganetzky B. Hum Mol Genet. 1994;3:1239–1243. doi: 10.1093/hmg/3.8.1239. [DOI] [PubMed] [Google Scholar]
- 30.Dworetzky S I, Trojnacki J T, Gribkoff V K. Mol Brain Res. 1994;27:189–193. doi: 10.1016/0169-328x(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 31.Shen V, Pfaffinger P. Neuron. 1995;14:625–633. doi: 10.1016/0896-6273(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Yu W, Jan Y N, Jan L, Li M. J Biol Chem. 1995;270:24761–24768. doi: 10.1074/jbc.270.42.24761. [DOI] [PubMed] [Google Scholar]
- 33.Fujiki Y, Hubbard A L, Fowler S, Lazarow P B. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurnett C A, De Waard M, Campbell K P. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 35.Andrews D W, Young J C, Mirels L F, Czarnota G J. J Biol Chem. 1992;267:7761–7769. [PubMed] [Google Scholar]
- 36.Shen K Z, Lagrutta A, Davies N W, Standen N B, Adelman J P, North R A. Pflügers Arch Eur J Physiol. 1994;426:440–445. doi: 10.1007/BF00388308. [DOI] [PubMed] [Google Scholar]
- 37.Hoshi T, Zagotta W N, Aldrich R W. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 38.Sipos L, Von Heijne G. Eur J Biochem. 1993;213:1333–1340. doi: 10.1111/j.1432-1033.1993.tb17885.x. [DOI] [PubMed] [Google Scholar]
- 39.Chen C Y, Sarnow P. Science. 1995;268:415–417. doi: 10.1126/science.7536344. [DOI] [PubMed] [Google Scholar]
- 40.Sewing S, Roeper J, Pongs O. Neuron. 1996;16:455–463. doi: 10.1016/s0896-6273(00)80063-x. [DOI] [PubMed] [Google Scholar]
- 41.Yu W, Xu J, Li M. Neuron. 1996;16:441–453. doi: 10.1016/s0896-6273(00)80062-8. [DOI] [PubMed] [Google Scholar]