

Abstract
We provide evidence that normal human presenilins can substitute for Caenorhabditis elegans SEL-12 protein in functional assays in vivo. In addition, six familial Alzheimer disease-linked mutant human presenilins were tested and found to have reduced ability to rescue the sel-12 mutant phenotype, suggesting that they have lower than normal presenilin activity. A human presenilin 1 deletion variant that fails to be proteolytically processed and a mutant SEL-12 protein that lacks the C terminus display considerable activity in this assay, suggesting that neither presenilin proteolysis nor the C terminus is absolutely required for normal presenilin function. We also show that sel-12 is expressed in most neural and nonneural cell types in all developmental stages. The reduced activity of mutant presenilins and as yet unknown gain-of-function properties may be a contributing factor in the development of Alzheimer disease.
Keywords: Alzheimer disease, sel-12, genetics, transgenic nematode, expression
Genetic linkage studies have identified a number of genetic loci associated with familial Alzheimer disease (1). Mutations in two genes, encoding the presenilins PS1 and PS2, are dominant and fully penetrant (1–5). PS1 and PS2 are related multipass transmembrane proteins that are about 67% identical in amino acid sequence. The presenilins are ubiquitously expressed (4, 5) and found in conjunction with intracellular membranes (6).
The normal function of presenilins and the mechanism by which mutant presenilins cause Alzheimer disease are not yet known. An understanding of the normal function of presenilins and of the nature of the dominant mutations is crucial to elucidating the role of mutant presenilins in Alzheimer disease. The fact that more than 30 dominant, fully penetrant mutations in PS1 and PS2 are all missense mutations has suggested that Alzheimer disease is associated with a gain-of-function activity of mutant proteins, although it remains possible that they partially lower activity of a dose-sensitive gene. Classical studies have indicated that gain-of-function mutations in principle fall into one of three classes: hypermorphic mutations, which elevate gene activity; antimorphic mutations, which reduce wild-type gene activity in trans (this category includes dominant-negative mutations); and neomorphic mutations, which create a novel activity (7). Thus, the mechanism by which hypermorphic and antimorphic mutations exert their effects is by altering the level of normal gene activity. Studies of neomorphic missense mutations suggest that they cause relatively subtle changes, so that at the biochemical level, the novel activity resulting from a neomorphic mutation still relates to the normal mechanism of gene function. For example, neomorphic mutations in the Drosophila abnormal wing discs (awd) gene appear to alter the substrate specificity of nucleoside diphosphate kinase and to reduce activity for its normal substrate (8), and mutations that cause familial amyotropic lateral sclerosis affect different activities of the normal protein, increasing the level of peroxidase activity (9), while in some cases reducing superoxide dismutase activity (10). It is notable that in these cases the neomorphic mutations have both gain-of-function and loss-of-function characteristics.
Genetic studies in simple organisms offer a powerful approach to understanding the normal role of presenilins. A Caenorhabditis elegans gene, sel-12, encodes a protein that displays about 50% amino acid sequence identity to PS1 and PS2 (11). sel-12 was identified by reverting a phenotype caused by constitutive activation of LIN-12, a member of the LIN-12/Notch family of receptors (sel is suppressor/enhancer of lin-12). Genetic analysis established that reducing or eliminating sel-12 activity reduces the activity of lin-12 and causes an egg-laying defective (Egl) phenotype. The Egl phenotype may be a direct consequence of reducing lin-12 activity (12) or an independent effect of reducing sel-12 activity. In this paper, we provide evidence that SEL-12 and the presenilins are functional homologs and that studies in C. elegans will be directly applicable to issues of presenilin structure and function in humans.
MATERIALS AND METHODS
General Methods and Mutations Used.
Methods for handling and culturing C. elegans have been described (13). The wild-type parent for all strains used was C. elegans var. Bristol strain N2 (13). sel-12(ar131) is described in ref. 11. All strains containing pLEX-based plasmids (see below) contained the smg-1(r861) and unc-54(r293) mutations (14). smg-1 mutations stabilize mRNAs with long 3′ untranslated regions (15), and unc-54(r293) is suppressed by smg-1(r861) (14).
pLEX-Based Constructs.
The pLEX vector has been described (16). It contains a 15.1-kb genomic region encompassing the lin-12 gene, in which the normal translational start ATG was destroyed and replaced with a NotI site. cDNAs containing stop codons but lacking polyadenylylation signals are inserted into the NotI site and are efficiently expressed in a smg-1 background. The following cDNAs were inserted into pLEX for this study.
sel-12.
The sel-12 cDNA is described in ref. 11 and, as described below, results in efficient rescue of a sel-12 mutant. We note that the C. elegans genome project has sequenced through the sel-12 region (C. elegans Sequencing Consortium, personal communication). By comparing the genomic sequence with that of the available sel-12 cDNA, we discovered that the cDNA has a frameshift mutation, beginning at codon 413, probably introduced by reverse transcription. This frameshift results in the substitution of 31 amino acids C terminal to the frameshift mutation by 49 amino acids. The 31-amino acid sequence deleted is 42% identical between SEL-12 and PS1 or PS2.
PS1.
Full-length human PS1 cDNA and cDNA encoding the PS1 A246E substitution were generated by reverse transcription-coupled PCR of cytoplasmic RNA isolated from skin fibroblasts of a patient harboring the A246E mutation (National Institute of Aging Cell Repository AG06848B) using a sense primer, hAD3-ATG-Kpn (GGGGTACCATGACAGAGTTACCTGCAC), and antisense primer, hAD3-R-3′UTR (CCGGGATCCATGGGATTCTAACCGC). PCR products were digested with Asp718 and BamHI and ≈1.4-kB hPS1 cDNAs were gel-purified and ligated to Bluescript KS+ vector (Stratagene) previously digested with Asp718 and BamHI, to generate phPS1 and phPS1A246E. The cDNAs were sequenced in their entirety using a Sequenase kit (United States Biochemical).
To generate human PS1 cDNA encoding the M146L, H163R, L286V, or C410Y substitutions (5), we used a four-way PCR strategy with two primer pairs and full-length PS1 cDNA as template. The inserts and junctions were sequenced using Sequenase.
For M146L, primer pairs were hAD3-M146LF (GTCATTGTTGTCCTGACTATCCTCCTG)/hAD3-R284 (GAGGAGTAAATGAGAGCTGG) and hAD3-M146LR (CAGGAGGATAGTCAGGACAACAATGAC)/hAD3-237F (CAGGTGGTGGAGCAAGATG). PCR products from each reaction were gel-purified, combined, and subject to a second round of PCR with primers hAD3-237F and hAD3-R284. The resulting product was digested with KasI and PflMI and an ≈300-bp gel-purified fragment was ligated to KasI/PflMI-digested phPS1 to generate phPS1M146L.
For H163R, primer pairs were hAD3-H163RF (CTAGGTCATCCGTGCCTGGC)/hAD3-R284 and hAD3-H163RR (GCCAGGCACGGATGACCTAG)/hAD3-237F. PCR products from each reaction were gel-purified, combined, and subject to a second round of PCR with primers hAD3-237F and hAD3-R284. The resulting products were digested with KasI and PflMI and a gel-purified ≈300-bp fragment was ligated to KasI/PflMI-digested phPS1 to generate phPS1H163R.
For L286V, primer pairs were hAD3-L286VF (CGCTTTTTCCAGCTGTCATTTACTCC)/hAD3-RL-GST (CCGGAATTCTCAGGTTGTGTTCCAGTC) and hAD3-L286VR (GGAGTAAATGACAGCTGGAAAAAGCG)/hAD3-F146 (GGATCCATTGTTGTCATGACTATC). PCR products from each reaction were gel-purified, combined, and subject to a second round of PCR with primers hAD3-F146 and hAD3-RL-GST. The resulting products were digested with PflMI and BbsI and a gel-purified ≈480-bp fragment was ligated to PflMI/BbsI-digested phPS1 to generate phPS1L286V.
For C410Y, primer pairs were hAD3-C410YF (CAACCATAGCCTATTTCGTAGCC)/LRT7 (GCCAGTGAATTGTAATACGACTCACTATAGGGC) and hAD3-C410YR (GGCTACGAAATAGGCTATGGTTG)/hAD3-243S (CCGGAATTCTGAATGGACTGCGTG). PCR products from each reaction were gel-purified, combined, and subject to a second round of PCR with primers hAD3-243S and LRT7. The resulting products were digested with BbsI and BamHI and an ≈300-bp fragment was gel-purified and ligated to BbsI/BamHI-digested phPS1 to generate phPS1C410Y.
The strategy for generating cDNA encoding hPS1 lacking exon 9 (amino acids 290–319) was as described (17).
PS2.
Full-length cDNA encoding human PS2 was generated by reverse transcription-coupled PCR of total human brain RNA using a sense primer, huAD4-ATGF (CCGGTACCAAGTGTTCGTGGTGCTTCC), and antisense primer, hAD4-stopR (CCGTCTAGACCTCAGATGTAGAGCTGATG). PCR products were digested with Asp718 and XbaI and ≈1.4-kB hPS2 cDNA were gel-isolated and ligated to a vector fragment from expression plasmid pCB6 (17) previously digested with Asp718 and XbaI to generate phPS2. The insert was sequenced in its entirety using a Sequenase kit.
Transgenic Lines and Rescue Assays.
Transgenic lines were established by microinjection of plasmid mixtures into the hermaphrodite germ line to create extrachromosomal arrays (18). By accepted convention, Ex is used to represent extrachromosomal arrays, and Is to represent integrated arrays (which can be generated from extrachromosomal arrays; see below).
pLEX and derivatives were injected at 20 μg/ml, 2 μg/ml, or other concentrations (data not shown) into recipient strains of genotype smg-1(r861) unc-54(r293); sel-12(ar131) or smg-1(r861) unc-54(r293). pRF4, a plasmid containing the cloned dominant rol-6(su1006) gene (18) was used as a cotransformation marker and coinjected at a concentration of 100 μg/ml. F1 Roller progeny were picked, and F2 Roller progeny were used to establish lines.
To assess rescue of sel-12(ar131), approximately 40 L4 Rol progeny from at least three lines generated in a smg-1(r861) unc-54(r293); sel-12(ar131) background were picked individually and scored daily for the ability to lay eggs. We note that rescue assays were performed using sel-12(ar131), a strong partial loss-of-function allele of sel-12, because the strongest existing sel-12 mutation, sel-12(ar171), is somewhat suppressed by smg-1 (data not shown). The sel-12(ar131) egg-laying defect is of variable penetrance (see Fig. 1) and expressivity. About 7% of smg-1 unc-54; sel-12(ar131) hermaphrodites have normal egg-laying, while the remainder bloat with retained eggs; some of these bloated hermaphrodites never lay eggs, whereas others lay eggs. However, the proportion of hermaphrodites that lay eggs normally appears to be reduced by the pLEX vector and/or the rol-6 cotransformation marker (see Fig. 1). We scored hermaphrodites as “Egl+” only if they displayed robust egg-laying characteristic of wild-type hermaphrodites for 2 days as adults, since pLEX-containing control hermaphrodites very rarely lay eggs after 2 days. However, we note that this criterion appears to underestimate rescuing activity, since many hermaphrodites containing human wild-type and mutant presenilins displayed improved egg-laying for 1 day relative to pLEX-containing controls (data not shown) but no longer laid eggs by the second day. The pLEX vector causes a low level of sterility, and sterile hermaphrodites were not scored.
Figure 1.
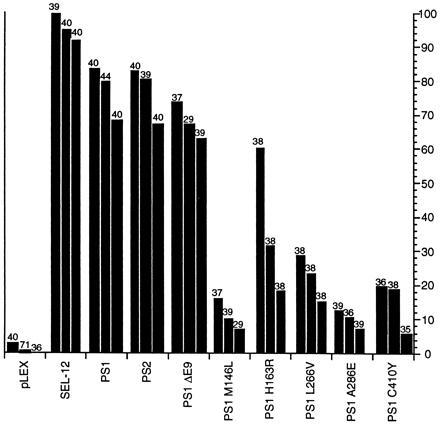
Rescue of the sel-12 Egl and abnormal vulva phenotypes by normal and mutant human presenilins. The data are shown for transgenic lines generated by injecting the construct being tested at a concentration of 20 μg/ml. Each line in the histogram represents data for an independent transgenic line; the number of hermaphrodites scored is shown above each line. The transgene is indicated on the horizontal axis. The percentage of Egl+ hermaphrodites is indicated on the vertical axis. Egl+ signifies robust egg-laying after 2 days; this criterion is very stringent and underestimates the degree of rescuing activity. The ability of PS1 point mutant proteins (data not shown) and the PS1ΔE9 mutant protein (see Fig. 2) to rescue sel-12(ar131) was further reduced when transgenic lines were generated by injecting DNA at a concentration of 2 μg/ml. Most PS1 mutations that cause Alzheimer disease affect amino acids that are identical in SEL-12. The N termini of PS1, PS2, and SEL-12 are not well conserved and are of different lengths. Therefore, for the mutations used herein, the amino acid corresponding to Met-146 in PS1 is Met-115 in SEL-12; PS1 His-163 is SEL-12 His-132; PS1 Ala-246 is SEL-12 Val-216; PS1 Leu-286 is SEL-12 Leu-255; PS1 Cys-410 is SEL-12 Cys-384. The ΔE9 mutation inhibits cleavage of PS1 (17); we note that SEL-12 is cleaved in a comparable position (24). Note that the sel-12 cDNA used (11) has a frameshift mutation, beginning at codon 413, resulting in the substitution of 31 amino acids C-terminal to the frameshift mutation by 49 amino acids.
Transgenic Lines and β-Galactosidase Staining.
pIB1Z17 [sel-12::lacZ] was made as follows: A unique BamHI site was inserted using the PCR at the second amino acid of a sel-12 rescuing genomic fragment containing 2.8 kb of 5′ flanking region. A lacZ gene encoding a β-galactosidase protein containing a nuclear localization signal was excised from plasmid pPD16.43 (19) and inserted in frame into the BamHI site to generate the plasmid pIB1Z17. The predicted transcript contains an abnormally long 3′ untranslated region, consisting of the sel-12 coding and 3′ untranslated region, and is expected to be stabilized in a smg-1 background (15). pIB1Z17 was injected at a concentration of 10 μg/ml into smg-1 unc-54 hermaphrodites. Nine independent lines containing extrachromosomal arrays were established. Four independent integrated lines were generated (using the method of C. Kari, A. Fire, and R. K. Herman, personal communication) from two of the extrachromosomal arrays. All integrated and seven of the nine extrachromosomal arrays displayed staining; all staining lines had similar expression patterns, but some lines carrying extrachromosomal arrays displayed more variability in intensity or penetrance of staining. The integrated lines all appeared to have comparable staining patterns, so we focused on analyzing the expression pattern of the line containing the integrated array arIs17 as a representative line.
Mixed stage populations were grown at 25°C, fixed using an acetone fixation protocol (20) and stained for β-galactosidase activity overnight at room temperature. Stained nuclei were identified based on their size, shape, and position (21, 22). Counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) allowed visualization of all nuclei in the animal by fluorescence microscopy, facilitating the unambiguous identification of stained nuclei. Pictures of the staining pattern were taken at ×1000 using TMAX400 (Kodak) film.
RESULTS
A Presenilin Functional Assay.
There are currently no biochemical assays for presenilin activity, so there has been no direct assay for the effects of mutations on presenilin function. The high level of similarity of SEL-12, PS1, and PS2 suggested that the ability to rescue the distinctive egg-laying defective (Egl) phenotype caused by mutations that reduce or eliminate sel-12 activity (11) could serve as an assay for presenilin function. The sel-12(ar131) mutation is a C60S change (11) in a cysteine that is conserved in SEL-12, PS1, and PS2. sel-12(ar131) is a strong hypomorph in the genetic background used for the rescue experiments (Fig. 1). The pLEX vector (16), which places inserted cDNAs under the control of lin-12 regulatory sequences, can direct sufficient expression of a full-length sel-12 cDNA to rescue the sel-12(ar131) Egl phenotype (Fig. 1). Thus, as described below, the activity of normal and mutant human presenilins can be assessed by creating transgenic C. elegans lines expressing human cDNAs in the pLEX vector and assaying hermaphrodites for their egg-laying ability.
Rescue is assessed in transgenic lines that are created by the microinjection of plasmid DNA into the hermaphrodite germ line. This procedure generates extrachromosomal arrays, and there is some inherent variability in expression from different arrays, in part due to different numbers of copies of plasmid incorporated into the array (18). However, variability can be controlled for by examining multiple independent lines for each construct. Furthermore, arrays generated at the same concentration of injected DNA are likely to have comparable numbers of plasmid copies and, therefore, comparable levels of transgene expression (18). In all of the experiments described below, we have examined three independent lines for each construct and compare the results for lines generated at the same concentration of injected DNA.
Rescue of a sel-12 Mutant by Wild-Type PS1 and PS2.
We have assessed the ability of wild-type human PS1 or PS2 cDNAs to rescue the Egl defect of sel-12(ar131) hermaphrodites (Fig. 1). We found that the human proteins can efficiently substitute for SEL-12 in this assay, despite the vast evolutionary distance between nematodes and humans. The human proteins seem to be slightly less efficient than the C. elegans protein, but this small difference might in principle result from inefficient translation of human presenilin RNA due to the different codon usage between C. elegans and humans, so that less presenilin protein may be produced even if a comparable level of mRNA is expressed from the extrachromosomal arrays. The dramatic increase in rescuing activity when PS1 or PS2 is expressed using lin-12 regulatory sequences, even at a relatively low concentration of injected DNA (Fig. 2), suggests that the human proteins are substituting for C. elegans SEL-12. An alternative interpretation is that the human protein functions in this assay by stabilizing the mutant endogenous SEL-12(ar131) protein. However, this interpretation seems less likely in view of the efficient rescue; furthermore, a corrective interaction of this sort would imply that a SEL-12 and PS1 or PS2 complex is functional, which in itself would be evidence for functional similarity of the C. elegans and human proteins.
Figure 2.

Rescue of the sel-12 Egl phenotype by PS1 and PS1ΔE9 expressed from arrays formed at a concentration of 2 μg/ml. Each line in the histogram represents data for an independent transgenic line; the number of hermaphrodites scored is shown above each line. The transgene is indicated on the horizontal axis. The percentage of Egl+ hermaphrodites (see Fig. 1) is indicated on the vertical axis. At 2 μg/ml of injected DNA, expression from arrays or representation of the plasmid in the arrays may be reduced, accounting for the reduced activity of SEL-12 (transgenic line 3) and PS1 (transgenic line 5) compared with arrays generated at 20 μg/ml (Fig. 1).
Activity of PS1 Point Mutants.
We expressed five different human mutant PS1 proteins, each containing a single amino acid alteration that causes Alzheimer disease, and found that they displayed reduced ability to rescue sel-12(ar131) relative to wild-type PS1 (Fig. 1). These data suggest that the mutations that cause Alzheimer disease may reduce but not eliminate normal presenilin activity. The variable loss of extrachromosomal arrays confounds any determination of steady-state protein levels, so we do not know if the apparently lower activity of mutant presenilins results from reduced protein stability or reduced function.
Activity of PS1ΔE9.
PS1 is subject to endoproteolysis in vivo, and the PS1ΔE9 mutant fails to be cleaved (17). We have found that the human mutant PS1ΔE9 retains a high level of activity, when arrays are formed at the injected DNA concentration of 20 μg/ml (Fig. 1). Since arrays generated at a concentration of 20 μg/ml of injected DNA are likely to contain many plasmid copies, which might mask a small difference in relative activity of PS1 and PS1ΔE9, we generated arrays at the injected DNA concentration of 2 μg/ml. At this concentration of injected DNA, the number of copies of plasmid present in the the arrays should be reduced roughly tenfold (18). At this lower concentration, PS1ΔE9 has reduced ability to rescue sel-12(ar131) as compared with wild-type PS1 (Fig. 2), suggesting that PS1ΔE9, like the PS1 missense mutations, has reduced activity.
Examination of PS1 Mutant Transgenes in a sel-12(+) Background.
In an attempt to reveal gain-of-function activity, we assayed the ability of transgenes encoding mutant presenilins to cause phenotypes in a sel-12(+) background. Antimorphic activity may in principle have caused transgenic hermaphrodites to display the Egl defect associated with reduced sel-12 activity or defects associated with reduced lin-12 activity; hypermorphic activity may in principle have caused egg-laying or vulval defects associated with elevated lin-12 activity (see ref. 11). However, we saw no evidence for gain-of-function activity by these criteria (data not shown). Since intrinsic limitations of the pLEX expression system may have masked moderate changes in sel-12 or lin-12 activity, a definitive assessment of the gain-of-function activity of mutant presenilins in C. elegans will not be possible until other expression systems or strategies are developed.
sel-12 Is Widely Expressed in Neural and Nonneural Cells.
We have examined the expression pattern of transgenic lines carrying a sel-12::lacZ reporter gene. Using this reporter gene, we have found that sel-12, like human presenilins (4, 5), is widely expressed in neural as well as nonneural cells (Fig. 3). Staining was seen in most cell types at all developmental stages from embryo to adult, with the notable exception of the intestine.
Figure 3.

Transgenic hermaphrodites expressing a sel-12::lacZ transgene. Expression is seen in neural and nonneural cells. (A) Adult. Large arrow indicates nerve ring; smaller arrows indicate muscle nuclei. (B) Adult. Arrows indicate ventral cord nuclei. (C) L3 larva. Arrows indicate nuclei of the vulval precursor cells P3.p–P8.p. (D). L2 larva. Arrows indicate the nuclei of the somatic gonadal cells Z1.ppp and Z4.aaa. sel-12 activity has been shown to influence the fates of P3.p–P8.p and Z1.ppp and Z4.aaa in sensitized genetic backgrounds (11). Compromised neural function associated with reduced activity has not yet been seen in the nerve ring or ventral cord, possibly because an appropriate sensitized genetic background has not been examined. Complete genotype: smg-1(r861) unc-54(r293); arIs17 [pRF4, pIB1Z17].
DISCUSSION
Sequence analysis revealed that SEL-12 is similar to human presenilins (11). Herein, we have provided experimental evidence that SEL-12 is a bona fide presenilin, since it may be functionally replaced by either of the two human presenilins. We have also shown that sel-12 is widely expressed in most neural and nonneural tissues of developing animals and adults, as has been observed for human presenilins (4, 5). Furthermore, SEL-12 and PS1 also appear to have similar membrane topology (23, 24). These striking parallels between C. elegans and human presenilins suggest that studies of SEL-12 in C. elegans will bear directly on fundamental issues of presenilin structure and function. In the absence of any description of proteins similar to presenilins in single-celled organisms, including Saccharomyces cerevisiae, it appears that C. elegans is the simplest practical system for studying issues relevant to the biology of presenilins in vivo.
Since PS1 and PS2 appear to be similar in their ability to substitute for SEL-12, they may also have overlapping functions in mammals. As a consequence, studies of normal and mutant PS1 proteins should be directly applicable to PS2 and vice versa. Furthermore, since PS1 and PS2 have broad and overlapping expression patterns (4, 5), the phenotype of mutants homozygous for null alleles of individual mouse presenilin genes may be less severe than the phenotype of double mutants, since there may be functional redundancy where the expression patterns overlap.
The rescue experiments also provide an indication that two regions of the presenilins are not essential for normal function. (i) A SEL-12 protein lacking the last 31 amino acids is highly functional (see Fig. 1), suggesting that the C terminus is dispensable for SEL-12 function. (ii) The PS1ΔE9 protein, which lacks 30 amino acids and fails to be proteolytically cleaved (17), retains considerable activity, suggesting that neither the deleted region nor cleavage is a prerequisite for presenilin activity in C. elegans. We note that our rescue experiments do not address the possibility that the various mutations we tested have gain-of-function activity. Although the nature of the hypothetical gain-of-function activity of mutant presenilins is not clear in humans, the mutant presenilins appear to increase the extracellular concentration of β-amyloid-(1–42 or –43) (25, 26) and hence may cause Alzheimer disease by fostering β-amyloid deposition.
By expressing human genes in C. elegans, we have obtained evidence that six different presenilin mutations that cause early-onset Alzheimer disease lower normal presenilin activity. Hypomorphic characteristics were manifested as reduced ability to rescue a C. elegans mutant defective in sel-12 presenilin function. In the absence of any other assays for normal presenilin function, this information may be useful in considering the pathogenesis of Alzheimer disease, and the development of mammalian models for the disease. It is possible that reduced presenilin activity may contribute to the development of Alzheimer disease, either directly or in conjunction with an as yet unknown gain-of-function activity associated with mutant presenilins.
Acknowledgments
We thank Richard Ruiz for plasmid preparations. This work was supported by National Institutes of Health Grants AG0514 and NS20471 and grants from the Adler Foundation, Alzheimer’s Association, and the Develbiss Fund (to S.S.S.). D.L. was a Postdoctoral Associate of the Howard Hughes Medical Institute. T.G.D. is a predoctoral fellow of the National Science Foundation, and I.G. is an Associate Investigator of the Howard Hughes Medical Institute.
Footnotes
Abbreviation: Egl, egg-laying defective.
References
- 1.Schellenberg G D. Proc Natl Acad Sci USA. 1995;92:8552–8559. doi: 10.1073/pnas.92.19.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark R F, Cruts M, Korenblat K M, He C, Talbot C, Van Broeckhaven C, Goate A M. Human Mol Genet. 1995;4:1347–1354. doi: 10.1093/hmg/4.8.1347. [DOI] [PubMed] [Google Scholar]
- 3.Levy-Lahad E, Wysman E M, Nemens E, Anderson L, Goddard K A B, Weber J L, Bird T D, Schellenberg G D. Science. 1995;269:973–977. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- 4.Rogaev E I, Sherrington R, Rogaeva E A, Levesque G, Ikeda M, et al. Nature (London) 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 5.Sherrington R, Rogaev E I, Liang Y, Rogaeva E A, Levesque G, et al. Nature (London) 1995;375:754–760. [Google Scholar]
- 6.Kovacs D M, Fausett H J, Page K J, Kim T-W, Moir R D, Merriam D E, Hollister R D, Hallmark O G, Mancini R, Felsenstein K M, Hyman B T, Tanzi R E, Wasco W. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 7.Muller H J. Proc Int Congr Genet. 1932;6:213–252. [Google Scholar]
- 8.Timmons L, Xu J, Hersperger G, Deng X F, Shearn A. J Biol Chem. 1995;270:23021–23030. doi: 10.1074/jbc.270.39.23021. [DOI] [PubMed] [Google Scholar]
- 9.Wiedau-Pazos, Goto J J, Rabizadeh S, Gralla E B, Roe J A, Lee M K, Valentine J S, Bredesen D E. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- 10.Borchelt D R, Lee M K, Slunt H S, Guarnieri M, Xu Z S, Wong P C, Brown R H, Jr, Price D L, Sisodia S S, Cleveland D W. Proc Natl Acad Sci USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levitan D, Greenwald I. Nature (London) 1995;377:351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- 12.Sundaram M, Greenwald I. Genetics. 1993a;135:755–763. doi: 10.1093/genetics/135.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodgkin J, Papp A, Pulak R, Ambros V, Anderson P. Genetics. 1989;123:301–313. doi: 10.1093/genetics/123.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pulak R, Anderson P. Genes Dev. 1993;7:1885–1897. doi: 10.1101/gad.7.10.1885. [DOI] [PubMed] [Google Scholar]
- 16.Struhl G, Fitzgerald K, Greenwald I. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- 17.Thinakaran G, Borchelt D R, Lee M K, Slunt H H, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey A I, Gandy S E, Jenkins N A, Copeland N G, Price D L, Sisodia S S. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 18.Mello C C, Kramer J M, Stinchcomb D T, Ambros V A. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fire A, Harrison S W, Dixon D. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- 20.Fire A. Genet Anal Tech Appl. 1993;9:151–158. doi: 10.1016/1050-3862(92)90042-4. [DOI] [PubMed] [Google Scholar]
- 21.Sulston J, Horvitz H R. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 22.Kimble J, Hirsh D. Dev Biol. 1979;81:208–221. doi: 10.1016/0012-1606(79)90035-6. [DOI] [PubMed] [Google Scholar]
- 23.Doan, A., Thinkaran, G., Borchelt, D. R., Slunt, H. H., Ratovitsky, T., et al. (1996) Neuron, in press. [DOI] [PubMed]
- 24.Li, X. & Greenwald, I. (1996) Neuron, in press. [DOI] [PubMed]
- 25.Scheuner D, Eckman C, Jensen M, Song X, Citron M, et al. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 26.Borchelt, D., Thinkaran, G., Eckman, C. B., Lee, M. K., Davenport, F., et al. (1996) Neuron, in press.