

Abstract
Results of transgenetic studies argue that the scrapie isoform of the prion protein (PrPSc) interacts with the substrate cellular PrP (PrPC) during conversion into nascent PrPSc. While PrPSc appears to accumulate primarily in lysosomes, caveolae-like domains (CLDs) have been suggested to be the site where PrPC is converted into PrPSc. We report herein that CLDs isolated from scrapie-infected neuroblastoma (ScN2a) cells contain PrPC and PrPSc. After lysis of ScN2a cells in ice-cold Triton X-100, both PrP isoforms and an N-terminally truncated form of PrPC (PrPC-II) were found concentrated in detergent-insoluble complexes resembling CLDs that were isolated by flotation in sucrose gradients. Similar results were obtained when CLDs were purified from plasma membranes by sonication and gradient centrifugation; with this procedure no detergents are used, which minimizes artifacts that might arise from redistribution of proteins among subcellular fractions. The caveolar markers ganglioside GM1 and H-ras were found concentrated in the CLD fractions. When plasma membrane proteins were labeled with the impermeant reagent sulfo-N-hydroxysuccinimide-biotin, both PrPC and PrPSc were found biotinylated in CLD fractions. Similar results on the colocalization of PrPC and PrPSc were obtained when CLDs were isolated from Syrian hamster brains. Our findings demonstrate that both PrPC and PrPSc are present in CLDs and, thus, support the hypothesis that the PrPSc formation occurs within this subcellular compartment.
The prion diseases include Creutzfeldt–Jakob disease (CJD) of humans, scrapie of sheep, and bovine spongiform encephalopathy (1, 2). The posttranslational conversion of cellular prion protein (PrPC) into the scrapie PrP isoform (PrPSc) is the fundamental process underlying both the transmission and pathogenesis of these fatal illnesses (3, 4). While no difference in the covalent structures of PrPSc and PrPC have been discerned (5), their conformations differ markedly (6).
PrPC is bound to the external surface of the plasma membrane by a glycosylphosphatidylinositol (GPI)-anchor (7) while PrPSc accumulates in late endosomes and lysosomes (8, 9). Studies on the localization of PrPC in mouse neuroblastoma (N2a) cells indicated that it clustered in caveolae or caveolae-like domains (CLDs) (10); in accord with these results, PrPC was found to be insoluble in ice-cold Triton X-100 (11, 12). Caveolae appear as raft-like membranous domains or as coated invaginations of plasma membranes; in the presence of Triton X-100, they form detergent-insoluble complexes that are enriched for GPI-anchored proteins, cholesterol, and glycosphingolipids (13). Using silica beads, detergent-insoluble complexes have been separated into caveolin-rich fractions resembling invaginated caveolae and GPI-protein enriched domains (52). The presence of such domains does not depend on the marker protein caveolin because cells lacking caveolin, such as N2a cells, also contain detergent-insoluble domains enriched for GPI-anchored proteins (11, 12), which we therefore called CLDs. Caveolae are involved in diverse cellular functions such as folate uptake (14), signal transduction (13, 15, 16), and down-regulation of blood clotting activity (17).
The localization of PrPC to CLDs and the finding that PrPSc formation was inhibited by lovastatin, which diminishes cellular cholesterol levels, suggested that glycosphingolipid- and cholesterol-rich CLDs might be the site where prions are propagated (12). Replacing the GPI-anchor addition signal sequence of PrP with the CD4 transmembrane C-terminal 62 residues targeted the chimeric PrP molecule to clathrin-coated pits and prevented PrPSc formation. Furthermore, C-terminal truncation of PrP that deleted the signal sequence for GPI-anchor addition substantially reduced PrPSc formation (18). Recent studies have extended those observations by showing that squalestatin, a more specific inhibitor of cholesterol biosynthesis, and three other transmembrane C-terminal segments inhibit PrPSc formation (K. Kaneko, M. Vey, M. Scott, S. Pilkahn, S. B. Prusiner, personal communication). Earlier investigations had shown that PrPSc interacts with PrPC during the formation of nascent PrPSc (19, 20) and that this process occurs after PrPC transits to the plasma membrane (21, 22).
On this background, we asked whether CLDs isolated from scrapie-infected cells and brain tissue contain both PrPC and PrPSc. The studies reported herein show that both PrPC and PrPSc are found in CLDs and, thus, argue that CLDs are likely to be the subcellular site where conversion of PrPC into PrPSc occurs.
METHODS
Isolation of CLDs Using Ice-Cold Triton X-100.
CLDs were isolated from ice-cold Triton X-100 lysates of N2a and ScN2a cells by flotation into sucrose gradients as described (13, 23). One-milliliter fractions were collected from the top, and the pellet was resuspended in 10% SDS. Fifty microliters of each fraction was subjected to SDS/PAGE and immunoblot analysis. Aliquots taken from the indicated fractions were adjusted to 0.5% Triton X-100 and 0.5% deoxycholate, supplemented with bovine serum albumin to give a final concentration of total protein of 1000 μg/ml, and treated with proteinase K (PK, 1 μg of PK per 100 μg of protein) for 45 min at 37°C prior to SDS/PAGE. CLDs were collected from pooled gradient fractions after diluting them 2.5-fold and centrifugation at 10,000 × g for 10 min. Five micrograms of CLD and lysate proteins were subjected to immunoblot analysis; 10 μg was loaded for silver staining. Five micrograms of CLD proteins from N2a and ScN2a cells was treated with 0.5 units of peptide-N-glycosidase F (PNGase F; Boehringer Mannheim) for 16 h prior to SDS/PAGE.
Detergent-Free Isolation of CLDs.
The detergent-free isolation of CLDs was performed as described (24). Lysates of N2a and ScN2a cells were prepared in buffer A (0.25 M sucrose/1 mM EDTA/20 mM Tricine, pH 7.8) by homogenization in a Wheaton tissue grinder. After low-speed centrifugation, postnuclear supernatants were obtained and 8 mg of total protein was fractionated on 30% Percoll gradients by centrifugation in Ti60 rotors (Beckman) for 30 min at 29,000 rpm. The plasma membranes appearing as a band ≈1.5 cm below the meniscus were isolated, then sonicated in a SW41 tube (Beckman) with a Vibra Cell sonicator (model VC60S, Sonics & Materials, Danbury, CT), and then adjusted to 4 ml containing 23% OptiPrep (GIBCO/BRL) in buffer A. A linear 10–20% OptiPrep gradient was formed on top and centrifuged at 52,000 × g for 90 min. The top 5 ml of the gradients was transferred to a new tube and mixed with 4 ml of buffer B (50% OptiPrep in 0.04 M sucrose/1 mM EDTA/20 mM Tricine, pH7.8). One milliliter of 5% OptiPrep was placed on top and centrifugation was carried out at 52,000 × g for 90 min. CLDs appeared as an opaque band in the top fraction. One-milliliter fractions were collected from top to bottom of the OptiPrep gradients, and proteins were precipitated with trichloroacetic acid (TCA) and subjected to immunoblot analysis. Five micrograms of CLD, plasma membrane, postnuclear supernatant, and cell lysate proteins from N2a and ScN2a cells was precipitated with TCA, separated on SDS gels, and transferred to poly(vinylidene difluoride) membranes (Millipore). Membranes were probed with the anti-PrP R073 antiserum or with cholera toxin B conjugated to horseradish peroxidase (HRP) (Transduction Laboratories, Lexington, KY) to detect ganglioside GM1; a mAb was used for detection of H-ras (Transduction Laboratories). HRP-conjugated secondary antibodies (Amersham) and enhanced chemiluminescence (Amersham) were used for visualization of bands.
Ultrastructure.
CLDs isolated by the detergent-based method and the detergent-free method were applied to Formvar-coated electron microscopy grids (Ted Pella, Redding, CA) and negatively stained with uranyl acetate. Specimens were viewed in a Jeol 100CX electron microscope at 80 keV.
Biotinylation of Membrane Proteins.
Plasma membrane proteins of N2a and ScN2a cells were biotinylated with sulfo-N-hydroxysuccinimide-biotin (Pierce) (25) prior to detergent-free isolation of CLDs. After isolation of biotinylated and nonbiotinylated CLDs, proteins were denatured in 3 M guanidinium hydrochloride and precipitated with methanol. Pellets were solubilized in DLPC buffer (2% Sarkosyl/0.4% l-α-lecithin/0.15 M NaCl/20 mM Tris·HCl, pH 7.5) and biotinylated proteins were precipitated with streptavidin-agarose (GIBCO/BRL). Streptavidin-precipitated proteins and unbound methanol-precipitated proteins of the supernatants were subjected to immunoblot analysis. To 5 μg of CLD proteins, 100 μg of BSA was added and the samples were treated with PK (1 μg of PK per 100 μg of protein) for 60 min at 37°C. After inactivation of PK with phenylmethylsulfonyl fluoride (Sigma), samples were processed for analyses as described above.
RESULTS
Ice-Cold Triton X-100 Isolation of CLDs.
ScN2a cells were lysed in buffer containing ice-cold Triton X-100 and the detergent-insoluble complexes were separated by flotation in sucrose gradients (Fig. 1) (13, 23). After ultracentrifugation, most PrP molecules were detected in the fractions of the sucrose gradients that contained the CLDs (Fig. 1A, lanes 3–6), whereas no PrP was found in the lysate fractions (Fig. 1A, lanes 8–11) and only minor amounts were recovered from the pellets that were formed by incompletely solubilized cell debris (Fig. 1A, lane P). The CLD fractions contained less than 1% of the total proteins, showing that PrP had been separated from the lysates (data not shown). There were no consistent differences in PrP distribution in the gradients of N2a and ScN2a cells, indicating that prion infection did not interfere with formation of CLDs and trafficking of PrP. Furthermore, the presence of protease-resistant PrP in the CLD fractions of ScN2a cells (Fig. 1B, lanes 8, 10, and 12) revealed that, in addition to PrPC, PrPSc was also associated with these membranous subdomains.
Figure 1.
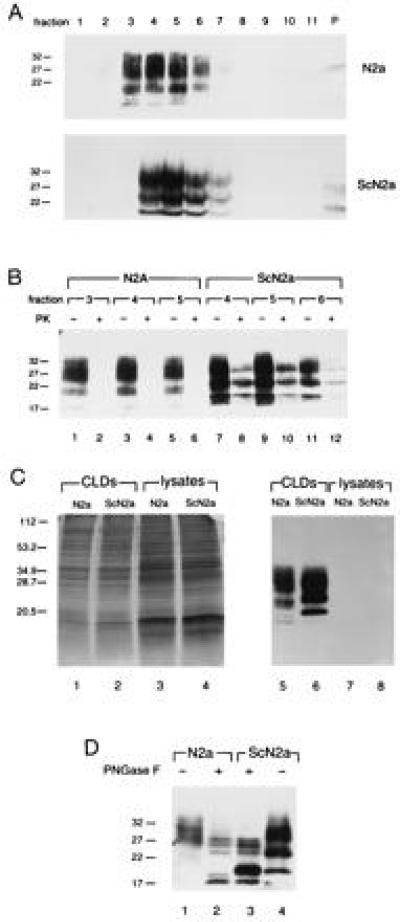
PrPC and PrPSc are concentrated in CLDs isolated from neuroblastoma cells using the ice-cold Triton X-100 detergent method. (A) Distribution of PrP after lysis of N2a and ScN2a cells in ice-cold Triton X-100 and separation of CLDs by flotation into sucrose gradients. Aliquots of gradient fractions were subjected to immunoblot analysis with the anti-PrP polyclonal R073 rabbit antiserum (26). Lanes: 1–7, fractions of the sucrose gradients; 8–11, lysate fractions; P, pellets. (B) Detection of PrPSc in gradient fractions. Immunoblot of gradient fractions from before (−) and after (+) treatment with PK. (C) Concentration of PrP and other proteins in CLDs. Immunoblot (Right) and silver stain (Left) of CLD proteins and cell lysate proteins from N2a and ScN2a cells. (D) Detection of PrPC degradation products in CLDs from N2a and ScN2a cells. Immunoblot of CLD proteins before (−) and after (+) treatment with PNGase F (27).
To collect additional evidence for a physical association of PrP with CLDs, we concentrated the CLDs from the sucrose gradients and found that isolated CLDs (Fig. 1C, lanes 5 and 6) are highly enriched for PrP when compared with cell lysates (Fig. 1C, lanes 7 and 8). We then analyzed the proteins present in CLDs and found identical protein patterns in CLDs from N2a and ScN2a cells (Fig. 1C, lanes 1 and 2) but when compared with the respective cell lysates (Fig. 1C, lanes 3 and 4), several proteins were found to be concentrated in CLDs.
We next asked whether CLDs might play a role in the metabolism of PrPC because recent studies have suggested that proteolytic cleavage of the N terminus of PrPC, the initial step in PrPC degradation, might take place in CLDs before the cleaved PrPC is completely degraded in acidic compartments of the cells (12). To detect this degradation product, which has a molecular mass of 17 kDa after removal of Asn-linked glycans, proteins present in CLDs were treated with PNGase F prior to immunoblot analysis. The degradation intermediate of 17 kDa could be detected in CLDs from N2a cells after deglycosylation (Fig. 1D, lane 2). In addition to PrPC, PrPSc appeared as a 19-kDa protein in CLDs from ScN2a cells after treatment with PNGase F (Fig. 1D, lane 3). These data strongly suggest that CLDs are important sites of PrPC degradation, which is also supported by earlier studies showing an inhibiting effect of cholesterol depletion on the production of the 17-kDa form of PrPC and on the formation of PrPSc (12).
Detergent-Free Isolation of CLDs.
Using the ice-cold Triton X-100 method, it was possible that the isolated CLDs acquired proteins from other subcellular sites (28). This is a particularly important consideration since the majority of PrPSc has been thought to accumulate in lysosomes in ScN2a cells (8, 29), even though we found most of the PrPSc in fractions where CLDs fractionate (Fig. 1A). To address this issue, we used a detergent-free method for the isolation of caveolae membranes (24). Postnuclear supernatants were prepared from N2a and ScN2a cells and fractionated on Percoll gradients. Those fractions enriched for plasma membranes were disrupted by sonication, and the CLDs were separated from nondisrupted membranes and membranous debris by flotation in linear OptiPrep gradients (Fig. 2A). PrP was detected near the top of the OptiPrep gradient (Fig. 2A, lanes 1–5), indicating its association with CLDs of low density. Determination of the protein concentrations revealed that less than 2% of the proteins loaded onto the gradient floated into these fractions (data not shown). These low-density membranes were concentrated in a 5% OptiPrep cushion by using step-gradient centrifugation (Fig. 2B). Almost all of the PrP molecules collected from the first gradient were found to be concentrated in the first fraction of the step gradient, indicating that they were associated with the CLDs (Fig. 2B, lanes 1). Comparison of the relative amounts of PrP present in the CLDs with equal amounts of proteins from cell lysates, postnuclear supernatants, and plasma membranes demonstrated that CLDs were highly enriched for PrP (Fig. 2C, lanes 1 and 5). With ScN2a cells, CLDs were found to be enriched for both PrPC and PrPSc (Fig. 2C, lane 9). These findings argue that both PrPC and PrPSc are constituents of the CLDs that are derived from the plasma membranes of ScN2a cells.
Figure 2.
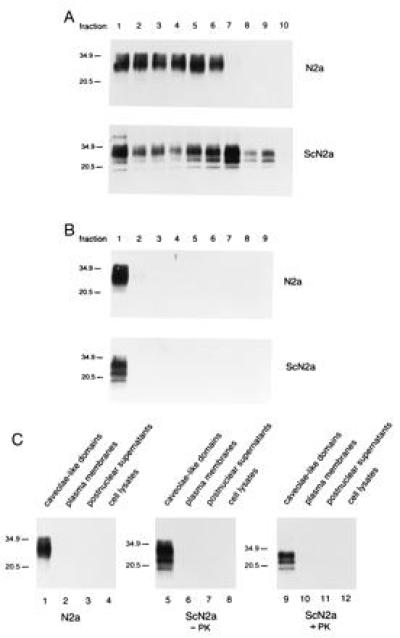
PrPC and PrPSc are concentrated in CLDs isolated from N2a and ScN2a cells isolated by a detergent-free method. (A) Distribution of PrP in gradients after disruption of isolated plasma membranes by sonication and separation of CLDs from plasma membrane debris by flotation into OptiPrep gradients. Proteins precipitated from gradient fractions were subjected to immunoblot analysis using the anti-PrP polyclonal R073 rabbit antiserum (26). (B) Distribution of PrP after concentration of CLDs by step-gradient centrifugation. Immunoblot of proteins precipitated from step-gradient fractions probed with the anti-PrP R073 antiserum. (C) Concentration of PrP in CLDs. Equal amounts of proteins derived from cell lysates, postnuclear supernatants, plasma membranes, and CLDs of N2a and ScN2a cells were subjected to immunoblot analysis with anti-PrP R073 antiserum.
Ultrastructure and Chemical Markers of CLDs.
When we compared the CLDs isolated by the detergent-free method (Fig. 3 C and D) with those produced using ice-cold Triton X-100 (Fig. 3 A and B), the samples were indistinguishable ultrastructurally. Each contained convoluted vesicle-like structures and membrane aggregates. The ganglioside GM1, a caveolar marker (30), was concentrated in CLDs from both cell lines as determined by binding of peroxidase-conjugated cholera toxin B to the ganglioside transferred to poly(vinylidene difluoride) membranes (Fig. 4, lanes 1 and 5). In addition, immunoblot analysis showed that the small GTP-binding protein H-ras, which is involved in signal transduction in caveolae (31), was also present in CLDs of ScN2a and N2a cells (Fig. 4, lanes 9 and 13). These results indicate that N2a and ScN2a cells contain subdomains in their plasma membranes which closely resemble caveolae.
Figure 3.
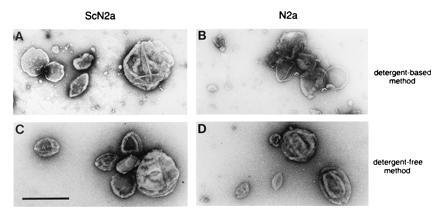
Ultrastructure of isolated CLDs. (A and B) CLDs were isolated by the ice-cold Triton X-100 detergent method. (C and D) CLDs isolated by the detergent-free procedure. Samples were prepared from ScN2a (A and C) and N2a cells (B and D). (Bar = 0.5 μm.)
Figure 4.
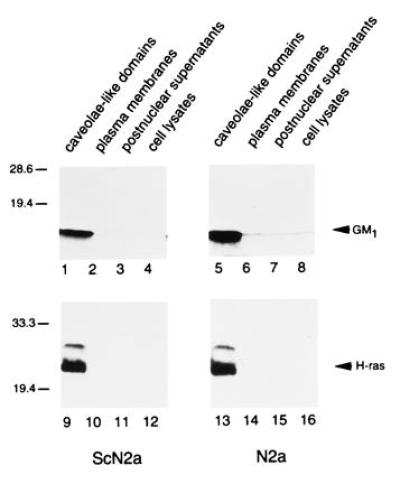
Concentration of ganglioside GM1 and H-ras in CLDs. Equal amounts of proteins from CLDs, plasma membranes, postnuclear supernatants, and lysates of ScN2a and N2a cells were probed with cholera toxin B conjugated to horseradish peroxidase (lanes 1–8) and a mAb against H-ras (lanes 9–16).
Biotinylation of PrPC and PrPSc.
PrPSc can be detected in the plasma membranes of ScN2a cells using surface biotinylation of membrane proteins (32, 33). Having found that CLDs contain PrPSc, we labeled N2a and ScN2a cells with the membrane-impermeant biotinylating reagent sulfo-N-hydroxysuccinimide-biotin (Fig. 5). Biotinylated PrPC (Fig. 5, lanes 3 and 7) and PrPSc (Fig. 5, lane 15) were both detected by streptavidin-agarose precipitation from the CLD fraction. These results argue that both PrP isoforms exist on the surface of cells in association with CLDs.
Figure 5.
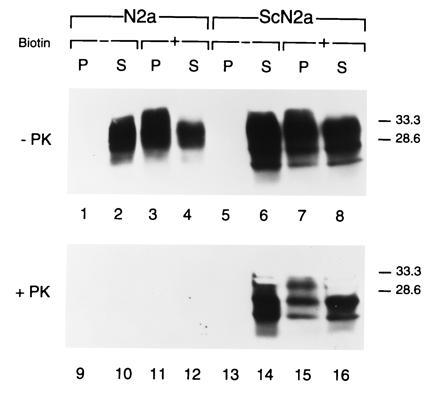
PrPC and PrPSc are concentrated in CLDs on the surface of ScN2a cells. Detection of streptavidin-agarose-precipitated PrP from CLDs derived from biotinylated plasma membranes of N2a and ScN2a cells isolated with the detergent-free method. Immunoblots of CLD proteins precipitated with streptavidin-agarose (lanes P) and unbound proteins of the respective supernatants (lanes S) using anti-PrP R073 antiserum without (− PK) or with (+ PK) limited digestion by proteinase K.
Isolation of CLDs from Brain.
To determine whether our findings with cultured cells are applicable to rodent brain, we isolated CLDs from the brains of normal and scrapie-infected Syrian hamsters. Brains were lysed in Triton X-100 and the majority of the prion proteins were found in the CLD fractions of sucrose gradients (data not shown). In addition to PrPC, protease-resistant PrPSc was also present in CLDs from scrapie-infected hamster brains (data not shown). Our results indicate that PrPC and PrPSc from brain distribute in an OptiSucrose gradient similar to CLD fractions from cultured cells.
DISCUSSION
The studies reported herein indicate that both PrP isoforms, like other GPI-anchored proteins, are physically associated with CLDs. Not only does the first step in PrPC degradation occur in CLDs but it is also likely that PrPC is converted into PrPSc within this subcellular compartment (12). These findings complement those obtained by other methods where redirecting PrPC from CLDs to clathrin-coated pits prevented PrPSc formation (ref. 12 and K. Kaneko et al., personal communication). Thus, PrPSc formation seems to be restricted to CLDs, which is consistent with the conclusions of transgenetic studies arguing that PrPSc formation requires the participation of auxiliary proteins (20).
CLDs seem to be the compartment in which PrPC is either cleaved to form PrPC-II or it is converted into PrPSc (12, 34, 35). In our studies, both full-length PrPC and N-terminally cleaved PrPC-II were found in CLDs (Fig. 2B). It may prove important to identify the specific protease(s) mediating the cleavage of PrPC to PrPC-II since such proteases might be useful in the therapy of prion infections where lower levels of the substrate PrPC would be expected to reduce formation of prions and thus extend the incubation time (19, 36, 37). GPI-anchored proteolytic enzymes have been described (38, 39) and it might be possible to find the specific protease(s) cleaving PrPC also in CLDs.
In contrast to our results and earlier localization studies showing mouse PrP in caveolae-like structures in N2a cells (10), clathrin-coated pits have been described as potential sites for PrPSc formation based on the presence of overexpressed chicken PrP (ChkPrP) in these organelles (40–42). Localization of recombinant ChkPrP to clathrin-coated pits may have resulted from the high-level expression of the ChkPrP in mouse N2a cells as well as the divergent sequence of this protein. ChkPrP is only ≈30% homologous with mammalian PrP (43); most mammalian species have PrPs that are >90% homologous with that of other species (44, 45). Biochemical analyses demonstrated that ChkPrP is highly concentrated in CLDs and not in clathrin-coated pits (11). In contrast, our studies have focused on endogenous mouse PrP in ScN2a cells where the association of both PrPC and PrPSc with CLDs was found using two procedures (Figs. 1 and 2).
Our findings showing that both PrP isoforms are concentrated in CLDs may open several new approaches to the study of prion diseases. (i) Isolation of CLDs may facilitate the identification of non-PrP proteins that function in the formation of PrPSc, as suggested from the results of transgenetic studies (20). Such proteins have been provisionally labeled protein X and suggested to act as molecular chaperones. If PrPSc formation proves to be restricted to CLDs, then the notion that PrPSc is formed by the binding of PrPC to aggregates of PrPSc will be increasingly unlikely (46, 47). (ii) Isolated CLDs might prove useful in developing a cell-free system for studies of PrPSc formation. Such a system might permit the generation of prion infectivity de novo (46–48) and permit the rapid screening of pharmacotherapeutics for the effective treatment of prion diseases.
The identification of PrPSc in CLDs derived from plasma membrane fractions may provide insights into the mode of the spread of prion infection through the central nervous system and other organs. The GPI anchor of PrPSc may facilitate its exchange between surfaces of cells as has been described for other GPI-anchored proteins (49). Accordingly, PrPSc might move between plasma membranes of infected and uninfected neurons or even astrocytes where PrPC is thought to be expressed albeit at levels much below that found in neurons (50, 51). PrPSc might also be delivered to uninfected neurons across membranes of cells lacking PrP expression but containing CLDs such as oligodendrocytes that are in close contact with neurons.
In summary, a persuasive body of data is beginning to emerge that argues that CLDs are the site of PrPSc formation. CLDs are rich in cholesterol, glycosphingolipids, and GPI-anchored proteins (15). Both PrPC and PrPSc have GPI anchors and both are found in CLDs. Reducing cellular cholesterol levels by treatment with either lovastatin or squalestatin blocked the initial proteolytic step in PrPC degradation and inhibited PrPSc formation (ref. 12 and K. Kaneko et al., personal communication). Although substantial amounts of PrPSc accumulate in lysosomes (8, 9, 29), PrPSc can be labeled by addition of the membrane impermeant reagent sulfo-N-hydroxysuccinimide-biotin added to the outside of ScN2a cells, indicating that some of the PrPSc is bound to the external surface of plasma membrane (Fig. 5). The biotinylated PrPSc was subsequently recovered in CLDs isolated by the detergent-free method; such results contend that PrPSc on the plasma membrane is concentrated in CLDs. The foregoing findings with results from other studies showing that PrPSc formation was abolished by replacing the GPI anchor of PrPC with a transmembrane segment that targets PrP to clathrin-coated pits (ref. 12 and K. Kaneko et al., personal communication) build a substantial case for the formation of PrPSc within CLDs.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NS14069, AG08967, AG02132, NS22786, AG10770, 42 20948, GM43169, and GM15631) and the American Health Assistance Foundation, as well as by gifts from Sherman Fairchild Foundation and Centeon. M.V. is supported by the “Stipendium zur Infektionsforschung” from the German Cancer Research Center. H.W. was supported by the Alexander von Humboldt Foundation. R.N. is supported by a National Institutes of Health training grant (NS07219).
Footnotes
Abbreviations: PrP, prion protein; PrPC, cellular PrP; PrPSc, scrapie PrP isoform; CLD, caveolae-like membrane domain; GPI, glycosylphosphatidylinositol; PNGase, peptide-N-glycosidase F; PK, proteinase K; ChkPrP, chicken PrP.
References
- 1.Gajdusek D C. Science. 1977;197:943–960. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 2.Wells G A H, Wilesmith J W. Brain Pathol. 1995;5:91–103. doi: 10.1111/j.1750-3639.1995.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 3.Prusiner S B, Bolton D C, Groth D F, Bowman K A, Cochran S P, McKinley M P. Biochemistry. 1982;21:6942–6950. doi: 10.1021/bi00269a050. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner S B. Annu Rev Microbiol. 1994;48:655–685. doi: 10.1146/annurev.mi.48.100194.003255. [DOI] [PubMed] [Google Scholar]
- 5.Stahl N, Baldwin M A, Teplow D B, Hood L, Gibson B W, Burlingame A L, Prusiner S B. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 6.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick R J, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stahl N, Borchelt D R, Hsiao K, Prusiner S B. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 8.McKinley M P, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond S J, Prusiner S B, Gonatas N. Lab Invest. 1991;65:622–630. [PubMed] [Google Scholar]
- 9.Arnold J E, Tipler C, Laszlo L, Hope J, Landon M, Mayer R J. J Pathol. 1995;176:403–411. doi: 10.1002/path.1711760412. [DOI] [PubMed] [Google Scholar]
- 10.Ying Y-S, Anderson R G W, Rothberg K G. Cold Spring Harbor Symp Quant Biol. 1992;57:593–604. doi: 10.1101/sqb.1992.057.01.065. [DOI] [PubMed] [Google Scholar]
- 11.Gorodinsky A, Harris D A. J Cell Biol. 1995;129:619–627. doi: 10.1083/jcb.129.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner S B. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sargiacomo M, Sudol M, Tang Z, Lisanti M P. J Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson R G W, Kamen B A, Rothberg K G, Lacey S W. Science. 1992;255:410–411. doi: 10.1126/science.1310359. [DOI] [PubMed] [Google Scholar]
- 15.Anderson R G W. Proc Natl Acad Sci USA. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smart E J, Ying Y, Anderson R G W. J Cell Biol. 1995;131:929–938. doi: 10.1083/jcb.131.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sevinsky J R, Rao L V M, Ruf W. J Cell Biol. 1996;133:293–304. doi: 10.1083/jcb.133.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers M, Yehiely F, Scott M, Prusiner S B. Proc Natl Acad Sci USA. 1993;90:3182–3186. doi: 10.1073/pnas.90.8.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prusiner S B, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson G A, Hoppe P C, Westaway D, DeArmond S J. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 20.Telling G C, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen F E, DeArmond S J, Prusiner S B. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 21.Caughey B, Raymond G J. J Biol Chem. 1991;266:18217–18223. [PubMed] [Google Scholar]
- 22.Borchelt D R, Taraboulos A, Prusiner S B. J Biol Chem. 1992;267:16188–16199. [PubMed] [Google Scholar]
- 23.Brown D A, Rose J K. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 24.Smart E J, Ying Y-S, Mineo C, Anderson R G W. Proc Natl Acad Sci USA. 1995;92:10104–10108. doi: 10.1073/pnas.92.22.10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lisanti M P, Sargiacomo M, Graeve L, Saltiel A R, Rodriguez-Boulan E. Proc Natl Acad Sci USA. 1988;85:9557–9561. doi: 10.1073/pnas.85.24.9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serban D, Taraboulos A, DeArmond S J, Prusiner S B. Neurology. 1990;40:110–117. doi: 10.1212/wnl.40.1.110. [DOI] [PubMed] [Google Scholar]
- 27.Tarentino A L, Gomez C M, Plummer T H. Biochemistry. 1985;24:4665–4671. doi: 10.1021/bi00338a028. [DOI] [PubMed] [Google Scholar]
- 28.Mayor S, Maxfield F R. Mol Biol Cell. 1995;6:929–944. doi: 10.1091/mbc.6.7.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taraboulos A, Serban D, Prusiner S B. J Cell Biol. 1990;110:2117–2132. doi: 10.1083/jcb.110.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parton R G. J Histochem Cytochem. 1994;42:155–166. doi: 10.1177/42.2.8288861. [DOI] [PubMed] [Google Scholar]
- 31.Mineo C, James G L, Smart E J, Anderson R G W. J Biol Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- 32.Stahl N, Borchelt D R, Prusiner S B. In: Molecular and Cell Biology of Membrane Proteins. Turner A J, editor. Chichester, U.K.: Ellis Horwood; 1990. pp. 189–216. [Google Scholar]
- 33.Lehmann S, Harris D A. J Biol Chem. 1996;271:1633–1637. doi: 10.1074/jbc.271.3.1633. [DOI] [PubMed] [Google Scholar]
- 34.Haraguchi T, Fisher S, Olofsson S, Endo T, Groth D, Tarantino A, Borchelt D R, Teplow D, Hood L, Burlingame A, Lycke E, Kobata A, Prusiner S B. Arch Biochem Biophys. 1989;274:1–13. doi: 10.1016/0003-9861(89)90409-8. [DOI] [PubMed] [Google Scholar]
- 35.Pan K-M, Stahl N, Prusiner S B. Protein Sci. 1992;1:1343–1352. doi: 10.1002/pro.5560011014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prusiner S B, Groth D, Serban A, Koehler R, Foster D, Torchia M, Burton D, Yang S-L, DeArmond S J. Proc Natl Acad Sci USA. 1993;90:10608–10612. doi: 10.1073/pnas.90.22.10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. Mol Med. 1994;1:19–30. [PMC free article] [PubMed] [Google Scholar]
- 38.Deddish P A, Skidgel R A, Kriho V B, Li X Y, Becker R P, Erdos E G. J Biol Chem. 1990;265:15083–15089. [PubMed] [Google Scholar]
- 39.Hooper N M, Hryszko J, Turner A J. Biochem J. 1990;267:509–515. doi: 10.1042/bj2670509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shyng S-L, Huber M T, Harris D A. J Biol Chem. 1993;21:15922–15928. [PubMed] [Google Scholar]
- 41.Shyng S-L, Heuser J E, Harris D A. J Cell Biol. 1994;125:1239–1250. doi: 10.1083/jcb.125.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shyng S-L, Moulder K L, Lesko A, Harris D A. J Biol Chem. 1995;270:14793–14800. doi: 10.1074/jbc.270.24.14793. [DOI] [PubMed] [Google Scholar]
- 43.Harris D A, Falls D L, Walsh W, Fischbach G D. Soc Neurosci. 1989;15:70.7. (abstr.). [Google Scholar]
- 44.Gabriel J-M, Oesch B, Kretzschmar H, Scott M, Prusiner S B. Proc Natl Acad Sci USA. 1992;89:9097–9101. doi: 10.1073/pnas.89.19.9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schätzl H M, Da Costa M, Taylor L, Cohen F E, Prusiner S B. J Mol Biol. 1995;245:362–374. doi: 10.1006/jmbi.1994.0030. [DOI] [PubMed] [Google Scholar]
- 46.Kocisko D A, Come J H, Priola S A, Chesebro B, Raymond G J, Lansbury P T, Jr, Caughey B. Nature (London) 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 47.Kaneko K, Peretz D, Pan K-M, Blochberger T, Gabizon R, Wille H, Griffith O H, Cohen F E, Baldwin M A, Prusiner S B. Proc Natl Acad Sci USA. 1995;92:11160–11164. doi: 10.1073/pnas.92.24.11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raeber A J, Borchelt D R, Scott M, Prusiner S B. J Virol. 1992;66:6155–6163. doi: 10.1128/jvi.66.10.6155-6163.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kooyman D L, Byrne G W, McClellan S, Nielsen D, Tone M, Waldmann H, Coffman T M, McCurry K R, Platt J L, Logan J S. Science. 1995;269:89–92. doi: 10.1126/science.7541557. [DOI] [PubMed] [Google Scholar]
- 50.Kretzschmar H A, Prusiner S B, Stowring L E, DeArmond S J. Am J Pathol. 1986;122:1–5. [PMC free article] [PubMed] [Google Scholar]
- 51.Moser M, Colello R J, Pott U, Oesch B. Neuron. 1995;14:509–517. doi: 10.1016/0896-6273(95)90307-0. [DOI] [PubMed] [Google Scholar]
- 52.Schnitzer J E, McIntosh D P, Dvorak A M, Liu J, Oh P. Science. 1995;269:1435–1439. doi: 10.1126/science.7660128. [DOI] [PubMed] [Google Scholar]