

Abstract
E-cadherin is a transmembrane protein that mediates Ca2+-dependent cell-cell adhesion. To study cadherin-cadherin interactions that may underlie the adhesive process, a recombinant E-cadherin lacking free sulfhydryl groups and its mutants with novel cysteines were expressed in epithelial A-431 cells. These cysteine mutants, designed according to various structural models of cadherin dimers, were constructed to reveal cadherin dimerization by the bifunctional sulfhydryl-specific cross-linker BM[PE0]3. Cross-linking experiments with the mutants containing a cysteine at strand B of their EC1 domains did show cadherin dimerization. By their properties these dimers correspond to those which have been characterized by coimmunoprecipitation assay. Under standard culture conditions the adhesive dimer is a dominant form. Calcium depletion dissociates adhesive dimers and promotes the formation of lateral dimers. Our data show that both dimers are mediated by the amino-terminal cadherin domain. Furthermore, the interfaces involved in both adhesive and lateral dimerization appear to be the same. The coexistence of the structurally identical adhesive and lateral dimers suggests some flexibility of the extracellular cadherin region.
Classic cadherins are transmembrane adhesive receptors that mediate Ca2+-dependent cell-cell adhesion in different types of cells. On the extracellular side of the plasma membrane, cadherins interact with one another, forming complexes that establish direct contacts between opposing cells. The intracellular cadherin portion, through interactions with catenins, anchors these adhesive complexes to the cortical cytoskeleton (4, 9, 23). Although these Ca2+-dependent intercellular adhesive structures are very important for various normal and abnormal morphogenetic events (10, 28, 30, 32, 33), the molecular processes underlying their assembly remain poorly understood.
Coimmunoprecipitation experiments have revealed two types of E-cadherin homodimers potentially important for cadherin-based adhesion (8, 20, 24). In these dimers cadherin molecules align in a lateral (e.g., they both belong to the same cell) or adhesive orientation. It is possible that these dimers represent detergent-resistant portions of larger multimeric cadherin complexes. Nevertheless, a number of observations suggest that the adhesive dimers are the simplest structural units of cadherin-mediated adhesion. Our previous data showed that under standard culture conditions the adhesive dimers appear to be a dominant form (14). The adhesive dimers immediately dissociate, however, and the lateral dimers become prevalent after the calcium concentration drops below 100 μM. This change is accompanied by the complete disruption of adherens junctions. An E-cadherin lacking either calcium-binding sites or its intracellular catenin-binding region can establish neither cell-cell adhesion nor adhesive dimers. In contrast, the E-cadherin point mutation D155A simultaneously increases both the amount of adhesive dimers and the recruitment of the E-cadherin into junctional sites (16). Finally, the amino-terminal domain of E-cadherin determines the specificity of both cell-cell adhesion and cadherin dimerization (13). Whether lateral dimers have any function in cell-cell adhesion is not so clear. Although adhesive and lateral dimers are remarkably stable in cell lysates, they have been suggested to be very dynamic in living cells. We have proposed that the continuous formation of short-lived adhesive dimers within cell-cell junctions is a basic mechanism of cadherin-mediated adhesion (14). To further evaluate this hypothesis, it is necessary to characterize in detail the binding sites involved in adhesive and lateral cadherin dimerization.
The classic cadherin extracellular region consists of five EC domains (numbered from the outermost N-terminal domain). Successive EC domains, each of which is folded into a seven-stranded (A to G) β-sandwich (5, 18, 25) are interconnected by three calcium ions. Structural studies suggest several alternative models for cadherin dimers. According to the “strand dimer” model, cadherin adhesive dimerization is driven by the reciprocal insertion of the EC1 domain residue Trp156 (amino acid numbering as in reference 6) located at the β-strand A into the hydrophobic pocket of the paired EC1 domain (5, 25). Controversy remains, however, regarding whether such Trp156-dependent strand dimer interaction does occur on the cell surface or if it is caused solely by the crystallization process (discussed in references 11, 15, and 17). Point mutagenesis experiments did show that the Trp156 residue is indispensable for adhesive dimerization (8), as well as for the adhesive activity of classic cadherins (29). One can argue, however, that the Trp156 mutation induces some conformational abnormalities in the EC1 domain and/or affects lateral cadherin dimerization, thereby precluding E-cadherin from adhesive dimerization which is mediated by a distinct mechanism. At least three alternative models of intercadherin interactions which, in theory, may produce lateral and/or adhesive dimers detected in coimmunoprecipitation assay are suggested. These models include: (i) cadherin dimerization via the “adhesive” interface of the EC1 domain containing His233 and Val235 residues (25); (ii) cadherin dimerization via EC1/EC2 calcium-binding sites (18, 21); and (iii) interactions between antiparallel cadherin molecules along their full length (7, 27). This uncertainty in the structure of adhesive and lateral dimers precludes an understanding of cadherin-based adhesion.
In the present study a new assay, a cysteine scanning mutagenesis of E-cadherin in conjunction with cysteine-specific cross-linking, was applied to study cadherin dimerization on the surface of epithelial A-431 cells. These experiments revealed adhesive and lateral cadherin dimers with features very similar to those obtained previously for cadherin dimers in a coimmunoprecipitation assay. Notably, this new approach confirmed that under standard culture conditions adhesive dimers are a dominant form. Furthermore, our experiments unexpectedly indicated that a single interface mediates both adhesive and lateral dimerization. Analysis of the cross-linking efficiency of various cysteine mutants suggested that this interface represents the strand dimer interface described elsewhere (5, 25). The coexistence of the structurally identical adhesive and lateral dimers on the cell surface suggests high flexibility of the extracellular cadherin region.
MATERIALS AND METHODS
The plasmids coding for the E-cadherin tagged either by myc (Ec1M) or by flag (Ec1F) epitopes and lacking the epitope for C20820 MAb were described (8). All mutants were constructed by using site-directed mutagenesis in the expression vector pRcCMV (Invitrogen). To design cysteine mutants suitable for site-specific cross-linking, the models of the N-cadherin EC1 domain strand dimer (Protein Data Bank ID code 1NCI), the N-cadherin EC1 adhesion dimer (code 1NCH), and the E-cadherin EC12 calcium dimer (codes 1EDH and 1FF5) were used. Molecular structures were analyzed by using RasMol2 and Cn3D4.1 programs. Correct plasmid construction was verified by endonuclease mapping and nucleotide sequencing.
Transfection, growth, and immunofluorescence microscopy of human A-431 cells were done as described previously (8). The following antibodies were used: anti-E-cadherin,clones HECD-1 (Zymed Laboratories) and C20820 (Transduction Laboratories); anti-myc, clone 9E10 (provided by R. Kopan, Washington University Medical School, St. Louis, Mo.); and anti-flag M2 (Sigma).
The immunoprecipitation assay was done as described previously (8). In brief, the confluent monolayer was washed and extracted at 4°C with 1.5 ml of immunoprecipitation lysis buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5 mM AEBSF [4-(2-aminoethyl)-benzenesulfonyl fluoride], 2 mM EDTA, 1% NP-40). NP-40-insoluble material was removed by centrifugation at 100,000 × g for 1 h. The lysates were subjected to immunoprecipitation by subsequent incubations with anti-myc antibody and protein A-Sepharose. Resulting precipitates were either boiled directly in sodium dodecyl sulfate (SDS)-gel sample buffer or cross-linked (see below).
The homobifunctional chemical cross-linker BM[PEO]3 with a spacer arm length of 1.47 nm (Pierce) was used for cell surface labeling. Confluent cultures were washed with phosphate-buffered saline containing 0.5 mM CaCl2 (PBS-C). Each plate was then incubated for 10 min at room temperature in PBS-C containing a 1 mM concentration of cross-linker. The reaction was stopped by washing the cells with PBS containing 10 mM dithiothreitol. Surface-cross-linked and control cells were either solubilized directly in the SDS-gel sample buffer or subjected to immunoprecipitation. For immunoprecipitation, cells were extracted in IP-SDS buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.5 mM AEBSF, 2 mM EDTA, 1% NP-40, 0.2% SDS) after cross-linking and then immunoprecipitated as described above. Aliquots of the total cell extracts or immunoprecipitates were separated by SDS-5% polyacrylamide gel electrophoresis and then analyzed by immunoblotting as described previously (8).
To cross-link proteins in the anti-myc immunoprecipitates, the protein A-beads were incubated for 5 min at room temperature after the final wash in PBS-T (PBS, 0.5% Triton X-100) with 100 μl of 1 mM BM[PEO]3 (in PBS-T). The reaction was stopped by adding an equal volume of PBS-T containing 10 mM dithiothreitol. The beads were then collected by centrifugation and boiled in an SDS-gel sample buffer.
In some experiments cell surface proteins were biotinylated with EZ-link PEO-Maleimide Activated Biotin (Pierce). Cells were washed two times with PBS-C and then incubated for 5 min with a biotinylation reagent solubilized immediately before use in the same buffer. Cells were then immunoprecipitated with anti-myc antibody as described above. Biotinylated proteins were visualized with streptavidin-horseradish peroxidase conjugate.
RESULTS
Adhesive and lateral Trp156-dependent E-cadherin dimers can be cross-linked via the EC1 domain on the surface of A-431 cells.
Previously, we showed that myc-tagged E-cadherin (Ec1M) forms adhesive and lateral homodimers in epithelial A-431 cells. Adhesive dimers are evident from the coimmunoprecipitation of flag-tagged E-cadherin (Ec1F) with an anti-myc antibody when cell lysates are obtained from a mixed culture of Ec1M- and Ec1F-expressing cells. The presence of lateral dimers can be demonstrated by the coimmunoprecipitation of endogenous cadherin with Ec1M after the complete disruption of adhesive dimers by calcium chelators (8). Since both types of dimers depend on the same set of EC1 residues, including Trp156 (16), we proposed that at least one of these dimers corresponds to the strand dimer suggested by Shapiro et al. (25). To confirm this hypothesis, the EC1 domain within Ec1M was modified so that E-cadherin molecules can be chemically cross-linked within the strand dimer by the cysteine-specific homobifunctional cross-linker BM[PE0]3 (Fig. 1A).
FIG. 1.

Strategy of cysteine-scanning mutagenesis. (A) Backbone structure of N-cadherin EC1 domain strand dimer (25). A view is given from carboxyl termini (arrows). Strands A of the paired molecules are colored red. The side chains of residues exposed in the dimer interface corresponding to E-cadherin Leu175 (yellow), Val176 (orange), Gln177 (green), and Lys179 (blue) are shown. (B) A-431 cells stably producing Ec1M (lane Ec1M), Ec1M-C163A (lane 163), Ec1M-C163A/L175C (lane 175), Ec1M-C163A/V176C (lane 176), Ec1M-C163A/Q177C (lane 177), and Ec1M-C163A/K179C (lane 179) were cocultured with Ec1F-expressing cells and then were immunoprecipitated by an anti-myc antibody. Immunoprecipitates were analyzed for the presence of either myc-tagged mutants (Myc), coimmunoprecipitated endogenous E-cadherin (Ec) derived from both lateral and adhesive dimers, or Ec1F (Flag) derived from adhesive dimers only. (C) A-431 cells expressing Ec1M mutants as in panel B were cross-linked by BM[PEO]3, and their total lysates were analyzed by Western blotting with anti-myc. Arrows indicate two cross-linked products of 220 and 280 kDa. The arrowhead indicates the monomeric form.
E-cadherin contains five cysteine residues. Four of them, present at the EC5 domain, form two disulfide bonds (5, 19). These cysteines were not biotinylated by the sulfhydryl-reactive reagent EZ-link PEO-maleimide activated biotin, which has the same reactive group as BM[PEO]3 (data not shown). The single unpaired cysteine (Cys163) which is present at the A strand of the EC1 domain also was not labeled by that same reagent, apparently because this residue is hidden inside an EC1 hydrophobic core (18). However, Cys163 becomes accessible for biotinylation after the depletion of Ca2+ ions (not shown). To eliminate the possible interference of Cys163 with our cross-linking assay, this residue was converted to Ala, and the resulting mutant (Ec1M-C163A) was examined for subcellular distribution (not shown) and the ability to produce adhesive dimers with Ec1F in our mixed-culture coimmunoprecipitation assay (Fig. 1B). No differences from Ec1M were found in either parameter. Furthermore, this mutant, in contrast to Ec1M, was not biotinylated at both the standard and low calcium levels (not shown). These experiments showed that the Ec1M-C163A mutant is suitable for cysteine-scanning mutagenesis.
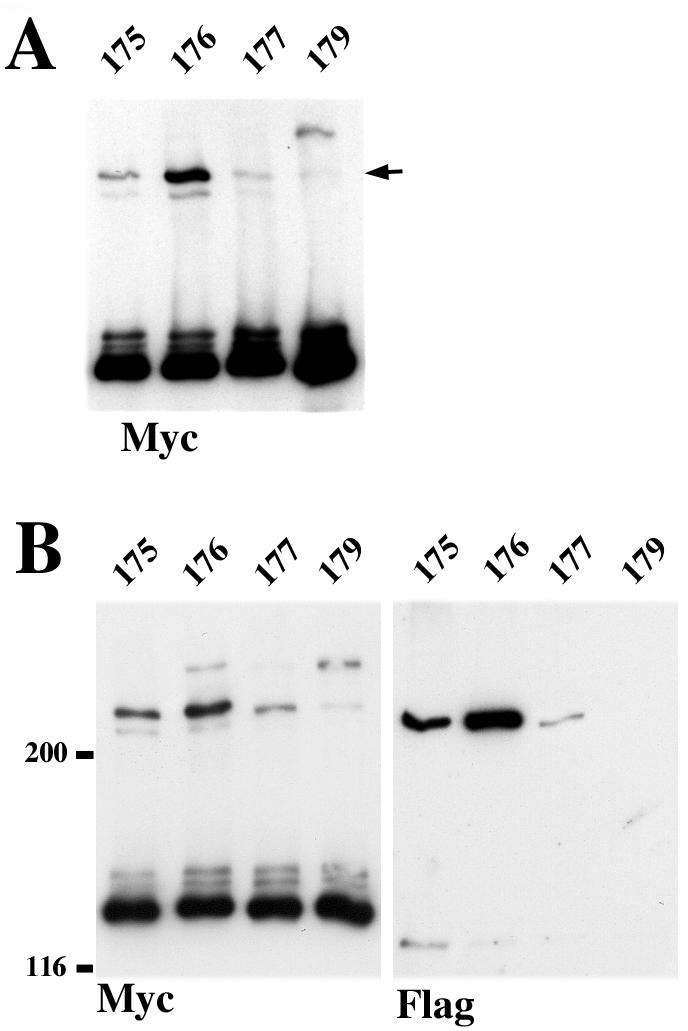
In the next step, the residues Leu175, Val176, Gln177, and Lys179, located at EC1 strand B and which protrude between two paired EC1 domains in the strand dimer interface model (25) (Fig. 1A), were individually substituted for cysteine. The resulting Ec1M mutants were stably expressed in A-431 cells and analyzed as described for Ec1M-C163A. Again, no abnormalities either in the subcellular distribution of the mutants or in the amounts of the adhesive dimers they formed (Fig. 1B) were found. When the corresponding cells were exposed to BM[PEO]3, all of these mutants, in contrast to Ec1M and Ec1M-C163A, formed a cross-linked product with a molecular mass of ∼220 kDa (Fig. 1C). The mutant Ec1M-C163A/K179C also produced a unique 280-kDa complex. The nature of this complex remains unknown.
The mutant Ec1M-C163A/V176C was studied in detail because it produced the highest level of the 220-kDa product. Figure 2A shows that this product forms equally well at both normal and low calcium concentrations, suggesting that all of its components may belong to the same cell. Inactivation of the EC1/EC2 calcium-binding sites by the additional mutation E165A did not abolish this product formation (Fig. 2A, lane E165A). In contrast, point mutation W156A completely inactivated this reaction (lane W156A). These experiments suggested that the 220-kDa product may correspond to the Trp156-dependent lateral dimer, which has been detected by coimmunoprecipitation assay and which has exactly the same features (8).
FIG. 2.
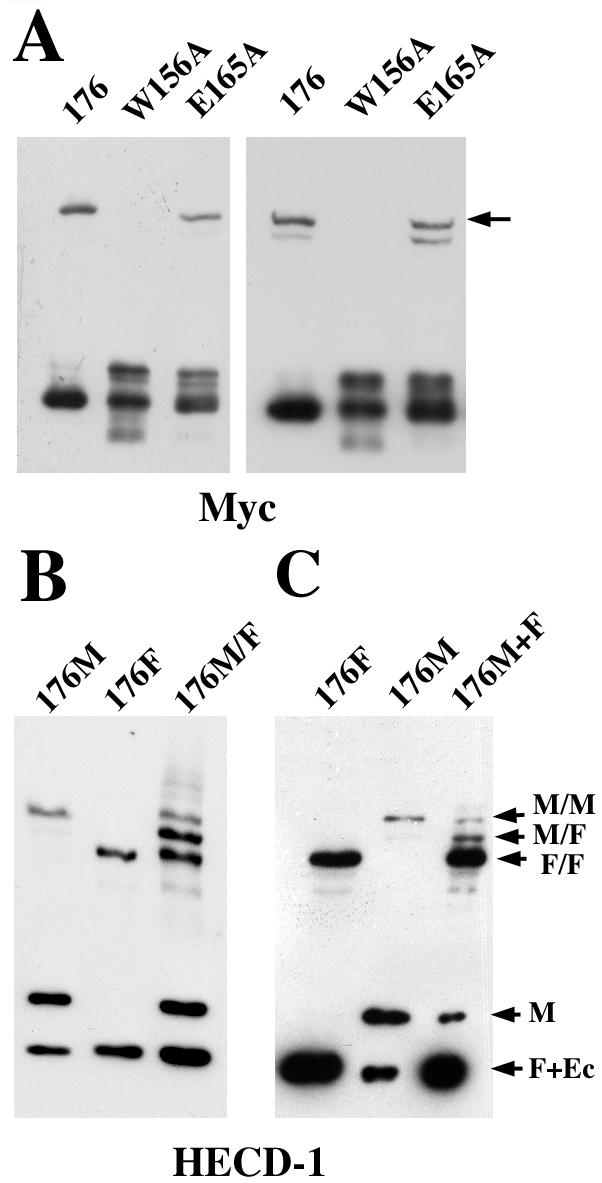
A 220-kDa product represents adhesive and lateral dimers. (A) A-431 cells expressing Ec1M-C163A/V176C (lane 176) or its point mutants Ec1M-C163A/V176C/W156A (lane W156A) or Ec1M-C163A/V176C/E165A (lane E165A) were cross-linked at 2 mM (left panel) or 50 μM (right panel) calcium by BM[PEO]3 and analyzed as in Fig. 1C. In the latter case, cells were preincubated in 50 μM calcium for 10 min at 37°C to ensure that they dissociated intercellular contacts. Note that the point mutation W156A completely abolished the formation of the 220-kDa product (arrow), whereas inactivation of the presumable EC1/EC2 calcium-binding sites by E165A mutation did not significantly change the amount of the product. The second band appearing in low calcium represents a cross-linked product between myc-tagged and endogenous E-cadherin. (B) BM[PEO]3 cross-linking assay with A-431 cells expressing only Ec1M-C163A/V176C (lane 176M), only Ec1F-C163A/V176C (lane 176F), or coexpressing both these mutants (lane 176M/F). The blot was stained with HECD-1 antibody, which equally recognizes all forms of E-cadherin. (C) Ec1F-C163A/V176C- and Ec1M-C163A/V176C-expressing cells were cross-linked either separately (lanes 176F and 176M, respectively) or in coculture (lane 176M+F). The ratio between flag- and myc-tagged cells was 4:1. Arrows: M/M, cross-linked dimer in which both E-cadherin molecules are tagged by myc; M/F, myc- and flag-tagged dimer; F/F, dimer with two flag-tagged molecules; M, myc-tagged monomer; F+Ec, the band containing both flag-tagged monomer and endogenous E-cadherin. Note that the M/F product in panel C represents exclusively adhesive dimers.
To confirm that the 220-kDa product is an E-cadherin dimer, the flag-tagged version of the Ec1M-C163A/V176C mutant was expressed either in wild-type A-431 cells (resulting V176-Flag cells) or in A-431 cells expressing Ec1M-C163A/V176C (V176-Myc cells). A cross-linking assay was performed with a homogeneous culture of double-transfected cells (Fig. 2B) or with a coculture of V176-Myc and V176-Flag cells (Fig. 2C). To maximize the rate of V176-Myc/V176-Flag interacting cells, the ratio between these cells in the coculture was 1 to 4. Since the myc- and flag-tagged forms of E-cadherin can be easily distinguished by their molecular masses, we expected that the hybrid dimer, if it forms, would have an intermediate size between only myc- and only flag-tagged dimers. The analysis of cross-linked products with anti-E-cadherin antibody HECD-1 showed that such an intermediate-size product was efficiently formed in both cultures (arrow M/F in Fig. 2B and C). Since the 220-kDa product was also found in the double-transfected cells after cell-cell contact dissociation by calcium depletion, these data showed that this product represents both adhesive and lateral dimers. Furthermore, since the amount of the hybrid (flag-/myc-tagged) dimers in the coculture exceeds the amount of only myc-tagged dimers, the adhesive dimer is a dominant form under standard culture conditions.
Adhesive and lateral dimers have similar structures.
We next studied whether adhesive and lateral dimers revealed by cross-linking experiments have similar structures. For this purpose, we compared the relative amounts of cross-linked lateral and adhesive dimers in cells producing various Cys mutants. Figure 3A (compare with Fig. 1C) shows that after calcium depletion, each mutant produced the same amount of dimers as in a standard culture. It indicated that the structure of lateral dimers cross-linked at low calcium and that of dimers present in standard conditions are very similar. To reveal exclusively adhesive dimers, the cells expressing a myc-tagged mutant were cocultured with the cells expressing the same mutant tagged with the flag epitope. Anti-myc immunoprecipitation and anti-flag analysis of the immunoprecipitates (Fig. 3B) showed that the relative amount of cross-linked adhesive dimers matched that of lateral dimers. These data suggested that the same binding site is responsible for the formation of lateral dimers in low calcium and adhesive dimers in standard conditions.
FIG. 3.
(A) BM[PEO]3 cross-linking assay with different B-strand cysteine mutants (indicated as in Fig. 1C) performed at 50 μM Ca2+. Note that the relative amounts of the cross-linked 220-kDa product (arrow) are the same as under the standard calcium concentration shown in Fig. 1C. (B) Cells expressing different myc-tagged cysteine mutants (indicated above the lane as in panel A) were cocultured with the cells expressing the same mutants but tagged by flag. The ratio between flag- and myc-tagged cells was 1:1. Cocultures were immunoprecipitated with anti-myc antibody and analyzed for myc (Myc) or for flag (Flag) epitopes. Note that the relative amounts of the cross-linked adhesive dimers revealed by anti-flag staining are the same as those of cross-linked lateral dimers shown in panel A.
To further clarify the structure of cross-linked dimers, a new set of Ec1F-C163A mutants was constructed. In these mutants the surface residues located at the EC1 strand G were individually replaced for cysteine (E247C, L249C, T251C, and T253C mutations). According to the strand dimer interface model, strands B and G of the paired EC1 domains are located in close proximity on the same side of the dimer (Fig. 4A). The distance between each pair of opposite residues (namely, Lys179-Glu247, Gln177-Leu249, Val176-Thr251, and Leu175-Thr253) is ca. 12 Å, which is close to the size of the BM[PEO]3 cross-linker (15 Å). To determine the pairs of the strand B/strand G residues in the adhesive dimer that are most favorable for cross-linking, an assay was performed with cocultures of cells expressing myc-tagged strand B cysteine mutants and cells expressing flag-tagged strand G mutants. This experiment showed that the most favorable pairs of mutants are those where two cysteines are located one in front of the other according to the strand dimer interface model (excepting only the pair Leu175-Thr253; Fig. 4B).
FIG. 4.

(A) Side view of N-cadherin EC1 domain strand dimer according to a previous study (25). The A strands of the paired molecules are red. The residues of the B (green or blue) and G (yellow or orange) strands that were subjected to mutagenesis are numbered. Note that all residues are on the same side of the dimer. In the experiment shown in panel B, cells expressing myc-tagged E-cadherin with cysteine in the B strand (vertical column) were cocultured with flag-tagged E-cadherin containing cysteine in the G strand. Cells were immunoprecipitated with anti-myc antibody, and the resulting immunoprecipitates were analyzed for myc- and flag-tagged proteins. Only anti-flag-reactive bands corresponding to cross-linked adhesive dimers are shown.
Only the strand dimer model is consistent with the site-specific cross-linking assay.
The cross-linking analysis described above showed that the interaction between two molecules in the E-cadherin dimer occurs along the β-strand A. To further clarify the structure of this dimer and to reveal alternative interactions, several additional Ec1M mutants were constructed and expressed in A-431 cells. Three mutants (Ec1 M-C163A/T199C, Ec1M-C163A-T229C, and Ec1M-C163A/F231C) were designed to reveal cadherin dimers mediated by the “adhesive” interface described by Shapiro et al. (25). The dimerization of these mutants according to this model would establish pairs of closely located cysteines (not shown). Coimmunoprecipitation assay confirmed that new cysteines did not prevent these mutants from homodimerization (not shown). However, no BM[PEO]3-cross-linked myc-positive products were detected in cells expressing either of these mutants (Fig. 5B). This result, in agreement with the previous alanine-scanning mutagenesis (12, 16, 26), showed that the “adhesive” interface described earlier (25) is not engaged in cadherin-cadherin interactions. In addition, this experiment demonstrated the high specificity of the site-specific cross-linking approach: efficient cross-linking occurs only when cysteines are located at very specific positions of the EC1 domain.
FIG. 5.
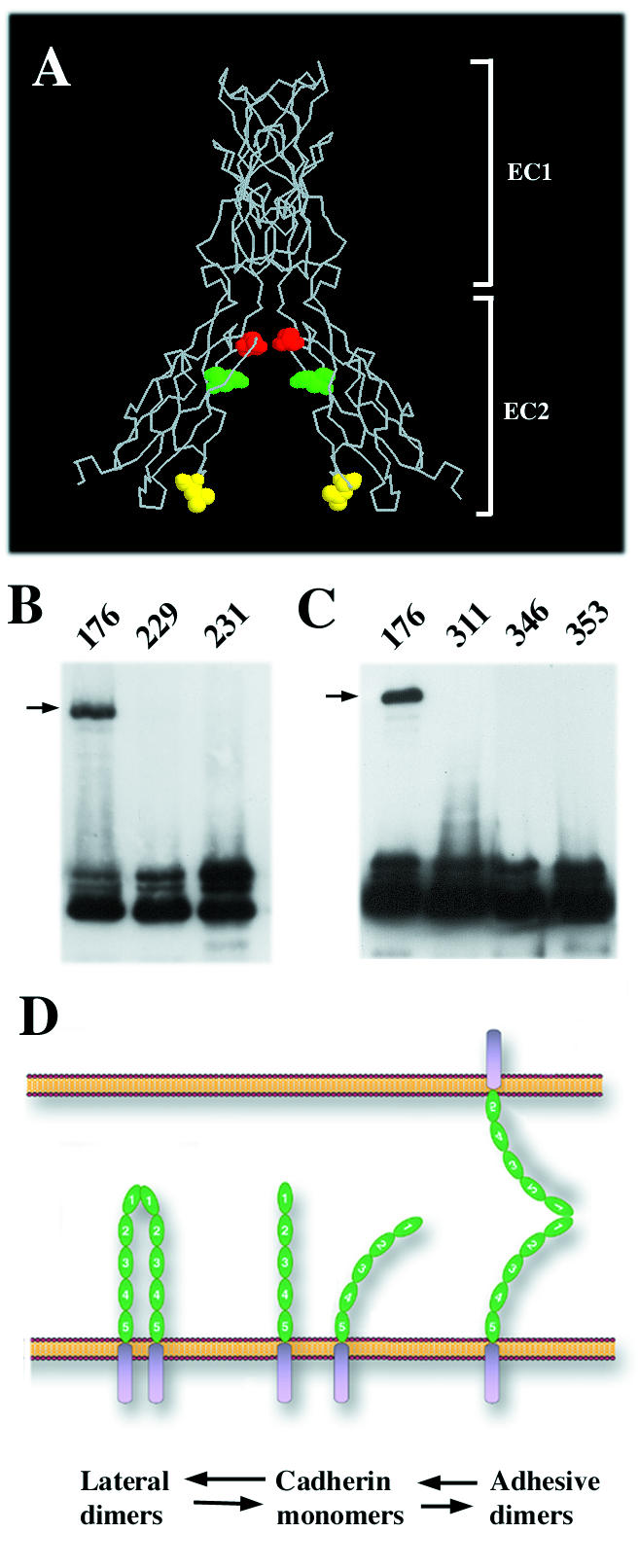
(A) Backbone structure of calcium site E-cadherin dimer (side view) according to an earlier study (21). The side chains of the EC2 domain residues Glu353 (red), Gln346 (green), and Leu311 (yellow) exposed in the dimer interface and subjected to cysteine replacement are shown. (B) A-431 cells stably producing Ec1M-C163A/V176C (lane 176), Ec1M-C163A-T229C (lane 229), and Ec1M-C163A/F231C (lane 231) were cross-linked by BM[PEO]3, and the total lysates were analyzed by Western blotting with anti-myc. (C) Ec1M mutants Ec1M-C163A/L311C (lane 311), Ec1M-C163A-Q346C (lane 346), and Ec1M-C163A/E353C (lane 353) containing cysteine mutations in the EC2 domain were analyzed as in panel B. Arrows indicate the 220-kDa cross-linked dimer. Note that cysteines in the mutants designed according to the calcium site model were unable to facilitate a cross-linking reaction. (D) The model shows formation of the adhesive and lateral dimers using the same binding site. According to this model, the extracellular cadherin region (green; EC domains are numbered) is flexible and may present a single dimerization site located at the EC1 domain in a conformation favorable either for lateral or adhesive dimerization.
A second group of cysteine mutants was constructed to identify the lateral dimer established by the calcium-binding site (calcium site dimer). This dimer was evident in the crystals produced by EC1/EC2 fragments of E-cadherin (18, 21). Similar calcium-dependent dimerization of the E-cadherin EC1/EC2 fragment was revealed in solution (11). Importantly, this type of lateral dimerization also proceeds along the axis of the EC1 strand A and, therefore, the cysteines located at the β-strands B or G may equally facilitate a cross-linking reaction in both strand and calcium site dimers. To distinguish calcium site dimers, the EC2 residues Leu311, Gln346, and Glu353 were separately replaced for cysteine, and the resulting mutants were expressed in A-431 cells as described above. Figure 5A shows that these cysteines would mediate E-cadherin cross-linking if E-cadherin dimerized according to the calcium site model. However, neither of these mutations enabled us to cross-link cadherin dimers (Fig. 5C).
As an alternative attempt to reveal a function of the Ca2+-binding interface, we constructed two additional mutants: Ec1M-Q255A and Ec1M-N297A. According to the calcium site model, the backbone carbonyl oxygen atoms of these residues coordinate calcium ions, while their side chains participate in dimerization. Therefore, we proposed that these point mutations would not change calcium binding but may have a dramatic effect on dimer formation. Expression of these mutants in A-431 cells was unable to reveal any specific effect of these mutations on the property of E-cadherin or cellular phenotype (not shown).
Identity of the dimers revealed by coimmunoprecipitation and site-specific cross-linking assays.
The E-cadherin dimerization detected by two alternative approaches—the BM[PEO]3 cross-linking of the living cells and the coimmunoprecipitation of myc/flag-tagged cadherins from cell lysates—shares similar features. In both cases dimerization is Trp156 dependent. Under standard culture conditions adhesive dimerization prevails over lateral dimerization, but the latter dimerization mode becomes dominant after calcium depletion. These similarities suggest that these two different approaches in fact detect the same cadherin-cadherin interaction. To further clarify this important issue, we studied whether E-cadherin cysteine mutants can be cross-linked by BM[PEO]3 in the anti-myc immunoprecipitates. Figure 6A shows that cross-linking of such an immunoprecipitate obtained from Ec1M- C163A/V176C-expressing cells results in formation of the same 220-kDa cross-linking product. Importantly, the cross-linking of this mutant in the immunoprecipitate and on the cell surface produced exactly the same (relative to the monomeric form) amounts of the 220-kDa product. As expected, the formation of the 220-kDa product was completely abolished after point mutation W156A (Fig. 6B). A high specificity of dimer cross-linking in vitro was further indicated by the fact that the mutants Ec1M-C163A/T229C and Ec1M-C163A/F231C cannot be cross-linked either on the cell surface (Fig. 5B) or in the immunoprecipitates (Fig. 6B). Taken together, these experiments demonstrated that E-cadherin dimers which can be cross-linked on the cell surface are stable during cell solubilization and in vitro manipulations.
FIG. 6.

Cross-linking of different cysteine Ec1M mutants in the anti-myc immunoprecipitates. (A) A-431 cells expressing the Ec1M-C163A/V176C mutant were exposed to BM[PEO]3. Their total lysate was then analyzed by Western blotting with anti-myc antibody (lane TL), or the same cells were first immunoprecipitated with anti-myc antibody; the resulting immunoprecipitate was then subjected to BM[PEO]3 cross-linking in the presence of 0.5 mM CaCl2 (lane IP). Note that the amount of the cross-linked product is approximately the same in both cases. (B) A-431 cells expressing Ec1M-C163A/V176C/W156A (lane 176W), Ec1M-C163A/V176C (lane 176), Ec1M-C163A/T229C (lane 229), and Ec1M-C163A/F231C (lane 231) were immunoprecipitated with anti-myc, and the immunoprecipitates were cross-linked by BM[PEO]3 in the absence of calcium. Samples were analyzed with myc- and E-cadherin (clone C20820)-specific antibodies. The latter recognizes endogenous cadherin coimmunoprecipitated with myc-tagged mutants. Note that the cysteine mutants unable to be cross-linked on the cell surface also cannot be cross-linked in the immunoprecipitates. Note also that, due to the Cys163 residue, endogenous E-cadherin forms a cross-linked dimer with Ec1M-C163A/V176C mutant (arrowhead) at a low calcium concentration (see also Fig. 2A).
DISCUSSION
During the last decade, numerous biochemical (1, 3, 11, 27) and structural (5, 18, 24, 21) studies have shown that extracellular cadherin regions can interact with one another in diverse ways. Uncertainty in intercadherin interactions has led to different, often controversial, models of cadherin-based adhesion (reviewed in references 7, 15, 17, and 31). In the present study, we analyzed cadherin complexes by using a site-specific cross-linking assay. This approach is based on our finding that E-cadherin lacking its single free sulfhydryl group is fully functional. This finding allowed us to design E-cadherin cysteine mutants that upon dimerization according to the particular structural models would expose closely located cysteine residues. Formation of such pairs of cysteines in vivo was verified by the homobifunctional SH-specific cross-linker BM[PEO]3. This approach allowed us to show that adhesive and lateral cadherin dimers which had been characterized previously by coimmunoprecipitation assay (8) have a very similar structure. Apparently both dimers are established by the same Trp156-dependent strand dimer interaction first shown for N-cadherin (25).
E-cadherin molecule contains a single unpaired cysteine, Cys163, located in close proximity to Trp156 (5, 19). We found that the sulfhydryl group of this cysteine cannot be efficiently biotinylated in standard culture conditions, apparently because it is buried within the EC1 hydrophobic core (18, 21). However, since this cysteine can be biotinylated after calcium depletion, it was replaced with Ala. The resulting E-cadherin mutant (Ec1M-C163A) showed no differences from its parent molecule, Ec1M, in subcellular distribution, binding to catenins (not shown), or adhesive and lateral dimerization. However, it could not be biotinylated, even at low calcium concentrations. Therefore, this mutant is ideal for site-specific cross-linking experiments.
In order to reveal cadherin dimers using BM[PEO]3 cross-linking, novel sulfhydryl groups were introduced to the strand B of the Ec1M-C163A mutant by point mutations L175C, V176C, Q177C, or K179C. The corresponding residues were selected because their side chains are exposed in the strand dimer interface described by Shapiro et al. (25). These substitutions did allow us to cross-link E-cadherin dimers. Notably, the major features of these dimers and those previously detected by coimmunoprecipitation assay (8, 14) are essentially the same. In both cases, (i) the combined amount of adhesive and lateral dimers is calcium independent; (iii) all dimers are lateral in a low calcium concentration, whereas adhesive dimers become a dominant form in standard calcium concentrations; and (iii) both the adhesive and lateral dimers require the integrity of Trp156 residue. Such prominent similarities between cadherin dimers detected in coimmunoprecipitation and cross-linking assays suggest that these two independent approaches reveal the same type of cadherin dimerization. This conclusion is further supported by the fact that cell extraction with the immunoprecipitation buffer and the following immunoprecipitation did not affect the pool of cadherin dimers available for cross-linking.
The V176C mutation resulted in the most efficient cross-linking of cadherin dimers. Perhaps this was due to the fact that in the strand dimer model the side chains of Val176 are facing each other (see Fig. 1A). Several additional considerations allow us to discard the possibility that the cross-linked products correspond to other types of dimers revealed by crystallography. It would be very improbable that cadherin dimerization in cross-linked dimers is mediated by the His233/Val235-containing “adhesive” interface discovered in the N-cadherin EC1 domain crystals (25). First, since the B strands in the dimer organized by this interface are located on opposite surfaces of the dimer, it is unlikely that they can be cross-linked in our experiments. Second, this interface was not evident in experiments with three different cysteine mutants specifically designed to verify this interaction. Therefore, the data obtained by our site-specific cross-linking assay is consistent with previous mutational and immunochemical analyses (12, 16, 26) which failed to reveal the function of this interface in cadherin-cadherin interactions.
The Trp156 but not Ca2+ dependence of the cross-linked dimers appears to exclude that they are organized according to the calcium-binding site model reported by Nagar et al. (18) and Pertz et al. (21). Moreover, all our attempts to reveal the calcium-binding site model by cysteine mutagenesis of the EC2 domain using this model as a blueprint failed. There are several explanations for why calcium-dependent cadherin dimerization documented for EC1/EC2 fragments of E-cadherin in crystals (18, 21) and in solution (11) was not detected in our cross-linking or in our coimmunoprecipitation assays. It is possible that calcium-binding site-mediated dimerization is more favorable than Trp156-dependent dimerization for some cadherin fragments. The presence of a foreign methionine at the E-cadherin N terminus may further have hampered strand dimer interaction in experiments reported by Haussinger et al. (11). This interpretation is supported by our recent work showing that such additional methionine prevents the assembly of Trp156-dependent E-cadherin dimers (16). Furthermore, E-cadherin extended from the cellular surface may utilize some specific mechanisms facilitating the assembly of strand dimers but not of calcium site dimers. An alternative possibility is that calcium site dimers, while present on the cell surface, cannot be detected by both of our assays: they are not stable during immunoprecipitation, and their lifetime is much shorter than the time needed for the cross-linking reaction. However, we regard the latter possibility as less likely since the point mutations Q255A and N297A, which according to the calcium-binding site model would affect the dimerization interface, did not change E-cadherin properties.
Our site-specific cross-linking experiments indicate that the same interface mediates both adhesive and lateral cadherin dimerization. Otherwise, it is difficult to explain the fact that neither one of the four cysteine strand B mutants allows preferential cross-linking of either adhesive or lateral dimers. The parallel alignment of two cadherin molecules in the adhesive dimer is also evident from the experiments with hybrid dimers consisting of strand B and strand G mutants. Our previous observation that the same set of mutations equally affects both adhesive and lateral dimers (16) further supports their structural identity. Therefore, our data, in complete agreement with a recent structural study (5), suggest that at a standard calcium concentration a curve of extracellular cadherin regions allows the EC1 domain to form an adhesive dimer via a strand dimer interface. Continual assembly-disassembly of such adhesive dimers within cadherin clusters may establish dynamic adhesive links between apposing cells (14). Whether cadherin clustering is achieved by catenins or by additional intercadherin interactions—as proposed by Boggon et al. (5)—remains to be determined.
The immediate assembly of lateral and the concomitant disassembly of adhesive dimers after calcium removal might be caused by two factors: (i) the short lifetime of preassembled adhesive dimers (14) and (ii) a change in the overall extracellular cadherin conformation, affecting a correct presentation of the EC1 domain and thereby inhibiting adhesive but permitting lateral dimerization. However, lateral Trp156-dependent dimerization (heterodimerization of two classic cadherins, in particular) was clearly documented in many experiments even in a standard calcium concentration (13, 20, 24). These observations indicate that at a high calcium concentration the cadherin extracellular region may also have some flexibility sufficient to form both types of dimers using the same dimer interface (see Fig. 5D). In agreement with this hypothesis, previous electron microscopic analysis demonstrated that, in the presence of 2 mM Ca2+, the cadherin ectodomain represents flexible rods (2, 22). Furthermore, it has been proposed that the flexibility of the cadherin ectodomain is necessary for adhesive interactions between apposing surfaces which are in constant motion (2). The molecular mechanisms and extent of this flexibility remain to be determined. Notably, an E-cadherin lacking a catenin-binding site preferentially forms lateral dimers (8). Therefore, catenins might have an important function in retaining the cadherin extracellular conformation required for adhesive dimerization. These catenin-induced changes in the cadherin extracellular region may also control the lifetime of adhesive dimers.
Acknowledgments
This work has been supported by grant AR44016-04 from the National Institutes of Health.
We thank J. Klingelhofer (Institute of Cancer Biology, Denmark) for help in the preparation of Fig. 5D and V. Mazurov for technical assistance in some experiments.
REFERENCES
- 1.Baumgartner, W., P. Hinterdorfer, W. Ness, A. Raab, D. Vestweber, H. Schindler, and D. Drenckhahn. 2000. Cadherin interaction probed by atomic force microscopy. Proc. Natl. Acad. Sci. USA 97:4005-4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becker, J. W., H. P. Erickson, S. Hoffman, B. A. Cunningham, and G. M. Edelman. 1989. Topology of cell adhesion molecules. Proc. Natl. Acad. Sci. USA 86:1088-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bibert, S., M. Jaquinod, E. Concord, C. Ebel, E. Hewat, C. Vanbelle, P. Legrand, M. Weidenhaupt, T. Vernet, and D. Gulino-Debrac. 2002. Synergy between extracellular modules of vascular endothelial cadherin promotes homotypic hexameric interactions. J. Biol. Chem. 277:12790-12801. [DOI] [PubMed] [Google Scholar]
- 4.Blaschuk, O. W., and T. M. Rowlands. 2002. Plasma membrane components of adherens junctions. Mol. Membr. Biol. 19:75-80. [DOI] [PubMed] [Google Scholar]
- 5.Boggon, T. J., J. Murray, S. Chappuis-Flament, E. Wong, B. M. Gumbiner, and L. Shapiro. 2002. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296:1308-1313. [DOI] [PubMed] [Google Scholar]
- 6.Bussemakers, M. J., A. van Bokhoven, S. G. Mees, R. Kemler, and J. A. Schalken. 1993. Molecular cloning and characterization of the human E-cadherin cDNA. Mol. Biol. Rep. 17:123-128. [DOI] [PubMed] [Google Scholar]
- 7.Chappuis-Flament, S., E. Wong, L. D. Hicks, C. M. Kay, and B. M. Gumbiner. 2001. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J. Cell Biol. 154:231-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chitaev, N. A., and S. M. Troyanovsky. 1998. Adhesive but not lateral E-cadherin complexes require calcium and catenins for their formation. J. Cell Biol. 142:837-846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gumbiner, B. M. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84:345-357. [DOI] [PubMed] [Google Scholar]
- 10.Hajra, K. M., and E. R. Fearon. 2002. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer 34:255-268. [DOI] [PubMed] [Google Scholar]
- 11.Haussinger, D., T. Ahrens, H. J. Sass, O. Pertz, J. Engel, and S. Grzesiek. 2002. Calcium-dependent homoassociation of E-cadherin by NMR spectroscopy: changes in mobility, conformation and mapping of contact regions. J. Mol. Biol. 324:823-839. [DOI] [PubMed] [Google Scholar]
- 12.Kitagawa, M., M. Natori, S. Murase, S. Hirano, S. Taketani, and S. T. Suzuki. 2000. Mutation analysis of cadherin-4 reveals amino acid residues of EC1 important for the structure and function. Biochem. Biophys. Res. Commun. 271:358-363. [DOI] [PubMed] [Google Scholar]
- 13.Klingelhofer, J., R. B. Troyanovsky, O. Y. Laur, and S. M. Troyanovsky. 2000. Amino-terminal domain of classic cadherins determines the specificity of the adhesive interactions. J. Cell Sci. 113:2829-2836. [DOI] [PubMed] [Google Scholar]
- 14.Klingelhofer, J., O. Y. Laur, R. B. Troyanovsky, and S. M. Troyanovsky. 2002. Dynamic interplay between adhesive and lateral E-cadherin dimers. Mol. Cell. Biol. 22:7449-7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koch, A. W., D. Bozic, O. Pertz, and J. Engel. 1999. Homophilic adhesion by cadherins. Curr. Opin. Struct. Biol. 9:275-281. [DOI] [PubMed] [Google Scholar]
- 16.Laur, O. Y., J. Klingelhofer, R. B. Troyanovsky, and S. M. Troyanovsky. 2002. Both the dimerization and immunochemical properties of E-cadherin EC1 domain depend on Trp156 residue. Arch. Biochem. Biophys. 400:141-147. [DOI] [PubMed] [Google Scholar]
- 17.Leckband, D., and S. Sivasankar. 2000. Mechanism of homophilic cadherin adhesion. Curr. Opin. Cell Biol. 12:587-592. [DOI] [PubMed] [Google Scholar]
- 18.Nagar, B., M. Overduin, M. Ikura, and J. M. Rini. 1996. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature 380:360-364. [DOI] [PubMed] [Google Scholar]
- 19.Ozawa, M., H. Hoschutzky, K. Herrenknecht, and R. Kemler. 1990. A possible new adhesive site in the cell-adhesion molecule uvomorulin. Mech. Dev. 33:49-56. [DOI] [PubMed] [Google Scholar]
- 20.Ozawa, M. 2002. Lateral dimerization of the E-cadherin extracellular domain is necessary but not sufficient for adhesive activity. J. Biol. Chem. 277:19600-19608. [DOI] [PubMed] [Google Scholar]
- 21.Pertz, O., D. Bozic, A. W. Koch, C. Fauser, A. Brancaccio, and J. Engel. 1999. A new crystal structure, Ca2+ dependence, and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 18:1738-1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pokutta, S., K. Herrenknecht, R. Kemler, and J. Engel. 1994. Conformational changes of the recombinant extracellular domain of E-cadherin upon calcium binding. Eur. J. Biochem. 223:1019-1026. [DOI] [PubMed] [Google Scholar]
- 23.Provost, E., and D. L. Rimm. 1999. Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr. Opin. Cell Biol. 11:567-572. [DOI] [PubMed] [Google Scholar]
- 24.Shan, W.-S., H. Tanaka, G. R. Phillips, K. Arndt, M. Yoshida, D. R. Colman, and L. Shapiro. 2000. Functional cis-heterodimers of N- and R-cadherins. J. Cell Biol. 148:579-590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapiro, L., A. M. Fannon, P. D. Kwong, A. Thompson, M. S. Lehmann, G. Grubel, J.-F. Legrand, J. Als-Neilsen, D. R. Colman, and W. A. Hendrickson. 1995. Structural basis of cell-cell adhesion by cadherins. Nature 374:327-337. [DOI] [PubMed] [Google Scholar]
- 26.Shimoyama, Y., H. Takeda, S. Yoshihara, M. Kitajima, and S. Hirohashi. 1999. Biochemical characterization and functional analysis of two type II classic cadherins, cadherin-6 and -14, and comparison with E-cadherin. J. Biol. Chem. 274:11987-11994. [DOI] [PubMed] [Google Scholar]
- 27.Sivasankar, S., B. Gumbiner, and D. Leckband. 2001. Direct measurements of multiple adhesive alignments and unbinding trajectories between cadherin extracellular domains. Biophys. J. 80:1758-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.St. Amand, A. L., and M. W. Klymkowsky. 2001. Cadherins and catenins, Wnts and SOXs: embryonic patterning in Xenopus. Int. Rev. Cytol. 203:291-355. [DOI] [PubMed] [Google Scholar]
- 29.Tamura, K., W. S. Shan, W. A. Hendrickson, D. R. Colman, and L. Shapiro. 1998. Structure-function analysis of cell adhesion by neural (N-) cadherin. Neuron 20:1153-1163. [DOI] [PubMed] [Google Scholar]
- 30.Tepass, U., K. Truong, D. Godt, M. Ikura, and M. Peifer. 2000. Cadherins in embryonic and neural morphogenesis. Nat. Rev. Mol. Cell. Biol. 1:91-100. [DOI] [PubMed] [Google Scholar]
- 31.Troyanovsky, S. M. 1999. Mechanism of cell-cell adhesion complex assembly. Curr. Opin. Cell Biol. 11:561-566. [DOI] [PubMed] [Google Scholar]
- 32.Wheelock, M. J., A. P. Soler, and K. A. Knudsen. 2001. Cadherin junctions in mammary tumors. J. Mammary Gland Biol. Neoplasia 6:275-285. [DOI] [PubMed] [Google Scholar]
- 33.Yap, A. S., W. M. Brieher, and B. M. Gumbiner. 1997. Molecular and functional analysis of cadherin-based adherens junctions. Annu. Rev. Cell Dev. Biol. 13:119-146. [DOI] [PubMed] [Google Scholar]