

Abstract
Whether persons with multiple chemical sensitivity syndrome (MCS) have immunological abnormalities is unknown. To assess the reliability of selected immunological tests that have been hypothesized to be associated with MCS, replicate blood samples from 19 healthy volunteers, 15 persons diagnosed with MCS, and 11 persons diagnosed with autoimmune disease were analyzed in five laboratories for expression of four T-cell surface activation markers (CD25, CD26, CD38, and HLA-DR) and in four laboratories for autoantibodies (to smooth muscle, thyroid antigens, and myelin). For T-cell activation markers, the intralaboratory reproducibility was very good, with 90% of the replicates analyzed in the same laboratory differing by ≤3%. Interlaboratory differences were statistically significant for all T-cell subsets except CD4+ cells, ranging from minor to eightfold for CD25+ subsets. Within laboratories, the date of analysis was significantly associated with the values for all cellular activation markers. Although reproducibility of autoantibodies could not be precisely assessed due to the rarity of abnormal results, there were inconsistencies across laboratories. The effect of shipping on all measurements, while sometimes statistically significant, was very small. These results support the reliability of fresh and shipped samples for detecting large (but perhaps not small) differences between groups of donors in the T-cell subsets tested. When comparing markers that are not well standardized, it may be important to distribute samples from different study groups evenly over time.
Multiple chemical sensitivity syndrome (MCS) is a chronic disorder characterized by symptoms that are elicited by exposure to diverse chemicals and involve three or more organ systems (usually neurological, immune, respiratory, cutaneous, gastrointestinal, and/or musculoskeletal) (1, 6, 33). The hallmark of MCS is hypersensitivity to low levels of inhaled, absorbed, or ingested chemicals, sometimes accompanied by hypersensitivity to light and sound. Typical symptoms include memory loss; migraines; nausea; abdominal pain; chronic fatigue; aching joints and muscles; difficulty in breathing, sleeping, or concentrating; and irritated eyes, nose, ears, throat, or skin (1). The pathogenesis of MCS is not well understood, and a variety of mechanisms have been proposed to explain it, including limbic kindling (2), neurogenic inflammation (22), toxicant-induced loss of tolerance (23), carbon monoxide poisoning (8), psychological factors (10) (for a critique, see reference 7), and immune abnormalities (21) (for an overall review, see reference 11).
Hyperreactivity of the immune system to environmental stimuli could explain both the diversity of symptoms in MCS and the very low levels of chemical exposures with which those symptoms have been associated. This hypothesis has been investigated in case series and controlled studies (12, 14, 18, 20, 26-28, 32, 34, 36) (for a review, see reference 24), but many of these studies have been controversial and/or difficult to interpret, for at least two reasons. First, the reliability of many of the immunological methods and tests used has not been demonstrated by standard epidemiological and laboratory criteria. While markers used for the diagnosis or management of known immunological diseases such as human immunodeficiency virus infection are now routinely validated and quality controlled (13, 17), this is not true for many of the immunological markers studied for MCS (J. B. Margolick and R. F. Vogt, Letter, Ann. Intern. Med. 220:249, 1994), particularly those related to lymphocyte phenotype and function. For example, one study (32) that found no immunological abnormalities in people with MCS generated considerable controversy, in part because it used methods whose reproducibility was questionable (31; Margolick and Vogt, letter). Second, diagnostic criteria and epidemiological case definitions of MCS have been inconsistent across studies, and many studies did not consider the possibility that some of the controls could have had MCS; this could be important given that up to 16% of those surveyed in recent population studies indicated that they had some degree of hypersensitivity to environmental chemicals (3, 15).
For immunological testing to be useful either in understanding the pathogenesis of MCS or, potentially, in its diagnosis, tests that reliably distinguish immunological differences of the magnitudes that might exist between people with and without MCS, and laboratories which can perform these tests reliably, must be available. The goal of this study was to address the reliability of some immunological methods and tests that have been used to investigate and evaluate MCS. To this end, we conducted a multiple-laboratory comparison of immunological markers commonly cited in the MCS literature. In particular, we evaluated the magnitudes of between- and within-laboratory differences in these measures and whether differences between individuals with and without well-defined immunological diseases could be identified. We focused primarily on cellular immunity, using measurements that have been widely studied in both MCS and related diseases such as chronic fatigue syndrome (16, 32), i.e., the major T-cell subsets (T-helper [CD4+] and T-cytotoxic [CD8+] cells) and cellular activation markers expressed by these cells. We also studied certain autoantibodies that have been reported to be elevated in persons with MCS, namely, antibodies to smooth muscle, myelin, and thyroid antigens (12, 32).
To evaluate the reproducibility of the tests as they would likely be performed in the assessment of people for possible MCS, replicate blood samples were tested in four to six laboratories, to which they were shipped from Baltimore, Md., by overnight mail. In addition, the effect of the shipment process on the collective results was examined by analyzing fresh samples in Baltimore on the day of phlebotomy.
MATERIALS AND METHODS
Study population.
The study participants (seen from November 1996 to June 1997) consisted of three groups. First, 19 healthy volunteers, who were likely to have no immunological disorder, were recruited from laboratory staff and students at the Johns Hopkins School of Public Health. Second, to provide participants similar to those who would seek MCS testing, 15 persons with a diagnosis of MCS were recruited from the private practice of one of us (G. Ziem) or by advertisements in a newsletter circulated to people with MCS. Third, to provide study participants exhibiting immunological abnormalities, 11 patients with established diagnoses of immune disease (6 patients with systemic lupus erythematosus [SLE] and 5 patients with Graves's disease) were recruited from outpatient clinics at The Johns Hopkins Hospital. As the study goal was to assess the reproducibility of analytic tests within and between laboratories rather than to identify laboratory abnormalities associated with MCS, the accuracy of the diagnosis of MCS among study participants was not verified. (A separate study designed to address this issue will be reported elsewhere.) All study participants gave informed consent, and study protocols were approved by the Institutional Review Boards of all participating centers.
Selection of tests and laboratories.
The cellular activation markers studied included CD25, CD26, and HLA-DR, which have been analyzed in past studies of MCS (12, 32, 36), and CD38, an activation marker which has not previously evaluated in MCS but which has been studied extensively in immune diseases, including AIDS (19) and chronic fatigue syndrome (16). Antibodies to smooth muscle, thyroid gland, and myelin (12, 32) were also studied. As previous studies of immunological testing in MCS used both commercial and research laboratories, we included two commercial laboratories that had been active in testing for MCS (Immunosciences, Inc., Los Angeles, Calif., and Specialty Laboratories, Inc., Los Angeles, Calif.) and four research-oriented laboratories, located at the Johns Hopkins School of Public Health (JHU) (Baltimore, Md.), Rutgers University (Piscataway, N.J.), Scripps Research Institute (La Jolla, Calif.), and the University of Washington (UW) (Seattle), that had longstanding interests in laboratory quality control for serological testing (4) and immunophenotyping. The JHU and UW laboratories participate in a flow cytometry proficiency control program sponsored by the National Institute of Allergy and Infectious Diseases, and the JHU and Rutgers laboratories had participated in the Immune Biomarkers Demonstration Project (35). One other commercial laboratory was approached but declined to participate.
Processing of specimens for laboratory testing.
Approximately 50 ml of blood was drawn from each study subject and processed as follows: 18 ml was drawn into a heparinized syringe and divided into 12 1.5-ml aliquots for lymphocyte subset analysis, and 30 ml was drawn into 10 3-ml serum separator tubes for autoantibody analysis. On the day the blood was drawn, two (duplicate) aliquots of heparinized blood from each person were sent to the flow cytometry laboratory at JHU for immediate (within 4 h) analysis of T-cell phenotypes, and two (duplicate) aliquots of serum were similarly sent to the autoantibody laboratory at JHU, according to protocols described below. Ten aliquots of heparinized blood and eight serum tubes were then transported by courier to FAST Systems, Inc. (Gaithersburg, Md.), for shipment in duplicate by overnight mail to all participating laboratories (including back to JHU), as illustrated schematically in Fig. 1. In one instance difficult venous access limited the amount of blood that could be drawn, and only one specimen was sent to each laboratory.
FIG. 1.

Processing of blood specimens from donor to laboratories. Dotted lines, serum; solid lines, whole blood. Univ., University; Inst., Institute.
All specimens were coded so that laboratories were unaware of their donors. This design allowed for (i) analysis of the effects of shipping by overnight mail, since JHU laboratories analyzed both fresh and shipped samples, and (ii) as-sessment of components of intra- and interlaboratory variability of each test performed, since multiple laboratories were used and replicate samples from the same person-visit were analyzed at each laboratory. The shipment of specimens by overnight mail simulated the manner by which clinical specimens are commonly sent to commercial laboratories by physicians evaluating patients with possible MCS. At each laboratory, specimens were processed according to the standard protocols for that laboratory. No attempt was made to delineate or control for differences in methods used to perform the same measurement in different laboratories, since our purpose was to evaluate “real-world” laboratory performance. One to three persons provided blood on any given study day, and all specimens (including both replicates) from these persons were sent together to each laboratory and were processed and analyzed on the same date.
For T-cell immunophenotypic analysis, the commercial laboratories (Immunosciences and Specialty Laboratories) used two-color analysis. The research laboratories (JHU, Rutgers, and UW) used a three-color antibody panel that was designed for this study and was configured by PharMingen Laboratories, San Diego, Calif. (now part of Becton-Dickinson Immunocytometry Systems, San Jose, Calif.). The panel included all antibodies studied here except anti-CD38. Antibodies were conjugated to either fluorescein isothiocyanate, phycoerythrin, or cychrome in the following combinations (fluorescein isothiocyanate/phycoerythrin/cychrome): HLA-DR/CD38/CD4, HLA-DR/CD38/CD8, CD25/CD26/CD4, and CD25/CD26/CD8. An anti-CD38 antibody from Becton Dickinson was added to appropriate antibody panel tubes when the specimen was stained, based on preliminary studies in which it gave superior staining compared to the PharMingen anti-CD38 antibody. The three-parameter data from JHU, Rutgers, and UW were reduced to the equivalent two-color analyses by using software supplied with the flow cytometers (ELITE cytometers and software; Beckman Coulter, Miami, Fla. [in all three laboratories]). As three-color data are not used in clinical practice, these data were not analyzed here. One laboratory measured expression of CD25 and CD26 only on CD3+ lymphocytes rather than on CD4+ or CD8+ lymphocytes. Data are reported as percentages of gated lymphocytes (identified by forward- and side-scatter gating at the three research laboratories).
For analysis of the autoantibodies selected for study (antithyroid, anti-smooth muscle, and antimyelin), replicates were shipped and fresh and frozen samples were compared by procedures similar to those described above for T-cell subsets (Fig. 1). The only difference was that performance of these measurements was batched in the research laboratories for both the fresh and frozen samples; i.e., samples were stored frozen until a sufficient number of samples were available for batch analysis. At the commercial laboratories, samples were tested as received. The comparison of fresh versus shipped samples at JHU was made between samples that were shipped without being frozen, but were then frozen when received, and samples that were frozen in the processing laboratory before being shipped. At each laboratory, serum was incubated with appropriate tissues, according to standard methods for that laboratory, using either freshly frozen serum or serum frozen after shipping.
Data analysis.
Summary statistics were analyzed by medians, means, standard deviations, and ranges. Box plots compared results by laboratory for each test. Since the distributions of measurements for antibodies were quite extreme, with the vast majority of person-tests having no antibodies detected and a few person-tests having very large values, descriptive statistics and more formal statistical comparison of these measures were not practical. However, as T-cell measures had more stable distributions, formal statistical comparisons of inter- and intralaboratory and intersubject variabilities for each T-cell marker were possible. The following (potential) effects on T-cell measures were tested through nested random/fixed effects analysis of variance models: (i) interlaboratory variation, (ii) interday variation (i.e., between date of analysis) within the same laboratory, (ii) difference between fresh versus shipped samples at JHU, (iv) variation due to disease group (healthy individuals, MCS, or other diagnosed autoimmune diseases), and (v) within-disease-group interperson variation. Specifically, two different models were fit, as described in the appendix.
As this was exploratory research, adjustments for multiple comparisons were not made (29), although for many of the comparisons reported, P values were so low (<0.0001) that this issue would not arise. Distributions of most of the laboratory measures were skewed to the right. However, logarithmic and square root transformations of these measures resulted in distributions that were skewed to the left. As residuals from the multivariate models fit to nontransformed measures were not skewed (or at least not more skewed than residuals of models fit to transformed measures), no transformations were made. In addition, the results of these analyses were not meaningfully affected by exclusion of extreme values.
RESULTS
Forty-five people enrolled in the study: 19 in the healthy group, 11 in the group with known immunological disease, and 15 in the MCS group.
T-cell measures. (i) Reproducibility of replicates.
Table 1 presents distributions of the absolute values of differences between T-cell measures among the replicate samples analyzed at the five laboratories, including the shipped samples (analyzed at all laboratories) and the fresh replicates (analyzed at JHU). The overall mean for all samples for each phenotype is also given, for comparison to the absolute differences. Except for one phenotype (CD26+ CD3+), the mean absolute differences between replicate samples analyzed at the same laboratory and condition were <2 percentage points, and the median differences were generally 1% or less. Moreover, as can be seen from the 90th percentile figures, for all phenotypes the vast majority of replicates analyzed at the same laboratory were within 3 percentage points of each other, which is considered to be acceptable variation among flow cytometric measurements of replicate specimens (5). This close agreement was observed even for dim markers such as CD25 and CD26, which are considered to be difficult to measure. The closeness of the replicates provides a firm basis for making the statistical inferences described below. No gross differences among laboratory performance in replicate reproducibility were evident, although we did not have the power (with only 40 subjects with replicates analyzed at each laboratory) to evaluate this statistically.
TABLE 1.
Absolute values of differences in replicate samples across the five laboratories
| Phenotype | Distribution of replicate differences
|
Phenotype mean (%) | ||
|---|---|---|---|---|
| Mean | Median | 90th percentile | ||
| CD4+ | 1.51 | 1 | 3 | 47.3 |
| CD8+ | 1.14 | 0.9 | 2 | 18.2 |
| CD25+ CD3+ | 1.84 | 1 | 4 | 18.5 |
| CD26+ CD3+ | 2.21 | 1.4 | 4.6 | 48.3 |
| CD26+ CD4+ | 1.57 | 1 | 3.2 | 36.0 |
| CD26+ CD8+ | 1.07 | 0.77 | 2.20 | 12.9 |
| CD38+ CD4+ | 1.29 | 1 | 2.50 | 25.7 |
| CD38+ CD8+ | 0.78 | 0.46 | 2 | 9.13 |
| CD25+ CD8+ | 1.10 | 0.47 | 2.54 | 6.84 |
| CD25+ CD4+ | 1.31 | 1 | 2.80 | 14.2 |
| DR+ CD4+ | 0.50 | 0.37 | 1 | 3.74 |
| DR+ CD8+ | 0.97 | 0.57 | 2 | 6.24 |
(ii) Effect of sample shipment.
We next examined differences between T-cell measures for fresh and shipped samples analyzed at JHU. Table 2 presents point estimates and confidence limits for these differences. Results from fresh and shipped samples had very close agreement, with mean differences of less than 1% for all phenotypes. Although some statistically significant differences were observed (i.e., for the CD25+ CD3+, CD25+ CD4+ and CD38+ CD4+ phenotypes), their magnitudes were not biologically important. Limits for 95% confidence intervals for the true mean differences between fresh and shipped samples never exceeded ±1.5 percentage points.
TABLE 2.
Comparison of T-cell subsets in 43 pairsa of fresh and shipped samples analyzed at the JHU site
| Phenotype | Mean % ± SD in:
|
Statistical analysis (shipped − fresh)
|
|||
|---|---|---|---|---|---|
| Shipped samples | Fresh samples | Main effectb | 95% CIc | P valued | |
| CD4+ | 47.1 ± 10.8 | 47.0 ± 11.0 | −0.02 | −0.35, 0.31 | 0.91 |
| CD8+ | 18.7 ± 6.74 | 18.2 ± 7.68 | 0.11 | −0.30, 0.52 | 0.56 |
| CD26+ CD3+ | 46.2 ± 9.94 | 45.9 ± 10.3 | 0.23 | −0.64, 1.10 | 0.58 |
| CD26+ CD4+ | 34.2 ± 9.61 | 34.3 ± 9.74 | 0.09 | −0.54, 0.72 | 0.75 |
| CD26+ CD8+ | 12.0 ± 4.98 | 11.6 ± 6.10 | 0.14 | −0.20, 0.48 | 0.41 |
| CD25+ CD3+ | 13.8 ± 7.28 | 14.4 ± 7.06 | −0.71 | −1.39, −0.03 | 0.04 |
| CD25+ CD4+ | 10.3 ± 4.38 | 10.8 ± 4.61 | −0.68 | −1.23, −0.13 | 0.02 |
| CD25+ CD8+ | 3.49 ± 4.33 | 3.60 ± 4.66 | −0.03 | −0.24, 0.18 | 0.74 |
| DR+ CD4+ | 3.04 ± 1.36 | 3.03 ± 1.23 | 0.02 | −0.08, 0.12 | 0.71 |
| DR+ CD8+ | 4.61 ± 5.26 | 4.42 ± 5.96 | 0.08 | −0.12, 0.28 | 0.38 |
| CD38+ CD4+ | 25.0 ± 8.67 | 26.1 ± 9.18 | −0.47 | −0.83, −0.11 | 0.01 |
| CD38+ CD8+ | 6.04 ± 3.82 | 6.30 ± 4.14 | −0.13 | −0.31, 0.05 | 0.16 |
In 40 cases, there were two replicates per subject for both fresh and shipped samples.
The main-effect estimates for each parameter are not identical to the differences between the overall means for shipped samples and the overall means for fresh samples, because the overall means are based on all subjects while the statistical comparisons consider only the 40 subjects who had replicates for both fresh and shipped samples.
CI, confidence interval.
Based on the 40 subjects with two fresh replicates and two shipped replicates.
(iii) Laboratory comparisons.
Table 3 presents the means and standard deviations of T-cell phenotype percentages for shipped samples tested by each laboratory. In addition to real laboratory differences, variability in these means across laboratories can be mediated by random (within-laboratory) date-of-analysis effects and to a minor extent via small differences in the numbers of subjects tested at each laboratory. The P values shown were based on an approach that attempts to conservatively adjust for the unbalanced study design, as explained in the appendix. Specifically, F tests were based on the ratio of type III sums of squares for laboratory and for day nested within laboratory.
TABLE 3.
Comparisons of phenotype percentages across laboratories for shipped samples
| Phenotype | Mean % ± SD (na) determined in laboratory:
|
Largest mean/ smallest mean | P value | ||||
|---|---|---|---|---|---|---|---|
| A | B | Cb | D | E | |||
| CD4+ | 47.1 ± 9.8 (44) | 46.9 ± 11.3 (44) | 48.8 ± 10.5 (44) | 46.7 ± 10.0 (43) | 47.1 ± 10.8 (43) | 1.04 | 0.06 |
| CD8+ | 24.1 ± 9.2 (44) | 20.1 ± 7.9 (44) | 24.9 ± 9.2 (44) | 19.7 ± 6.85 (43) | 18.7 ± 6.7 (43) | 1.33 | <0.0001 |
| CD26+ CD3+ | 45.6 ± 10.7 (44) | 54.4 ± 10.1 (41) | 45.2 ± 17.0 (44) | 52.7 ± 13.0 (43) | 46.2 ± 9.94 (43) | 1.20 | 0.001 |
| CD26+ CD4+ | 35.1 ± 9.1 (44) | 38.7 ± 10.0 (41) | 38.0 ± 10.2 (43) | 34.2 ± 9.61 (43) | 1.13 | <0.0001 | |
| CD26+ CD8+ | 10.5 ± 4.7 (44) | 15.7 ± 6.2 (41) | 14.7 ± 6.27 (43) | 12.0 ± 4.98 (43) | 1.50 | <0.0001 | |
| CD25+ CD3+ | 3.4 ± 1.6 (44) | 30.7 ± 11.3 (41) | 10.8 ± 7.4 (44) | 38.5 ± 12.4 (43) | 13.8 ± 7.28 (43) | 11.4 | <0.0001 |
| CD25+ CD4+ | 3.1 ± 1.5 (44) | 21.0 ± 9.8 (41) | 25.8 ± 8.97 (43) | 10.3 ± 4.38 (43) | 8.4 | <0.0001 | |
| CD25+ CD8+ | 1.0 ± 0.2 (18) | 9.7 ± 5.4 (41) | 12.6 ± 6.71 (43) | 3.49 ± 4.33 (43) | 12.1 | <0.0001 | |
| DR+ CD4+ | 4.03 ± 1.4 (44) | 4.0 ± 1.7 (41) | 4.60 ± 1.75 (43) | 3.04 ± 1.36 (43) | 1.51 | <0.0001 | |
| DR+ CD8+ | 5.6 ± 4.6 (43) | 7.0 ± 7.1 (41) | 9.5 ± 10.1 (44) | 6.35 ± 4.51 (43) | 4.61 ± 5.26 (43) | 2.05 | <0.0001 |
| CD38+ CD4+ | 22.6 ± 7.7 (44) | 24.7 ± 12.0 (41) | 30.3 ± 9.05 (43) | 25.0 ± 8.67 (43) | 1.34 | 0.0005 | |
| CD38+ CD8+ | 9.94 ± 4.4 (44) | 8.56 ± 4.9 (41) | 14.2 ± 6.2 (43) | 9.60 ± 5.32 (43) | 6.04 ± 3.82 (43) | 2.35 | <0.0001 |
Number of patients analyzed at the laboratory. In almost all cases, two replicates per subject were analyzed for each phenotype at each laboratory; occasionally, only one replicate per patient was analyzed.
Laboratory C did not measure CD26+ CD4+, CD26+ CD8+, CD25+ CD8+, CD25+ CD4+, DR+ CD4+, and CD38+ CD4+ cells.
The between-laboratory differences for all phenotypes studied were quite statistically significant, except for the CD4+ phenotype, which had little laboratory variation (the largest laboratory mean was only 5% greater than the smallest [48.8 versus 46.6 percentage points]). This probably reflects the fact that CD4+ is the most commonly measured phenotype clinically and thus has the best quality control. For the CD8+ phenotype, the between-laboratory variation was larger, with the largest laboratory mean (24.9%) being 33% larger than the smallest mean (18.7%). The CD26+ CD3+ and CD26+ CD4+ phenotypes also had relatively low between-laboratory differences, with the highest laboratory mean being ≤20% greater than the lowest laboratory mean. This level of agreement across all laboratories in measurement of CD26 expression was surprisingly good, as there are no reference standards for this measurement. For the CD38+ CD4+, DR+ CD4+, and CD26+ CD8+ phenotypes, the between-laboratory variation was somewhat greater, with differences of 34 to 51% between the highest and lowest laboratory means, and for the DR+ CD8+ and CD38+ CD4+ phenotypes, the ratio of largest to smallest laboratory means was >2. Generally, the ratios of largest to smallest laboratory means were highest in the activated T-cell subsets, especially CD8+ DR+ lymphocytes and CD25+ cells, among all T-cell subsets. Specifically, the ratio of largest to smallest laboratory mean was up to 11.4 for the CD25+ CD4+, CD25+ CD8+, and CD25+ CD3+ phenotypes. Much of this variation was due to one laboratory (laboratory A in Table 3), which obtained CD25+ values that were lower than those of the other laboratories and were implausible based on reported studies of T cells from healthy humans (25, 37). However, even if data from this laboratory were omitted, the largest laboratory means for phenotypes including CD25+ were 3.5- to 4.6-fold greater than the smallest laboratory means.
(iv) Disease group comparisons.
Table 4 presents T-cell phenotype summary statistics for all samples tested (across day and laboratory) for healthy subjects and those in the group with immunological disease. We have not presented summary statistics for individuals identified with MCS because these cases were not clinically confirmed, and a study with confirmed MCS cases will be reported separately. In Table 4, however, P values for differences among all three disease groups are reported (consistent with the models fit). Disease group differences (among all three groups) were statistically significant for the CD8+, CD25+ CD3+, CD26+ CD8+, CD38+ CD8+, and DR+ CD8+ phenotypes. The magnitudes of the differences between healthy individuals and those with immunological diseases in Table 4 were generally far smaller than the magnitudes of laboratory differences in Table 3. Except for the DR+ CD8+ phenotype, where the mean for individuals with immunological disease (10.6%) was 2.7-fold greater than that for healthy individuals (3.9%), the ratio of the mean of the higher group to that of the lower group was never more than 2.
TABLE 4.
Comparisons of T-cell subsets by disease group
| Phenotype | Mean % ± SD (na) in:
|
Largest mean/smallest mean | P valueb | |
|---|---|---|---|---|
| Healthy group | Group with immunological disease | |||
| CD4+ | 47.9 ± 7.13 (225) | 43.6 ± 17.0 (130) | 1.13 | 0.13 |
| CD8+ | 19.6 ± 5.67 (225) | 24.6 ± 12.0 (130) | 1.29 | 0.03 |
| CD26+ CD3+ | 45.9 ± 10.7 (221) | 46.7 ± 17.8 (127) | 1.13 | 0.47 |
| CD26+ CD4+ | 35.8 ± 7.98 (183) | 31.8 ± 14.2 (103) | 1.25 | 0.83 |
| CD26+ CD8+ | 11.0 ± 3.78 (183) | 14.9 ± 8.23 (104) | 1.35 | 0.04 |
| CD25+ CD3+ | 15.9 ± 13.6 (221) | 18.5 ± 15.7 (127) | 1.40 | 0.03 |
| CD25+ CD4+ | 12.9 ± 10.5 (183) | 13.8 ± 10.8 (103) | 1.26 | 0.07 |
| CD25+ CD8+ | 4.74 ± 5.50 (157) | 7.61 ± 7.60 (89) | 1.93 | 0.15 |
| DR+ CD4+ | 3.25 ± 1.13 (183) | 4.30 ± 2.26 (104) | 1.10 | 0.76 |
| DR+ CD8+ | 3.90 ± 2.82 (218) | 10.6 ± 10.7 (125) | 2.71 | 0.007 |
| CD38+ CD4+ | 27.3 ± 9.13 (183) | 21.8 ± 11.8 (104) | 1.25 | 0.84 |
| CD38+ CD8+ | 8.70 ± 4.67 (221) | 12.2 ± 7.11 (125) | 1.68 | 0.006 |
Number of patient replicates analyzed.
Based on comparison of all three study disease groups, including putative MCS.
(v) Importance of differences between patients and days.
There were 45 subjects nested within three disease groups. Since samples were taken from the 45 subjects on 17 different dates (i.e., two or three subjects were sampled on the same date in the clinic, and all samples taken on the same date were analyzed on the same date in each laboratory), there were 85 different laboratory dates of analysis (17 dates nested within five laboratories). When we designed this study, we anticipated that there might be a patient effect on measures (i.e., one patient in the healthy group could have higher CD8+ percentage than a different patient in the healthy group). We did not anticipate that there might be a within-laboratory date-of-analysis effect (i.e., samples analyzed on a given date in a laboratory would tend to have higher CD8+ values than those analyzed on a different date in the same laboratory).
For all of the measures, both the nested patient effects (45 patients nested in three disease groups) and the date-of-analysis differences (85 dates nested in 17 laboratories) were highly statistically significant (P < 0.0001, with large F values), strongly suggesting that these effects were statistically real. However, the fact that the study design was unbalanced with respect to date-of-analysis and patient effects (because the same sampled patients were always nested together in the same analysis date across all five laboratories) made it impossible to completely separate, and quantify the magnitude of, the patient effect and the date of analysis effect.
Analysis of autoantibody data.
In most cases, the antibodies tested were not detected or were within the laboratory's normal limits, with occasional large values for some subject-visits. The reproducibility of the tests was again very good on the replicates run by all laboratories. Antibody tests determined by titer (i.e., dilution of the specimen serially until the reactivity can no longer be detected) conventionally are allowed to differ by up to two serial dilutions if twofold dilutions are done. The laboratories participating in this study had this level of reproducibility for almost all specimens (data not shown). However, the prevalences of autoantibodies that were detectable were much less than anticipated in all study groups, even in the group with immune diseases, perhaps because these patients were generally being treated (with glucocorticoids and/or other immunosuppressive agents for systemic lupus erythematosus and with thyroid-suppressive therapy for Graves's disease). Overall, one laboratory had the greatest number of positive tests, detecting abnormal autoantibody levels in six subjects, mostly in the immunologically abnormal group. Only one laboratory found detectable antimyelin antibody in any of the specimens, and these results were highly reproducible in that laboratory. Still, virtually all of the results for this antibody were in the stated normal range. Any effects of shipment on the antibody titers or concentrations measured were very small and not clinically important, with titers being reduced by one dilution at most in some tests.
DISCUSSION
This study evaluated whether certain immunological measures, including some which have been commonly reported as abnormal in MCS, meet the minimal requirement for validity: namely, that they are reproducible as performed in patient testing. It should be emphasized that the goal of this study was not to determine the actual utility of these tests in discriminating subjects with and without MCS but rather to evaluate whether the tests' performance characteristics would support the use of the tests for this purpose. Since our ability to evaluate antibody testing was limited by the rarity of positive tests, this discussion focuses primarily on T-cell phenotypes.
The basic findings for T cells were reassuring. Participating laboratories generally had very strong within-date-of-analysis reproducibility, as reflected in the close agreement with the vast majority of replicate samples. Similarly, shipping of blood by overnight express mail did not materially affect any of the measures; while some statistically significant differences between the means for fresh and shipped samples were observed, the magnitudes of these differences were small. The effect of shipping has not been well studied for most of these markers, although the major T-cell subsets (i.e., CD4+ and CD8+) are known not to be affected by shipping as long as the shipping time and temperature are not extreme. The variability of expression of CD38 and HLA-DR expression by shipped CD8+ T cells analyzed in several laboratories was found to be substantial in one study (17), although fresh unshipped samples were not analyzed in that study. Those authors emphasized the need for quality control in the analysis of shipped specimens. The findings of the present study extend this by directly supporting the validity of using individual laboratories to analyze shipped samples of whole blood for the immunological markers assessed in this study, if the analysis is carefully done and validated, as described below.
The picture with respect to interlaboratory agreement was more mixed. Overall, there was good agreement on most of the measurements performed, including many which might have been expected to be more variable, such as those of CD26, HLA-DR, and CD38. However, variability was much greater across laboratories than within laboratory replicates for these markers, and for CD25 the between-laboratory variation was very wide indeed, due in part, although not entirely, to an apparently consistent falsely low determination of this marker in one laboratory. Nevertheless, taken as a whole, these data support the value of laboratory test validation and quality control in studies of MCS and other diseases. They also emphasize the need for the careful selection and validation of tests to be performed, since test validation may require substantial effort. In this connection, we have employed the markers that were validated for shipping and multilaboratory analysis in the present study in a separate, clinically rigorous study to determine if the T-cell subsets identified by these markers actually distinguish persons with and without MCS, in terms of either percentages of lymphocytes (as done in this study) or absolute cell counts. The results of that study will be reported separately (C. S. Mitchell et al., unpublished data).
For all measures, there was statistically significant within-laboratory date-of-analysis variation. While the magnitude of this variation was difficult to quantify, it appeared to be smaller than the between-laboratory differences. This source of variation can influence statistical comparisons among groups if it is not incorporated into the analysis and the groups are not balanced across dates. Indeed, Simon et al. (32) observed that a (systematic) laboratory measurement trend in interleukin-1 generation at a single laboratory in their study of MCS influenced comparisons of disease groups, because a higher portion of one disease group was tested earlier in the study. Although the phenomenon of within-laboratory date-of-analysis variation has not been well studied, potential reasons for within-laboratory measurement differences by date of analysis include (i) day-to-day variation in sample processing, (ii) variation in laboratory practices by the technician (or by different technicians on different days), and (iii) changes in ambient temperatures and other conditions for shipping and storing samples. Moreover, consistent performance over one time period, even one as long as the 7 months of this study, does not guarantee long-term consistency.
This potential effect of date of test, together with potential intraday collinearities of samples processed and analyzed together, complicated the statistical analysis in this study. In particular, associations due to disease group (control, MCS, or autoimmune disease) could not be easily separated from associations due to date of analysis, because in many cases all individuals analyzed on a given day were from the same disease group. For the same reason, interaction between laboratory and disease group could not be tested, because higher measures for one disease group at a specific laboratory could have been be mediated by date-of-analysis effects in ways that were would be difficult to include in a linear model.
These considerations suggest that studies with several treatment or patient groups should maintain a temporal balance in the distribution of samples from the disease groups tested, with (roughly) the same portion of samples from each disease group tested at each date of analysis. While this may be logistically difficult, other approaches, such as incorporating date of analysis into multivariate comparisons (as was done by Simon et al. [32]) or running known standards in laboratory testing at regular time intervals, may help to mitigate the influence of date-of-analysis effects on disease group comparisons. Further, given that laboratory performance may change over time, the participation of laboratories in ongoing quality assurance programs (13, 17) is important, and such participation should be described in studies of these markers. Where no quality assurance program is available (e.g., for exploratory studies of new markers or markers that have not been well standardized across laboratories), it may be desirable to have the measurements performed in more than one laboratory. If cost permits, duplicate samples from the same individual should be stored and tested on different days to improve the precision of estimates and identify components of variance. Further research to characterize and quantify the intralaboratory date-of-analysis effect that was observed in the present study may help in development of techniques to minimize its influence on study results.
The data presented here have some important limitations. Not all immunological markers that may be pertinent to MCS were studied. Although no major effects of shipping on autoantibody measurement were detected, the number of specimens with detectable levels of autoantibodies was smaller than expected. Therefore, further studies will be needed to determine the magnitude and importance of shipping and laboratory variations in these measurements. The fact that substantial variation was observed across laboratories even in this small number underscores this need.
Understanding whether immunological mechanisms play a role in MCS, or in subsets of individuals diagnosed with MCS, remains an important research goal. The results of this study support the potential ability of the immunophenotypic tests evaluated to detect strong associations between immunological parameters and diseases of interest, such as MCS. At the same time, however, they show that in order to have complete confidence in studies of this nature, attention must be paid to quality assurance for all tests conducted, including balanced testing of subjects in different disease groups across different dates.
Acknowledgments
We thank Michelle Petri, Paul Ladenson, and Marge Evert for help with recruitment of study participants. We thank Ellen Taylor, Nicole Carpenetti, Karen Eckert-Kohl, Stacey Meyerer, Emily Christman, Elvia Ramirez, and Donna Ceniza for laboratory assistance and Dongguang Li and Joshua Shinoff for assistance with data analysis.
This study was funded by a grant from the Washington State Department of Labor and Industries. Immunosciences, Inc., and Specialty Laboratories, Inc., provided discounted rates for the tests performed in this research. D.R.H. was supported by NIH grant MH43450 and NSF grant EIA 02-05116.
APPENDIX
A nested fixed/random effects model (9) was fit to test for all factors studied on shipped samples. The specific model was Yijklm = u + ai + bij(ai) + ck + dkl(ck) + eijklm, where Yijklm is the mth replicate (m = 1,2) from the lth person within the kth disease group (k = 1,2,3) measured on the jth day (j = 1,…,17) at the ith laboratory (i = 1,…,5); u is the main effect; ai for i = 1,…,5 is the random effect of laboratory i, which has mean 0 and variance σa2; bij(ai) is the random effect of day j nested within laboratory i, which has mean 0 and variance σb2; ck for k = 1,2,3 is the effect of disease group k with 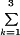 ck = 0; dkl(ck) is the random effect of patient k nested within disease group l, which has mean 0 and variance σd2; and eijklm is the random effect of replicate m nested within all of all the other groups, which has mean 0 and variance σe2. Note that σe2 consists of both sampling error and within-day laboratory testing error.
ck = 0; dkl(ck) is the random effect of patient k nested within disease group l, which has mean 0 and variance σd2; and eijklm is the random effect of replicate m nested within all of all the other groups, which has mean 0 and variance σe2. Note that σe2 consists of both sampling error and within-day laboratory testing error.
Since the design was not balanced (primarily due to the inseparability of date-of-analysis effect from disease group effect), hypotheses were tested by using type III sums of squares, which tend to conservatively exclude nonidentifiable variance from directed sums of squares. Hypotheses about day effect (σb2 = 0) and patient effect (σd2=0 = 0) were tested by F tests with comparison of type III sums of squares for days and patients to type III sums of squares for residual error σe2 (30). Hypotheses for the group effect being 0 (ck ≡ 0) were tested by F tests comparing type III sums of squares for groups to type III sums of squares for nested patients. Hypotheses for the laboratory effect being 0 (σa2 = 0) were tested by F tests comparing type III sums of squares for laboratory to type III sums of squares for nested days.
A simpler model was fit to test the effect of fresh versus shipped samples evaluated at JHU. All terms were the same as in the previous model, except that ai corresponds to fresh (i = 1) versus shipped (i = 2) with 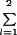 ai = 0. In order to obtain a balanced design, we excluded a disease group effect from the model and restricted inclusion to the 40 (of 45) patients who had both fresh and shipped samples tested for each laboratory parameter. We further reduced the F test to a paired t test of differences of corresponding date-of-analysis means to facilitate construction of confidence limits.
ai = 0. In order to obtain a balanced design, we excluded a disease group effect from the model and restricted inclusion to the 40 (of 45) patients who had both fresh and shipped samples tested for each laboratory parameter. We further reduced the F test to a paired t test of differences of corresponding date-of-analysis means to facilitate construction of confidence limits.
REFERENCES
- 1.Bartha, L., W. Baumzweiger, D. S. Buscher, T. Callender, K. A. Dahl, A. Davidoff, A. Donnay, S. B. Edelson, B. D. Elson, E. Elliott, D. P. Flayhan, G. Heuser, P. M. Keyl, K. H. Kilburn, P. Gibson, L. A. Jason, J. Krop, R. D. Mazlen, R. G. McGill, J. McTamney, W. J. Meggs, W. Morton, M. Nass, L. C. Oliver, D. D. Panjwani, L. A. Plumlee, D. Rapp, M. B. Shayevitz, J. Sherman, R. M. Singer, A. Solomon, A. Vodjani, J. M. Woods, and G. Ziem. 1999. Multiple chemical sensitivity: a 1999 consensus. Arch. Environ. Health 54:147-149.10444033 [Google Scholar]
- 2.Bell, I. R. 1975. A kindling model of mediation for food and chemical sensitivities: biobehavioral implications. Ann. Allergy 35:206-215. [PubMed] [Google Scholar]
- 3.Black, D. W., B. N. Doebbeling, M. D. Voelker, W. R. Clarke, R. F. Woolson, D. H. Barrett, and D. A. Schwartz. 2000. Multiple chemical sensitivity syndrome: symptom prevalence and risk factors in a military population. Arch. Intern. Med. 160:1169-1176. [DOI] [PubMed] [Google Scholar]
- 4.Burek, C. L., and N. R. Rose. 1995. Autoantibodies. Diagn. Immunopathol. 13:207-229. [Google Scholar]
- 5.Calvelli, T., T. N. Denny, H. Paxton, R. Gelman, and J. Kagan. 1993. Guideline for flow cytometric immunophenotyping: a report from the National Institute of Allergy and Infectious Diseases, Division of AIDS. Cytometry 14:702-715. [DOI] [PubMed] [Google Scholar]
- 6.Cullen, M. R. 1987. The worker with multiple chemical sensitivities: an overview. Occupat. Med. 2:655-661. [PubMed] [Google Scholar]
- 7.Davidoff, A. L., and L. Fogarty. 1994. Psychogenic origins of multiple chemical sensitivities syndrome: a critical review of the research literature. Arch. Environ. Health 49:316-323. [DOI] [PubMed] [Google Scholar]
- 8.Donnay, A. 1999. On the recognition of multiple chemical sensitivity in medical literature and government policy. Int. J. Toxicol. 18:383-392. [Google Scholar]
- 9.Dunn, O. J., and V. A. Clark. 1974. Applied statistics: analysis of variance and regression. John Wiley & Sons, New York, N.Y.
- 10.Giardino, N. D., and P. M. Lehrer. 2000. Behavioral conditioning and idiopathic environmental intolerance, p. 519-528. In P. J. Sparks (ed.), Occupational medicine: multiple chemical sensitivity/idiopathic environmental intolerance. Hanley & Belfus, Inc., Philadelphia, Pa. [PubMed]
- 11.Graveling, R. A., A. Pilkington, P. K. George, M. P. Butler, and S. N. Tannahill. 1999. A review of multiple chemical sensitivity. Occupat. Environ. Med. 56:73-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heuser, G., A. Wojdani, and S. Heuser. 1992. Multiple chemical sensitivities: addendum to biologic markers in immunotoxicology, p. 117-138. National Academy Press, Washington, D.C.
- 13.Kagan, J., R. Gelman, M. Waxdal, and P. Kidd. 1993. Quality assessment program. Ann. N.Y. Acad. Sci. 677:50. [DOI] [PubMed] [Google Scholar]
- 14.Kipen, H., N. Fiedler, C. Maccia, E. Yurkow, J. Todaro, and D. Laskin. 1992. Immunologic evaluation of chemically sensitive patients. Toxicol. Ind. Health 8:125-135. [PubMed] [Google Scholar]
- 15.Kreutzer, R., R. R. Neutra, and N. Lashuay. 1999. Prevalence of people reporting sensitivities to chemicals in a population-based survey. Am. J. Epidemiol. 150:1-12. [DOI] [PubMed] [Google Scholar]
- 16.Landay, A., C. Jessop, E. T. Lennette, and J. A. Levy. 1991. Chronic fatigue syndrome: clinical condition associated with immune activation. Lancet 338:707-712. [DOI] [PubMed] [Google Scholar]
- 17.Landay, A. L., D. Brambilla, J. Pitts, G. Hillyer, D. Golenbock, J. Moye, S. Landesman, J. Kagan, and the NIAID/NICHD Women and Infants Transmission Study Group. 1995. Interlaboratory variability of CD8 subset measurements by flow cytometry and its applications to multicenter clinical trials. Clin. Diagn. Lab. Immunol. 2:462-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levin, A. S., J. J. McGovern, Jr., J. B. Miller, L. Lecam, and J. Lazaroni. 1992. Multiple chemical sensitivities: a practicing clinician's point of view. Clinical and immunologic research findings. Toxicol. Ind. Health 8:95-109. [PubMed] [Google Scholar]
- 19.Liu, Z., W. G. Cumberland, A. H. Kaplan, L. E. Hultin, R. Detels, and J. V. Giorgi. 1998. CD8+ lymphocyte activation in HIV disease reflects an aspect of pathogenesis distinct from viral burden and immunodeficiency. J. Acquir. Immune Defic. Syndr. 18:332-340. [DOI] [PubMed] [Google Scholar]
- 20.McGovern, J. J., Jr., J. A. Lazaroni, M. F. Hicks, J. C. Adler, and P. Cleary. 1983. Food and chemical sensitivity: clinical and immunologic correlates. Arch. Otolaryngol. Head Neck Surg. 109:292-297. [DOI] [PubMed] [Google Scholar]
- 21.Meggs, W. J. 1992. Multiple chemical sensitivities and the immune system. Toxicol. Ind. Health 8:203-214. [PubMed] [Google Scholar]
- 22.Meggs, W. J. 1993. Neurogenic inflammation and sensitivity to environmental chemicals. Environ. Health Perspect. 101:234-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller, C. S. 1997. Toxicant-induced loss of tolerance—an emerging theory of disease? Environ. Health Perspect. 105:445-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell, C. S., A. Donnay, D. R. Hoover, and J. B. Margolick. 2000. Immunologic parameters of multiple chemical sensitivity, p. 647-665. In P. Sparks (ed.), Occupational medicine: state of the art reviews. Hanley & Belfus, Inc., Philadelphia. Pa. [PubMed]
- 25.Prince, H. E., V. Kermanni-Arab, and J. L. Fahey. 1984. Depressed interleukin-2 receptor expression in acquired immunodeficiency. J. Immunol. 133:1313-1317. [PubMed] [Google Scholar]
- 26.Rea, W. J., I. R. Bell, C. W. Suits, and R. E. Smiley. 1978. Food and chemical susceptibility after environmental chemical overexposure: case histories. Ann. Allergy 41:101-110. [PubMed] [Google Scholar]
- 27.Rea, W. J., A. R. Johnson, S. Youdim, E. J. Fenyves, and N. Samadi. 1986. T and B lymphocyte parameters measured in chemically sensitive patients and controls. Clin. Ecol. 4:11-14. [Google Scholar]
- 28.Rea, W. J., Y. Pan, A. R. Johnson, and E. J. Fenyves. 1987. T and B lymphocytes in chemically sensitive patients with toxic volatile organic hydrocarbons in their blood. Clin. Ecol. 5:171-175. [Google Scholar]
- 29.Rothman, K. J. 1990. No adjustments are needed for multiple comparisons. Epidemiology 1:43-46. [PubMed] [Google Scholar]
- 30.Searle, S. R. 1971. Linear models. John Wiley & Sons, New York, N.Y.
- 31.Simon, G. E. 1994. Question and answer session 3 from the Conference on Low-Level Exposure to Chemicals and Neurobiologic Sensitivity, Baltimore, Md., 6-7 April 1994. J. Toxicol. Ind. Health 10:526-527. [PubMed] [Google Scholar]
- 32.Simon, G. E., W. Daniell, H. Stockbridge, K. Claypoole, and L. Rosenstock. 1993. Immunologic, psychological, and neuropsychological factors in multiple chemical sensitivity: a controlled study. Ann. Intern. Med. 119:97-103. [DOI] [PubMed] [Google Scholar]
- 33.Sparks, P. J. 2000. Idiopathic environmental intolerances: overview, p. 497-510. In P. Sparks (ed.), Occupational medicine: state of the art reviews. Hanley & Belfus, Inc., Philadelphia, Pa. [PubMed]
- 34.Terr, A. I. 1986. Environmental illness: a clinical review of 50 cases. Arch. Intern. Med. 146:145-149. [DOI] [PubMed] [Google Scholar]
- 35.Vogt, R. F., Henderson, L. O., Straight, J. M., and Orti, D. L. 1990. Immune biomarkers demonstration project: standardized data acquisition and analysis for flow cytometric assessment of immune status in toxicant-exposed persons. Cytom. Suppl. 4:103. (Abstract.) [Google Scholar]
- 36.Ziem, G., and J. McTamney. 1997. Profile of patients with chemical injury and sensitivity. Environ. Health Perspect. 105S:417-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zola, H., L. Y. Koh, B. X. Mantzioris, and D. Rhodes. 1991. Patients with HIV infection have a reduced proportion of lymphocytes expressing the IL2 receptor p55 chain (TAC, CD25). Clin. Immunol. Immunopathol. 59:16-25. [DOI] [PubMed] [Google Scholar]