

Abstract
Stimulation of dopamine D1 receptors has profound effects on addictive behavior, movement control, and working memory. Many of these functions depend on dopaminergic systems in the striatum and D1–D2 dopamine receptor synergies have been implicated as well. We show here that deletion of the D1 dopamine receptor produces a neural phenotype in which amphetamine and cocaine, two addictive psychomotor stimulants, can no longer stimulate neurons in the striatum to express cFos or JunB or to regulate dynorphin. By contrast, haloperidol, a typical neuroleptic that acts preferentially at D2-class receptors, remains effective in inducing catalepsy and striatal Fos/Jun expression in the D1 mutants, and these behavioral and neural effects can be blocked by D2 dopamine receptor agonists. These findings demonstrate that D2 dopamine receptors can function without the enabling role of D1 receptors but that D1 dopamine receptors are essential for the control of gene expression and motor behavior by psychomotor stimulants.
Dopamine receptors have been centrally implicated in the behavioral responses generated by psychoactive drugs. Such drugs include psychomotor stimulants, which increase motor behavior and produce addictive behavior, as well as neuroleptic drugs, which are given therapeutically to treat symptoms of schizophrenia. A striking feature of the neural responses to such drugs is a rapid induction of immediate-early genes in the forebrain (1, 2). These rapid responses are thought to constitute an early phase of transcriptional regulation leading to long-term neural effects of the drugs. Paralleling this early wave of transcriptional regulation, there is marked regulation of neuropeptides in many of the target neurons (3–5). In the striatum, where these neural responses have been most intensively studied, the patterns of gene regulation are exquisitely specific for drug type and target cell type. Psychomotor stimulants regulate transcription factors and dynorphin mainly in striatal projection neurons giving rise to the movement-enhancing “direct pathway” of the basal ganglia and in striatonigral neurons of striosomes (2, 6, 7). By contrast, neuroleptics induce increases in transcription factor and enkephalin expression in different striatal cell populations, including projection neurons giving rise to the movement diminishing “indirect pathway” of the basal ganglia (8, 9). These selective actions of psychomotor stimulants and neuroleptics on striatal neurons are thought to underlie the movement-related effects of the drugs and aspects of the addictive response.
Both D1-class and D2-class dopamine receptors have been implicated in this immediate-early gene and peptide regulation. Pharmacologic studies point to D1-class dopamine receptors as essential to the induction of bZip and NGFI-A family transcription factors and to the regulation of dynorphin expression (3, 10–12). D2-class dopamine receptors have been shown to regulate these transcription factors also and to regulate enkephalin expression in the striatum (2, 4, 13). Strong interactive and synergistic effects of D1-class and D2-class receptors have been reported to affect these neural responses, however, and such interactive effects have been emphasized as clinically important (14–19).
The pharmacological agents so far available for dissecting the molecular specificity of these effects lack full selectivity for the subsets of D1-class (D1, D5) and D2-class (D2, D3, D4) receptors (20). However, mutant mice in which particular D1- or D2-class receptors have been deleted by homologous recombination provide a means for genetic analysis of receptor-specific drug effects and for determining receptor function (21–23). We have reported (21, 24) marked abnormalities in motor responses of D1 mutant mice in the absence of major anatomical changes in the striatum or its nigrostriatal afferents. This raised the question, addressed herein, of whether there are more subtle abnormalities in the striatum that could be related to these abnormal drug responses.
To test for such changes, we carried out experiments of three types. (i) We found in our original study that there is a marked loss of expression of dynorphin in the D1-deficient mutants. As dynorphin is a principal target of psychomotor stimulant regulation in the striatum, we asked whether, despite low basal levels of dynorphin in the mutant striatum, the expression of this neuropeptide could nonetheless be induced in striatal neurons by massive doses of psychomotor stimulants. (ii) We asked whether, in the absence of functional D1 receptors, psychomotor stimulants could induce immediate-early genes of the bZip family in the D1-deficient mice and, if so, whether they could do so in the specialized distributions characteristic for these drugs in normal animals. (iii) To examine the enabling role of D1 receptors in D2-mediated responses to psychoactive drugs, we studied striatal Fos/Jun induction by haloperidol, a widely used neuroleptic drug with high affinity for D2-class receptors, and we also asked whether behavioral catalepsy would occur in the D1-deficient mice in response to such treatment. Our findings demonstrate that the dopamine D1 receptor is necessary for the basal expression and cocaine-stimulated regulation of dynorphin in the striatum. Furthermore, in the absence of the D1 receptor, there is a complete loss of inducibility of cFos and JunB in striatal neurons in response to psychostimulants. Despite these regulatory abnormalities in striatal responses to psychomotor stimulants, however, haloperidol remained capable of D2-sensitive induction of bZip transcription factor at normal and even slightly augmented levels relative to those in wild-type mice and remained capable of inducing behavioral catalepsy. These findings suggest that functional D1 receptors are essential to the normal response of striatal neurons to psychomotor stimulants but that neuroleptic drugs can activate these neurons and motor behavior despite the absence of D1–D2 receptor synergies.
MATERIALS AND METHODS
Animals and Drug Treatments.
Experiments were carried out with male dopamine D1 receptor mutant mice (−/−) genetically engineered at the Massachusetts Institute of Technology (21) and their wild-type (+/+) littermates. Drugs were administered intraperitoneally (i.p.) at the following doses: 40 mg/kg, cocaine hydrochloride (Sigma); 10 mg/kg, d-amphetamine sulfate (Sigma); 10 mg/kg, SKF 81297 hydrobromide (Research Biochemicals); 2 or 5 mg/kg, haloperidol (Sigma); 1 mg/kg, quinelorane dihydrochloride (Research Biochemicals), a D2 class receptor agonist. Quinerolane was given alone or 15 min before haloperidol (2 mg/kg). Separate groups of mice were injected with cocaine (25 mg/kg) or saline five times at 1-hr intervals. Survival times after the drug treatment were 2 hr.
Immunohistochemistry.
Immunocytochemistry was performed on free-floating, 20-μm, frozen sections from 4% paraformaldehyde perfusion-fixed brains with avidin-biotin protocols (Vectastain, Vector Laboratories) as described (21, 25). Primary incubations were carried out with polyclonal rabbit antisera raised against (i) rat c-Fos (diluted 1:1000, Oncogene Science), (ii) rat JunB (diluted 1:6000, provided by R. Bravo, Bristol-Myers Pharmaceutical Research Institute, Princeton, NJ), (iii) rat dynorphin B1–29 peptide (leumorphin, diluted 1:10 000, provided by S. Watson, University of Michigan, Ann Arbor, MI).
In Situ Hybridization.
Brains from mice euthanized by decapitation were removed, frozen, and cut into 10-μm-thick cryostat sections. Thaw-mounted sections were fixed with 4% paraformaldehyde, rinsed in phosphate-buffered saline, and sequentially treated in 10-min steps with proteinase K (1 μg/ml) and with 0.25% acetic anhydride. Sections were hybridized with an 35S-labeled UTP-labeled RNA probe specific for the rat prodynorphin gene [provided by J. Douglass (26)] as described (11) and autoradiography was carried out with Hyperfilm βmax films (Amersham).
Ligand Binding.
Fresh-frozen sections were prepared as described for in situ hybridization, and autoradiography for dopamine transporter binding sites was carried out with 15 μM [3H]mazindol (DuPont/NEN) for 40 min (27) or with 5 nM [3H]WIN 35,428 (DuPont/NEN) for 2 hr (28). Films (Hyperfilm, Amersham) were exposed for 2 weeks for [3H]mazindol and 8 weeks for [3H]WIN 35,428, developed in Kodak D-19, and analyzed with the aid of an image analyzer (Biocom, Les Ulis, France). Density measurements were made with respect to tritium standards (3H Microscales, Amersham) applied to the same films to convert optical density values generated by the tritiated ligands bound to the tissue into nCi/g of tissue (1 Ci = 37 GBq).
RESULTS
Basal and Psychomotor Stimulant-Regulated Expression of Dynorphin Is Abnormal in D1 Receptor Mutant Striatum.
Dynorphin in the striatum is a major phenotypic marker for striatal projection neurons of two types: neurons of striosomes, which project to the dopamine-containing region of the substantia nigra, and neurons of the striatal matrix, which project to the internal pallidum and substantia nigra pars reticulata (3, 4). Evidence further suggests that dynorphin-containing projection neurons of both types express D1-class dopamine receptors (4, 19). Changes in dopaminergic stimulation strongly influence levels of dynorphin expression in these neurons. Removal of dopaminergic input lowers dynorphin expression, and stimulation of D1-class receptors augments its expression (4). We have shown (21) with antisera directed to amino acids 15–29 of the C-terminal end of the dynorphin B1–29 peptide that dynorphin is nearly undetectable by immunochemistry in the cell bodies of these neurons in D1-deficient mutant mice and that levels of dynorphin are reduced in their axonal projection fields as well. To determine whether the decrease in dynorphin immunoreactivity was due to a decrease in mRNA levels, we carried out in situ hybridization with a probe for the rat prodynorphin gene (26) in D1 mutant mice and their littermate controls.
There was a massive loss of prodynorphin mRNA in the D1 mutant striatum relative to that in wild-type mice (Fig. 1). In the wild types, prodynorphin mRNA was most densely concentrated in the ventral striatum, in the medial and basal caudoputamen and in striosomes throughout the caudoputamen. In the mutants, prodynorphin mRNA was nearly undetectable in the caudoputamen except along its medial and basal edges, and levels were lowered in the ventral striatum. In the neocortex of the mutants, where cells expressing dynorphin and prodynorphin mRNA are scattered in wild-type mice, mRNA levels per cell appeared slightly reduced, but there were still many neurons expressing the mRNA (Fig. 1). These defects in the expression of prodynorphin mRNA closely parallel patterns of loss of dynorphin peptide detected by immunohistochemistry (21).
Figure 1.
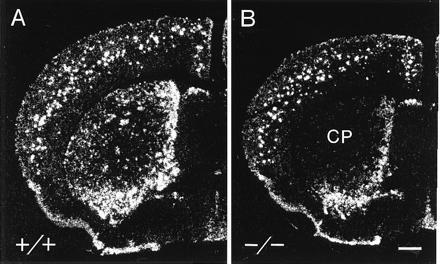
Prodynorphin mRNA levels are severely down-regulated in the striatum of the D1 mutant mouse. In situ autoradiograms illustrating the distribution of prodynorphin mRNA in wild-type control mice (+/+) (A) and mutant (B) mice lacking the D1 dopamine receptor. CP, caudoputamen. (Bar = 0.5 mm.)
These results indicate that the loss of dynorphin expression in the D1 mutant mice is likely at the level of transcription and that, under normal conditions, tonic D1 receptor activation maintains the basal levels of prodynorphin mRNA and dynorphin peptide expression present. Thus, even though other dopamine receptors may be coexpressed with D1 receptors in some or most dynorphin-containing striatal neurons (see ref. 29), these other dopamine receptors are unable to maintain normal basal levels of dynorphin in the striatum. Nor, apparently, are other nondopamine receptors expressed by striatal neurons.
Regulated Expression of Striatal Dynorphin Is Abnormal in D1 Receptor Mutants.
To determine whether striatal dynorphin levels could be increased in the D1 mutants despite the abnormality in basal expression of the neuropeptide, we administered cocaine in a “binge” protocol, a strategy that has been shown to increase manyfold dynorphin expression in the rat (12, 30). Administration of cocaine (25 mg/kg) at five 1-hr intervals greatly increased dynorphin immunoreactivity in the dorsal and ventral striatum as compared with levels in control mice given saline instead of cocaine (Fig. 2 A and C). The increase in dynorphin expression was robust both in perikarya and in the neuropil around them in the striosomal compartment. A much smaller increase occurred in neurons of the matrix. Only scattered neurons were strongly immunostained for dynorphin. Moderate patchy increases in dynorphin levels also occurred in the ventral striatum.
Figure 2.
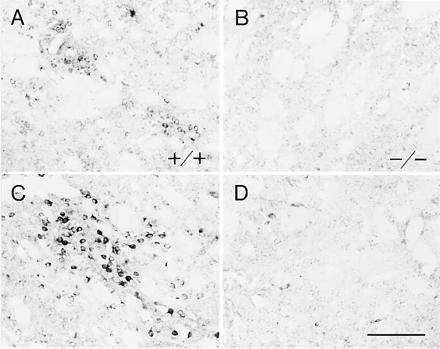
Binge administration of cocaine fails to upregulate expression of dynorphin in the striatum of D1 mutant mice. (A–D) Photomicrographs of equivalent parts of the caudoputamen of wild-type (+/+) (A and C) and mutant (−/−) (B and D) mice. In untreated wild-type mice (A), dynorphin is expressed in patchy clusters of neurons (striosomes) and in scattered neurons in the matrix. Dynorphin expression is nearly nil in the D1 mutants (B). Administration of cocaine in a repeated-dose (binge) protocol strongly up-regulates dynorphin expression in striosomes of the wild-type mice (C) but fails to do so in the D1 mutants (D). (Bar = 100 μm.)
In sharp contrast to these results for the wild-type mice, the same binge administration of cocaine failed to increase dynorphin levels in the caudoputamen of the mutant mice (Fig. 2 B and D). Nor was any marked increase visible in the nucleus accumbens. These experiments strongly suggest that the up-regulation of dynorphin expression in the dorsal and ventral striatum after cocaine exposure requires D1 receptor activation.
Psychomotor Stimulants Fail to Induce cFos and JunB Proteins in the Striatum of D1 Mutant Mice.
Our results for dynorphin expression suggest that striatal neurons in the D1 mutants cannot compensate for the loss of D1 receptors either in regulating basal expression of this neuropeptide or in mediating increases in its expression in response to high-dose psychomotor stimulant exposure. This absence of regulation raised the possibility that the striatum of the D1 mutants might have a generalized regulatory defect in responding to these drugs. If so, this might account for some or all of the behavioral abnormalities found in these mice, which include basal hyperactivity (21) and loss of cocaine-induced hyperactivity and stereotyped behavior (ref. 24 and present results). The expression of both hyperactivity and stereotypic behaviors in response to psychomotor stimulants depend on the striatum. The ventral striatum is required for the expression of locomotor hyperactivity and the dorsal striatum (caudoputamen) is required for the expression of stereotypy (31).
To test for the ability of striatal cells to regulate immediate-early genes in response to these drugs, we administered acute high doses of amphetamine (10 mg/kg) or of cocaine (40 mg/kg) to D1 mutants and to their wild-type controls and assayed the expression of cFos and JunB immunoreactivities in striatal neurons. This assay has the advantage of testing for the normality of the patterns as well as the levels of induction of these transcription factors (1, 25). In the rat, amphetamine across a large range of doses induces cFos and JunB expression preferentially in the striosomal compartment of the anterolateral striatum, whereas cocaine induces cFos and JunB expression nearly equally in striosome and matrix compartments.
In the wild-type mice, amphetamine and cocaine induced cFos and JunB immunoreactivity largely in the drug-specific patterns described previously for the rat (Figs. 3 and 4): the induction was patchy for amphetamine but relatively homogeneous for cocaine. In sharp contrast, the amphetamine and cocaine treatments failed to induce expression of either cFos or JunB in the striatum of the mutants (Figs. 3B and 4B). Nor was there detectable induction in the neocortex of the mutants, again in contrast to the results in wild-type mice. To determine whether this lack of induction was simply a threshold effect, we treated mutants and their wild-type controls with massive amounts of cocaine in the binge protocol used to study dynorphin regulation. There was no detectable induction in the D1 mutants, in contrast to their littermate controls.
Figure 3.
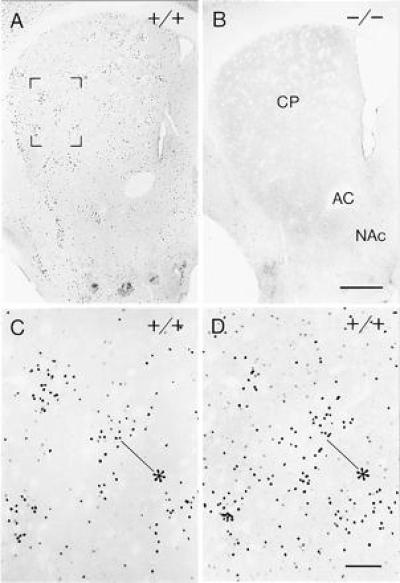
Acute amphetamine treatment fails to induce cFos in the D1 mutant mouse. (A) In wild-type (+/+) mice, amphetamine induces patchy striosome-predominant expression of cFos in the anterolateral caudoputamen. (B) No such induction occurs in the D1 mutant (−/−) caudoputamen. (Bar = 0.5 mm.) Region shown in brackets in A is illustrated at higher magnification in C. (D) An equivalent field from adjacent section immunostained for JunB. Asterisks in C and D point to corresponding striosomes. (Bar = 100 μm.)
Figure 4.
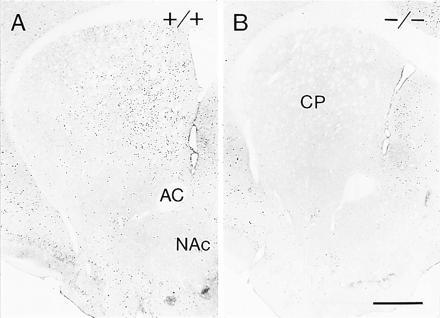
Acute cocaine treatment induces expression of cFos in the striatum of wild-type mice but fails to regulate cFos expression in the striatum of D1 mutants. A shows robust expression of cFos in the centromedial caudoputamen of a wild-type (+/+) mouse induced by administration of cocaine at 40 mg/kg 2 hr before perfusion. B shows that the same treatment given to a D1 mutant mouse does not induce striatal expression of cFos above constitutive levels. (Bar = 0.5 mm.)
Amphetamine and cocaine affect the amount of dopamine available for receptor binding rather than binding directly to the receptors themselves (see ref. 25) and, therefore, are classified as indirect receptor agonists. We also tested whether a direct D1-class receptor-selective agonist, SKF 81297, could provoke activation of cFos or JunB in the D1 mutants. It is with this drug that our earlier experiments on direct agonist induction of locomotor activity in the D1 mutants were performed (21). There was a complete absence of visible induction of either cFos or JunB in the D1 mutants, whereas their wild-type controls showed induction of both transcription factors in the striatum (Fig. 5), neocortex, and other brain sites as described for this and other D1-class agonists in the rat (16, 18). Thus the abnormal behavioral responses to psychomotor stimulants and to direct D1-class agonist drugs that we found in the D1 mutants were paralleled by abnormal immediate-early gene regulation in the mutant striatum.
Figure 5.
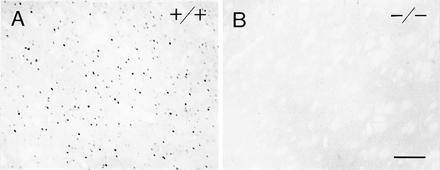
Selective D1 dopamine receptor agonist SKF 81297 fails to induce cFos in the striatum of D1 mutant mice. (A) Sparse but widespread induction of cFos in the caudoputamen of a wild-type (+/+) mouse after acute administration of SKF 81297 (10 mg/kg). (B) Absence of such induction in the caudoputamen of a D1 mutant (−/−) mouse. (Bar = 100 μm.)
Levels of Dopamine Transporter Are Normal in the Striatum of D1 Mutant Mice.
Previous analysis of the D1 mutant striatum showed the maintenance of normal immunoreactivity for tyrosine hydroxylase, the synthetic enzyme for dopamine and other catecholamines and a marker for dopamine-containing mesostriatal afferents. Even with intact mesostriatal systems in the D1 mutants, however, the D1 mutation might have affected the dopamine transporters normally expressed by dopamine-containing neurons. The converse situation, in which the dopamine transporter was deleted by homologous recombination, resulted in massive down-regulation of tyrosine hydroxylase (32). Changes in the dopamine transporter could result in the failure of amphetamine and cocaine to activate striatal systems normally in the mutants and could account for their behavioral abnormalities as well. Therefore, we carried out a quantitative analysis of striatal transporter expression, only qualitatively examined in our previous experiments (21).
Autoradiographic ligand binding was carried out with two different ligands that bind to the dopamine transporter, [3H]mazindol and [3H]WIN 35,428 (see ref. 28). For each ligand, computer-assisted densitometry showed nearly identical values for dopamine transporter binding sites in the caudoputamen of the mutants and wild types (Fig. 6). The mutants did have lower numbers of binding sites in the nucleus accumbens, but the differences between the mutants and controls did not reach statistical significance (Fig. 6).
Figure 6.

Preservation of dopamine transporter binding sites in the striatum of D1 mutant mice. Quantitative analysis showing levels of autoradiographic binding obtained in the striatum of wild-type (+/+) and mutant (−/−) mice with two selective radioligands, [3H]mazindol (A) and [3H]WIN 35,428 (B) (n = 2 wild-type and 2 mutant for [3H]mazindol and n = 3 wild-type and 3 mutant for [3H]WIN 35, 428).
These results suggest that the failure of amphetamine and cocaine to influence gene expression in the D1 mutant caudoputamen and the changes in caudoputamen-dependent behaviors in the mutants are not attributable to changes in numbers of dopamine transporters expressed in the caudoputamen. It seems unlikely that the small decreases in transporter numbers observed in the nucleus accumbens could account for the lack of response of ventral striatal neurons either.
Haloperidol-Induced Immediate-Early Gene Expression in D1 Mutant Mice.
A large series of previous electrophysiological and behavioral studies have shown that activation of D1-class dopamine receptors is required for D2-mediated functions and that activation of D1-class receptors can strongly modulate responses mediated by D2-class dopamine receptors (14, 17, 24, 33, 34). To test directly for such synergistic actions, we asked whether D2 receptor function coupled either to agonist or to antagonist actions could be elicited in striatal neurons in the D1 mutant mice. We first examined the effects of the neuroleptic haloperidol on the induction of Fos/Jun proteins in the mutant mice and their wild-type controls. Haloperidol and other D2-preferring dopamine receptor antagonists induce the expression of cFos and JunB in the rat striatum, and pharmacological evidence suggests that this induction requires D2-class receptors. Pretreatment with a D2-selective agonist blocks the capacity of haloperidol to induce cFos-like immunoreactivity in the striatum (13). Haloperidol also elicits D2-dependent behavior, including catalepsy.
Haloperidol (2 or 5 mg/kg) induced marked catalepsy in both the wild-type and the D1 mutant mice. In the wild types, haloperidol at each dose also induced both cFos and JunB immunoreactivities in a distribution pattern typical of that found in the rat. Immunopositive neurons were broadly distributed in the caudoputamen at anterior levels, but they were more selectively expressed in the dorsolateral rim of the middle and caudal caudoputamen (Fig. 7A). Haloperidol induced cFos and JunB in the striatum of the D1 mutants as well (Fig. 7B). Surprisingly, the induction appeared to be even greater than in the wild-type mice, particularly in the centromedial zone of the caudoputamen, where induction levels were low in the wild types (Fig. 7B). To compare the levels of induction in the mutants and wild types, we counted the numbers of positive nuclei in the striatum at two (more rostral and more caudal) levels. The values for the mutants were greater than those for the wild types for every mutant/wild-type pair (102–285%; P < 0.05 by 2-tailed t test for paired data).
Figure 7.
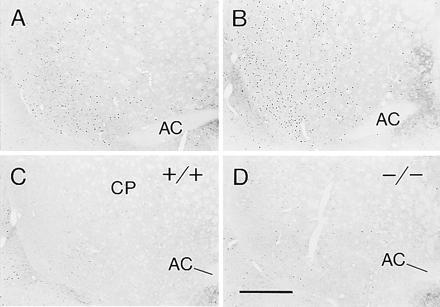
Preservation of D2 receptor responses leading to the induction (A and B) and the blockade (C and D) of cFos expression in the striatum of D1 mutant mice. Photomicrographs through the ventrocaudal caudoputamen of wild-type (+/+) (A and C) and mutant (−/−) (B and D) mice. A and B illustrate cFos induction by haloperidol, an antagonist at D2-class receptors (2 mg/kg, i.p.). C and D illustrate the ability of quinelorane (1 mg/kg, i.p.), an agonist at D2-class receptors, given 15 min before haloperidol, to block the induction of cFos expression. Note the increase of cFos expression induced by haloperidol in the mutant mice compared with the wild-type mice.
In a second set of mice, we tested whether the D2-class dopamine receptor agonist quinelorane could block the immediate-early gene induction. Quinelorane (1 mg/kg), administered 15 min prior to haloperidol, blocked the striatal cFos and JunB expression induced by haloperidol both in the D1 mutants and in the wild-type mice (Fig. 7 C and D). These experiments demonstrate the maintenance of cellular responses to D2 agonist and antagonist treatments despite the absence of D1 receptors and thus the absence of D1–D2 synergism.
DISCUSSION
The results reported herein explicitly demonstrate that D1 dopamine receptors are required to trigger the intracellular cascades leading to two well-characterized responses of striatal neurons to psychomotor stimulants: the induction of the bZip genes c-fos and junB and the induction of the neuropeptide dynorphin. D1 receptors normally are colocalized in the striatal neurons that express dynorphin and that respond to psychomotor stimulants. The neural defect thus may relate to the deficit shown by D1 receptor mutants in striatum-based behavioral responses to psychomotor stimulants. We show further that D1 receptors are necessary for psychomotor stimulant induction of cFos and JunB in the cerebral cortex, where D1 receptor function has been implicated in working memory (35). Our experiments thus suggest that D1 receptor function critically modulates corticobasal ganglia loops at both cortical and subcortical levels. Finally, although previous studies have suggested an essential enabling role of D1 receptors on functions mediated by D2 receptors, our results show that in the absence of D1 receptors, D2-class receptor antagonists (neuroleptics) and D2-class receptor agonists can interact functionally with dopamine receptors as measured by the induction of cFos and JunB expression in the striatum and the elicitation of D2-mediated behavior. The fact that this neural response was not only present but even augmented in the D1 mutants supports the view that the D1 dopamine receptor has regulatory control over neural responses to a wide range of psychoactive drugs including not only stimulants but also neuroleptics.
D1 Dopamine Receptors Are Essential for Fos–Jun Protein Expression in Response to Cocaine and Amphetamine.
Previous pharmacological studies have suggested that the effects of cocaine and amphetamine on locomotor activity, behavioral stereotypy, and immediate-early gene activation in the striatum all are mediated by D1-class receptor activation. More recent work, however, has directly implicated D2-class dopamine and serotonin receptors in cocaine- and amphetamine-induced effects on striatal immediate-early gene expression and behavior (36, 37). It has been shown that eticlopride (a selective D2-class receptor antagonist) at 0.5 mg/kg blocks the induction by cocaine and amphetamine of Fos-like immunoreactivity in striatal neurons by as much as 90% (37), and pharmacologically induced lesions of serotonin-producing neurons leads to a roughly 30% decrease in striatal Fos induction by cocaine (36). More recently, in genetically engineered mice lacking 5-HT1B receptors, a 70% reduction in intrastriatal Fos induction by cocaine has been reported (38). The results singling out D1 receptors in mediating these responses were based mainly on the pharmacological manipulation of administering SCH23390, a selective D1-class receptor antagonist, prior to or during the psychomotor exposures. Even at low dose levels, SCH23390 effectively blocks behavioral and neural responses to the psychostimulants. However, it has been argued that SCH23390 does not have full functional selectivity for D1 receptors, as it also can bind to D2 dopamine receptors and 5HT2 serotonin receptors (39).
In the experiments reported here, we show that both cocaine and amphetamine fail to induce cFos and JunB expression in the D1 mutant striatum, and, as we reported (24), also fail to induce locomotor activity or stereotypy. Thus our data unequivocally establish D1 dopamine receptor activation as essential to the expression of cFos and JunB proteins as well as to the expression of heightened motor behavior in response to representatives of the two major classes of psychomotor stimulants. The degree to which other receptors contribute to these responses to psychomotor stimulants could not be specified in our experiments, but we did test for and find D2-class receptor functionality in the D1 mutants. This preserved efficacy suggests that lack of D2-class dopamine receptor function could not have produced the complete blockage of cocaine- and amphetamine-induced gene expression and behavior that we found. The lack of neural response to cocaine and amphetamine in the D1 mutant mice also cannot be attributed to large-scale loss of dopamine transporters in the caudoputamen, as our binding experiments with [3H]mazindol and [3H]WIN 35,428 demonstrated similar numbers and distributions of dopamine transporter binding sites in the mutant and wild-type mice.
As expected, SKF 81297, a D1-selective receptor direct agonist, failed to induce cFos and JunB in brains of the mutant mice. Unexpectedly, blockade of the effects of amphetamine and cocaine was also widespread in the mutants: neither drug induced cFos or JunB expression in the cerebral cortex or in other sites such as the habenula. The results for the neocortex were surprising, because in the rat, the D1-selective antagonist SCH23390 does not fully block immediate-early gene induction in the neocortex after stimulation with cocaine (11, 36). These pharmacologic results with SCH23390 have been interpreted as suggesting that the cFos expression induced in the neocortex by amphetamine and cocaine is attributable to other monoamine systems, such as serotonin or noradrenaline, rather than to the dopaminergic neocortical system (36). Our results with the D1 mutants indicate that cortical expression of cFos and JunB induced by cocaine and amphetamine does require dopamine D1 receptors, as this expression cannot be induced in the D1 mutant mice.
The Expression of Dynorphin in the Striatum Requires D1 Dopamine Receptors.
Our results demonstrate that dynorphin regulation in the striatum is coupled to D1 dopamine receptor activation and show that the striking up-regulation in dynorphin expression induced by exposure to cocaine requires D1 dopamine receptors. These results extend previous pharmacological evidence for the coupling of dynorphin and D1 receptor function in the striatum (4, 6, 12, 40). Our results also suggest, however, that the D1 receptor regulation is likely coordinated with other monoaminergic signaling in the striatum. Dynorphin up-regulation in the wild-type mice was more pronounced in striosomes than in the matrix of the striatum after binge administration of cocaine, as has been shown for rats (12). Even with this intense treatment protocol, dynorphin expression in the matrix neurons was only slightly above control levels. This pattern is significant given that D1 receptors are primarily expressed by dynorphin-containing striatal neurons (4), which are widely distributed through the matrix as well as in striosomes. If dynorphin up-regulation were simply controlled by D1 receptors, it would be reasonable to expect that psychomotor stimulants would induce a similarly widespread expression of dynorphin in the wild-type caudoputamen, just as there was a widespread deficit in its induction in the D1 mutants.
One factor that could lead to these differential effects of psychomotor stimulants on dynorphin expression is that D1 receptors expressed in the striatum may not all be equally responsive functionally, in spite of their similar abilities to bind dopamine. The D1-bearing neurons in striosomes may respond promptly to the psychomotor stimulant treatment by activating dynorphin expression (12, 40), whereas D1-bearing neurons in the matrix may be inhibited from such responses by concomitant D2 receptor activation. Such D2 effects on D1 receptor function could occur whether D1- and D2-class receptors are colocalized or not (ref. 4; see ref. 29). If there were extensive colocalization, as claimed by some (29), there could be a straightforward explanation if colocalization were particularly extensive in the matrix. Dynorphin could be coupled to D1 receptors throughout the caudoputamen but could fail to be up-regulated or could be up-regulated to a much lesser extent in the matrix neurons bearing D1 and D2 receptors, because D2 receptor activation could inhibit a D1 receptor–cAMP–protein kinase A–CRE-binding protein (CREB) pathway (41). In the event that D1- and D2-class receptors are present mainly in different populations of neurons, they could lead to interactive effects through the effects of interneurons or collaterals (19, 42). Interestingly, under certain conditions, the expression of dynorphin in the striatum does follow the widespread distribution pattern predicted by D1 receptor expression. In D1 receptor hypersensitization conditions, following lesions of the nigrostriatal tract with 6-hydroxydopamine, both apomorphine and SKF 38393 treatments increase dynorphin mRNA expression broadly in striosomes and matrix (4).
Haloperidol-Induced Expression of Fos–Jun Proteins in the Striatum Does Not Require D1 Dopamine Receptors.
An important finding in our experiments was the maintenance of D2-class receptor responses in the absence of D1 receptors in the mutant mice. An enabling function for D1-class dopamine receptor activation on D2-class receptor effects has repeatedly been demonstrated (14, 17, 18, 24, 33, 37). With clear evidence for D1 and D2 receptor interaction coming from pharmacological, behavioral and electrophysiological work, the functional boundaries between the effects of these two classes of dopamine receptors are not well established.
In our experiments, we found that D2 receptor agonist and antagonist drugs (respectively, quinelorane and haloperidol) were able to induce functional effects in D1 mutant mice. Unexpectedly, we further found that in the absence of D1 receptors, haloperidol induced an even slightly larger than normal field of immediate-early gene response in the striatum and that this induction was effectively blocked by quinelorane, suggesting that the response was D2 receptor-mediated. The apparent increase in the induction of cFos and JunB expression did not occur throughout the caudoputamen. There was an obvious increase in the numbers of nuclei immunopositive for cFos and JunB in the centromedial part of the mutant caudoputamen but not in the lateral caudoputamen. Neurons in the lateral caudoputamen have been found to be more responsive electrophysiologically to haloperidol than those in other parts of the striatum in freely moving rats (43). D2-class receptor binding, D2 receptor mRNA, and dopamine transporters are more concentrated laterally than medially in the caudoputamen (44, 45). This pattern is almost the opposite of cFos and JunB expression induced by cocaine, which occurs largely in the centromedial part of the caudoputamen, requires D1 receptors, and is absent in the D1 mutants.
These two specific and contrasting anatomical patterns of cFos and JunB induced by haloperidol and by cocaine suggest that D2-class dopamine receptor function is predominant in the lateral caudoputamen and that D1-class dopamine receptor function is predominant in the centromedial caudoputamen, regardless of any interaction between the two receptor families. Our results with D1 mutant mice suggest that D1 receptors might be able in part to suppress or to override D2 receptor signaling in the centromedial caudoputamen. This could account for the greater than normal cFos and JunB induction by haloperidol in the centromedial caudoputamen of the D1 mutant mice. More generally, our results with the D1 mutants suggest that D1–D2 synergism is not obligatory for D2 receptor function and that D1- and D2-class dopamine receptors may interact synergistically or in opposition in striatal neurons, depending on the neural subpopulations engaged.
Acknowledgments
We thank Mr. Glenn Holm for his help with the image analysis; Mr. Henry Hall, who is responsible for the photography, Miss Diane Major, and Mr. Andrew Tan for their help in the histology; and Drs. R. Bravo and S. Watson for their generous gifts of antisera. This work was supported by National Institute on Drug Abuse Grant 5R01 DA08037 (A.M.G.) and by the Howard Hughes Medical Institute and a grant from the Shionogi Institute for Medical Science (S.T.).
References
- 1.Graybiel A M, Moratalla R, Robertson H A. Proc Natl Acad Sci USA. 1990;87:6912–6916. doi: 10.1073/pnas.87.17.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes P, Dragunow M. Pharmacol Rev. 1995;47:133–178. [PubMed] [Google Scholar]
- 3.Graybiel A M. Trends Neurosci. 1990;13:244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 4.Gerfen C R, Engber T M, Mahan L C, Susel Z, Chase T N, Monsma F J, Jr, Sibley Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 5.Angulo J A, McEwen B S. Brain Res Rev. 1994;19:1–28. doi: 10.1016/0165-0173(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 6.Sivam S P. J Pharmacol Exp Ther. 1989;250:818–824. [PubMed] [Google Scholar]
- 7.Berretta S, Robertson H A, Graybiel A M. J Neurophysiol. 1992;68:767–777. doi: 10.1152/jn.1992.68.3.767. [DOI] [PubMed] [Google Scholar]
- 8.Robertson G S, Vincent S R, Fibiger H C. Neuroscience. 1992;49:285–296. doi: 10.1016/0306-4522(92)90096-k. [DOI] [PubMed] [Google Scholar]
- 9.Hiroi N, Graybiel A M. J Comp Neurol. 1996;374:70–83. doi: 10.1002/(SICI)1096-9861(19961007)374:1<70::AID-CNE5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 10.Young S T, Porrino L J, Iadarola M J. Proc Natl Acad Sci USA. 1991;88:1291–1295. doi: 10.1073/pnas.88.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moratalla R, Vickers E A, Robertson H A, Cochran B H, Graybiel A M. J Neurosci. 1993;13:423–433. doi: 10.1523/JNEUROSCI.13-02-00423.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daunais J B, McGinty J F. Mol Brain Res. 1995;29:201–210. doi: 10.1016/0169-328x(94)00246-b. [DOI] [PubMed] [Google Scholar]
- 13.Miller J C. J Neurochem. 1990;54:1453–1455. doi: 10.1111/j.1471-4159.1990.tb01983.x. [DOI] [PubMed] [Google Scholar]
- 14.Walters J R, Bergstrom D A, Carlson J H, Chase T N, Braun A R. Science. 1987;236:719–722. doi: 10.1126/science.2953072. [DOI] [PubMed] [Google Scholar]
- 15.Seeman P, Niznik H B, Guan H-C, Booth G, Ulpian C. Proc Natl Acad Sci USA. 1989;86:10156–10160. doi: 10.1073/pnas.86.24.10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaHoste G J, Jen Y, Marshall J F. Proc Natl Acad Sci USA. 1993;90:7451–7455. doi: 10.1073/pnas.90.16.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cepeda C, Buchwald N A, Levine M S. Proc Natl Acad Sci USA. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wirtshafter D, Asin K E. Brain Res Bull. 1994;35:85–91. doi: 10.1016/0361-9230(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 19.Gerfen C R, Keefe K A, Gauda E B. J Neurosci. 1995;15:8167–8176. doi: 10.1523/JNEUROSCI.15-12-08167.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gingrich J A, Caron Annu Rev Neurosci. 1993;16:299–321. doi: 10.1146/annurev.ne.16.030193.001503. [DOI] [PubMed] [Google Scholar]
- 21.Xu M, Moratalla R, Gold L H, Hiroi N, Koob G F, Graybiel A M, Tonegawa S. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 22.Drago J, Gerfen C R, Lachowicz J E, Steiner H, Hollon T R, Love P E, Ooi G T, Grinberg A, Lee E J, Huang S P, Bartlett P F, Jose P A, Sibley D R, Westphal H. Proc Natl Acad Sci USA. 1994;91:12564–12568. doi: 10.1073/pnas.91.26.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baik J-H, Picetti R, Salardi A, Thirlet G, Dierich A, Depaulls A, Le Meur M, Borrelli E. Nature (London) 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- 24.Xu M, Hu X, Cooper D C, Moratalla R, Graybiel A M, White F J, Tonegawa S. Cell. 1994;79:945–955. doi: 10.1016/0092-8674(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 25.Moratalla R, Elibol B, Vallejo M, Graybiel A M. Neuron. 1996;17:147–156. doi: 10.1016/s0896-6273(00)80288-3. [DOI] [PubMed] [Google Scholar]
- 26.Civelli O, Douglass J, Goldstein A, Herbert E. Proc Natl Acad Sci USA. 1985;82:4291–4295. doi: 10.1073/pnas.82.12.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graybiel A M, Moratalla R. Proc Natl Acad Sci USA. 1989;86:9020–9024. doi: 10.1073/pnas.86.22.9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufman M J, Spealman R D, Madras B K. Synapse. 1991;9:177–187. doi: 10.1002/syn.890090304. [DOI] [PubMed] [Google Scholar]
- 29.Surmeier D J, Reiner A, Levine M S, Ariano M A. Trends Neurosci. 1993;16:299–305. doi: 10.1016/0166-2236(93)90103-s. [DOI] [PubMed] [Google Scholar]
- 30.Unterwald E M, Ho A, Rubenfeld J M, Kreek M J. J Pharmacol Exp Ther. 1994;270:1387–1397. [PubMed] [Google Scholar]
- 31.Cador M, Bijou Y, Stinus L. Neuroscience. 1996;65:385–395. doi: 10.1016/0306-4522(94)00524-9. [DOI] [PubMed] [Google Scholar]
- 32.Giros B, Jaber M, Jones S R, Wightman R M, Caron M G. Nature (London) 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 33.Hu X-T, Wachtel S R, Galloway M P, White F J. Neurosci. 1990;10:2318–2329. doi: 10.1523/JNEUROSCI.10-07-02318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freedman S B, Patel S, Marwood R, Emms F, Seabrook G R, Knowles M R, McAllister G. J Pharmacol Exp Ther. 1994;268:417–426. [PubMed] [Google Scholar]
- 35.Williams G V, Goldman-Rakic P S. Nature (London) 1995;376:572–575. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- 36.Bhat R V, Baraban J M. J Pharmacol Exp Ther. 1993;267:496–505. [PubMed] [Google Scholar]
- 37.Ruskin D N, Marshall J F. Synapse. 1994;18:233–240. doi: 10.1002/syn.890180309. [DOI] [PubMed] [Google Scholar]
- 38.Lucas J J, Koester J, Hen R. Soc Neurosci Abstr. 1995;21:751. [Google Scholar]
- 39.Bischoff S, Heinrich M, Sonntag J M, Krauss J. Eur J Pharmacol. 1986;129:367–370. doi: 10.1016/0014-2999(86)90449-8. [DOI] [PubMed] [Google Scholar]
- 40.Cole R L, Konradi C, Douglass J, Hyman SEM. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kebabian J W, Calne D B. Nature (London) 1979;277:93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- 42.Kawaguchi Y, Wilson C J, Augood S J, Emson P C. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 43.Rebec G V, Haracz J L, Tschanz J T, Wang Z, White I. In: The Basal Ganglia. Bernadi G, Carpenter M B, DiChiara G, Morelli M, Stanzione P, editors. Vol. 3. New York: Plenum; 1991. pp. 463–470. [Google Scholar]
- 44.Marshall J F, O’Dell S J, Navarrete R, Rosenstein A J. Neuroscience. 1990;37:11–21. doi: 10.1016/0306-4522(90)90187-9. [DOI] [PubMed] [Google Scholar]
- 45.Cline E O, Adams C A, Larson G A, Gerhardt G A, Zahniser N R. Exp Neurol. 1995;134:135–149. doi: 10.1006/exnr.1995.1044. [DOI] [PubMed] [Google Scholar]