

Abstract
A Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitor that is potent (KI = 15 nM) and species-selective (>5,000-fold over the human enzyme) was identified by high-throughput screening. The substituted triazolopyrimidine and its structural analogs were produced by an inexpensive three-step synthesis and the series showed good association between PfDHODH inhibition and parasite toxicity. This study has identified the first nanomolar PfDHODH inhibitor with potent antimalarial activity in whole cells (EC50 = 79 nM).
Introduction
Malaria infects up to 900 million people and causes as many as 2.7 million deaths worldwide each year.1,2 Nearly 40% of the world’s population is at risk for contracting the disease, which has been a major cause of mortality throughout history. Antimalarial drugs have been the mainstay for managing new infections as well as established disease. In recent decades wide-spread drug resistance has been encountered for chloroquine, as well as for almost every other available antimalarial agent.3 Further, there are indications that drug-resistance may be appearing at faster rates in some parts of the world.4 Multi-drug combinations offer temporary relief 5,6 but given current trends, it is clear that the disease will continue to have an unacceptable impact on global health unless novel drugs are developed. A significant portion of the current arsenal of malaria drug therapies is rooted in natural remedies, starting with the discovery of quinine nearly 350 years ago.7,8 The current challenge is to couple our knowledge of malaria genomics and biochemistry with modern platforms for drug discovery.
Pyrimidines are essential metabolites, required for DNA and RNA biosynthesis, as well as the biosynthesis of phospholipids and glycoproteins. Unlike mammalian cells, the malaria parasite cannot salvage preformed pyrimidine bases or nucleosides, and pyrimidines must be acquired through the de novo biosynthetic pathway.9-12 These biochemical results have been confirmed by the genome sequence showing pyrimidine salvage enzymes are missing from the parasite.13 Dihydrofolate reductase is a validated target for malaria treatment14 and inhibitors of thymidylate synthase have potent antimalarial activity,15-17 illustrating the importance of the pyrimidine biosynthetic pathway to parasite survival.
The fourth and rate-limiting step of pyrimidine biosynthesis is catalyzed by DHODH, a flavin mononucleotide (FMN)-dependent enzyme. Both the human and malarial enzymes are localized to the inner mitochondrial membrane and utilize ubiquinone (CoQ) as the physiological oxidant in the reaction.18,19 Recent studies have suggested that the sole function of the parasite mitochondrial electron transport chain is to provide oxidized CoQ to DHODH for the synthesis of pyrimidines, confirming the essential role that DHODH plays in the biology of the parasite.20 Inhibitors of human DHODH have proven efficacy for the treatment of rheumatoid arthritis,21,22 with an approved compound on the market for this application (2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide 1 (A77 1726)23 (Figure 1A), the active metabolite of 5-methyl-N-[4-(trifluoromethyl)phenyl]isoxazole-4-carboxamide), thus demonstrating that DHODH is a “druggable” target. The X-ray structures of both human and malarial DHODH have been determined.24,25 Orotate and FMN stack against each other in the center of the β/α barrel and the inhibitor-binding site is formed adjacent to this site by two α-helices, which lie between the predicted α-terminal transmembrane domain and the canonical β/α barrel domain (Figure 1B). The inhibitor binding-pocket has extensive variation in amino acid sequence between the human and malarial enzymes, providing the structural basis for the identification of species-specific inhibitors.
Figure 1.

A. Chemical structure of compound 1. B. Structure of PfDHODH active site. Residues conserved between P. falciparum and human DHODH are displayed in grey, variable residues are displayed in purple. Orotic acid (Oro), FMN and 1 are displayed in yellow. The figure was generated using PyMol from the file 1TV5.pdb.
We previously utilized a high-throughput screen (HTS) to identify a series of halogenated phenyl benzamide/naphthamides that are potent and species-selective inhibitors of PfDHODH.26 Tricyclic aromatic amines have also been reported to be parasite specific inhibitors of PfDHODH.27 However the inhibitors described in these previous studies had poor antimalarial activity in whole cell assays. Thus, while DHODH has received significant attention as a promising new target for the development of antimalarials, chemical validation of DHODH as a target in malaria had not been fully established.
Results and Discussion
HTS-based discovery of an antimalarial
Here we describe the identification of a PfDHODH inhibitor, (5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine 7 (DSM 1)28 (Figure 2A), that shows potent and species-selective anti-proliferative effects against the P. falciparum malaria parasite. Compound 7 was discovered by HTS of a 220,000 compound library of “drug-like” molecules using a colorometric enzyme assay.26 P. falciparum DHODH was inhibited by Compound 7 with an IC50 = 0.047 ± 0.022 μM, and it is >5,000-fold selective when compared to the human enzyme (Figure 3A; Table 1). It inhibits the proliferation of P. falciparum parasites in whole cell assays with similar potency (EC50 = 0.079 ± 0.048 μM for clone 3D7; Figure 3B) and it does not inhibit the growth of a mouse cell line (L1210) (EC50 > 10 micromolar). It is also highly active against drug resistant strains of P. falciparum, including the multiple-drug-resistant Dd2 (EC50 = 0.14 ± 0.05 μM).
Figure 2.

Compound 7. A. Chemical structure. B. X-ray crystal structure. C. 1H NMR in DMSO-d6 and D. Mass Spectra.
Figure 3.

Selective and potent inhibition of PfDHODH and P. falciparum cells by compound 7. A. Inhibition profile for PfDHODH (orange circles; IC50 0.047 ± 0.022 μM; n=6) compared to human DHODH (black diamonds; IC50 >200 μM); [E]T = 10 nM. B. Activity in whole cell assays against P. falciparum 3D7 (orange circles) or mouse L1210 (black diamonds) cells (EC50 = 0.079 ±0.045; n=9). C. Relationship between IC50 and substrate concentration ([E]T = 5 nM). KI was determined by fitting the data to Eq. 2 (KI = 0.015 ± 0.0011 μM). D. Rapid kinetic analysis showing compound 7 inhibits the CoQD-dependent oxidative half-reaction (bottom panel) but not the DHO-dependent reductive half-reaction (top panel). Blue trace (1) - no compound 7 and orange trace (2) - compound 7 (50 μM).
Table 1.
Structure and activity of the triazolopyrimidine-based series against PfDHODH and P. falciparum in whole cell assays
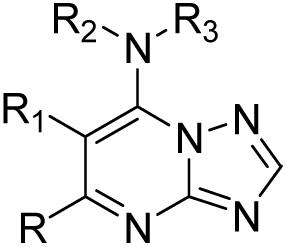 | ||||||
|---|---|---|---|---|---|---|
| compd | R | R1 | R2 | R3 | IC50PfDHODH | EC50(μM) Pf3D7 cells |
| 7 | CH3 | H | H | 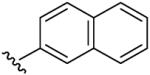 |
0.047±0.022 | 0.079±0.045 |
| 8 | CF3 | H | H | 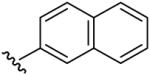 |
0.21±0.07 | 3.3±0.0 |
| 9 | C2H5 | H | H | 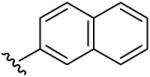 |
0.19±0.073 | 0.31±0.32 |
| 10 | CH3 | CH3 | H | 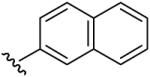 |
0.16±0.096 | 0.55±0.22 |
| 11 | CH3 | H | CH3 | 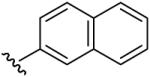 |
3.0±0.84 | 16±4.0 |
| 12 | CH3 | H | C6H5CH2 | 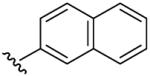 |
93±9 | 35±35 |
| 13 | CH3 | H | H | 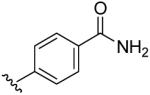 |
>200 | >50 |
| 14 | CH3 | H | H | 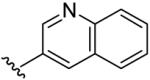 |
1.7±0.49 | 1.6±0.35 |
| 15 | CH3 | H | H | 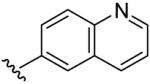 |
1.2±0.28 | 2.2±0.5 |
| 16 | CH3 | H | H | 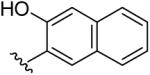 |
0.33 ±0.1 | 1.7±0.56 |
| 17 | CH3 | H | H | 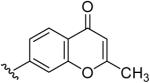 |
2.0±0.07 | 0.41±0.18 |
| 18 | CH3 | H | H | 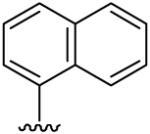 |
45±6.0 | 4.4±1.8 |
| 19 | CH3 | H | H | 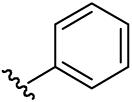 |
>100 | >100 |
| 20 | CH3 | H | H | 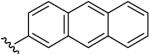 |
0.056±0.024 | 0.19±0.12 |
Errors represent the standard error of the mean. The IC50 for inhibition of human DHODH was >200 μM for all listed compounds. Enzyme data were collected with the DCIP assay. Growth inhibition by compound 7 was also tested on additional P. falciparum cell lines (EC50 values for FCR3, K1, Dd2, HB3 and D6 were 0.18±0.018, 0.14±0.008, 0.14±0.05, 0.10±0.044 and 0.058±0.001 respectively).
The initial structural annotation of compound 7 in the HTS chemical library data-base was incorrect as judged by high-resolution mass spectroscopy. The chemical identity of compound 7 was elucidated using NMR analysis of the original material and this data was consistent with the structure depicted in Figure 2A. Compound 7 was resynthesized using a simple 3-step synthetic method (Scheme 1). The absolute structure of resynthesized compound 7 was determined by x-ray crystallography (Figure 2B), and further confirmed by mass spectroscopy and NMR (Figure 2C and 2D).
Scheme 1. Synthesis of the triazolopyrimidine-based seriesa.
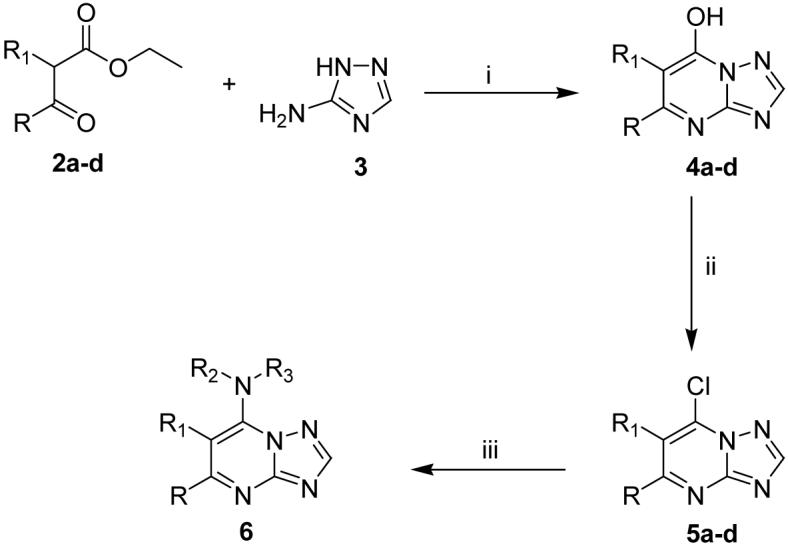
aReagents and conditions: (i) AcOH, 3.5-8 h, reflux, 40-58%; (ii) POCl3, 30-60 min., reflux, 43-65%; (iii) R2R3NH, EtOH, 8-15 h, rt, 80-87%.
Structure activity relationships (SAR)
A series of triazolopyrimidine analogs of compound 7 were synthesized and tested against the enzyme and against parasites in whole cell assays (Table 1). These compounds show a wide range of IC50 values against PfDHODH (0.05 μM - >200 μM), and importantly the inhibitory activity against the enzyme shows strong association with potency on the parasites in whole cell assays. These results are consistent with DHODH being the cellular target of compound 7 and its derivatives. The compound series also allowed us to establish an emerging and preliminary SAR for inhibitory activity on both PfDHODH and parasites. From this data we now know that: (i) R and R1 alkyl substituients can be modified with only a modest decrease in activity (8, 9, 10); (ii) substitutions at R2 resulted in a loss of potency (11, 12); (iii) the introduction of heteroatoms on, or in, the napthyl ring reduced activity (13, 14, 15, 16, 17); (iv) the naphthalene attached at the 2-position is optimal, whereas naphthalene attached at the 1-position showed reduced activity (18); (v) replacement of naphthalene with the smaller aniline group significantly reduced activity (19), while the larger anthracene moiety was well tolerated (20).
Binding mode and species selectivity
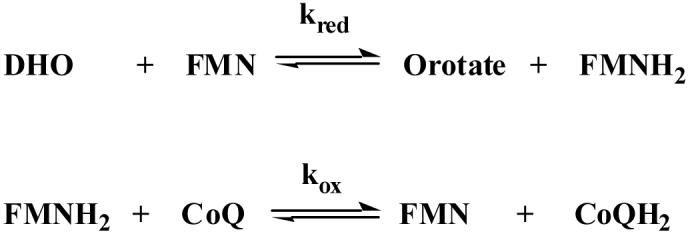
The biochemical mechanism of inhibition by compound 7 was studied in detail to gain insight into the structural basis for the selective and potent binding of this compound to PfDHODH. Steady-state kinetic analysis shows that the IC50 for compound 7 increases linearly with increasing CoQD concentration (Figure 3C) as expected for a competitive tight binding inhibitor.29 These data were fitted to Eq. 2 yielding a KI in the low nanomolar range (KI = 0.015 ± 0.001 μM). Pre-steady state stopped flow spectroscopy was performed to characterize the effect of compound 7 on the individual oxidative and reductive half-reactions catalyzed by PfDHODH. Compound 7 inhibited the oxidative half reaction (kox), preventing the transfer of electrons from FMN to CoQ, while it did not affect the DHO dependent reductive half reaction (kred) (Scheme 2, Figure 3D). We previously observed a similar pattern of inhibition for both compound 1 and the biphenylamide inhibitors from our HTS screen.30 Thus, these data suggest all three inhibitor classes utilize the same mechanism to inhibit DHODH. Finally, site-directed mutagenesis of residues in the inhibitor binding-site (F227A and R265A) of PfDHODH increased the IC50 of compound 7 by 940-fold and 130-fold respectively (IC50 = 44 ± 10 μM for F227A, and IC50 = 6.1 ± 1.1 μM for R265A), providing strong evidence that compound 7 is also bound in this site. As the inhibitor binding-site is not conserved in amino acid composition between P. falciparum and human DHODH this binding mode explains the profound species-selective binding of compound 7 and its derivatives (Figure 1).
Scheme 2.
Significance
Malaria is one of the most pressing medical problems in the developing world. Target-based drug discovery has been put forth as a promising mechanism for the discovery of new drugs, however it is often difficult to translate potency on the enzyme target to activity in whole cell assays. Our discovery of DHODH inhibitors by HTS screen that have potent antimalarial activity provides a successful example of this approach. The triazolopyrimidine-based series exhibits good association between PfDHODH inhibitory activity and antimalarial potency in the infected erythrocyte model, while showing no appreciable activity against the human enzyme or a mouse cell line. These data provide the first example of a DHODH inhibitor with low nanomolar activity in malaria whole cell assays, and the SAR analysis provides the best chemical evidence to date validating PfDHODH as a target for the discovery of new antimalarial compounds. Compound 7 is drug-like (as defined by Lipinski’s rule of 5),31 simple, and inexpensive to synthesize. Thus our study has identified a highly promising candidate for a lead optimization program to develop an antimalarial drug that exploits this new target.
Experimental Section
HTS screen
Compound 7 was identified by HTS based on a colorimetric endpoint assay of PfDHODH activity. The details of the HTS assay, the compound library and the screen method have been previously published.26
Protein purification and steady-state kinetic analysis
P. falciparum DHODH, mutant PfDHODHs and human DHODH were expressed in their truncated soluble form in E. coli, and purified as previously described.26,32 Steady-state kinetic assays were performed as previously described.26,32,33 The reduction of 2,6-dichloroindophenol (DCIP; 0.12 mM) was followed at 600 nm (ε=18.8 mM-1cm-1) using enzyme (ET = 5 - 10 nM) and substrates (0.2 mM L-dihydroorotate and 0.02 mM CoQD) in assay buffer (100 mM HEPES, pH 8.0, 150 mM NaCl, 10% Glycerol, 0.1%Triton) at 20°C. Background CoQ-independent oxidase activity (<3% of activity) was subtracted from the data prior to analysis. For potent compounds activity was confirmed by the direct detection of orotic acid at 296 nm (ε296 = 4.30 mM-1cm-1), which was also used for KI determination. Substrate and buffer conditions were as above except oxygen was depleted by the inclusion of an oxidase/catalase system (0.1 mg/ml glucose oxidase, 0.02 mg/ml catalase, and 50 mM glucose). Data were fitted to Eq. 1 to determine IC50 values and to Eq. 2 to determine KI, where Eq. 2 describes the relationship between IC50 and substrate concentration for a tight-binding competitive inhibitor.29
| Eq. 1 |
| Eq. 2 |
Pre-steady-state kinetic analysis by stopped flow spectroscopy
Rapid kinetic analysis was performed as described previously.30 The transition of FMN between the oxidized and reduced state was monitored at 485 nm on a Bio-Logic SFM-3 stopped-flow instrument. For DHO-dependent reactions, enzyme (final concentration 20 μM) was mixed with DHO (125 μM final concentration) in assay buffer at 4 °C. For CoQD-dependent reactions, oxygen was depleted from the reactions using the oxidase/catalase system (described above) and by bubbling with nitrogen. Oxidized enzyme (45 μM) was reduced with a limiting amount of DHO (30 μM) before loading on to the stopped-flow instrument. Enzyme reduced in this manner (final concentration 10 μM) was then mixed with CoQD (100 μM final concentration). For inhibitor analysis enzyme (10 or 20 μM final concentration) was pre-mixed with inhibitor (50 μM final concentration) prior to reaction with substrates and monitoring of the reaction.
P. falciparum cell culture
Parasite clones 3D7, FCR3, K1, Dd2, HB3 and D6 were propagated in Gibco-Invitrogen RPMI-1640 supplemented with 20% human type A+ plasma and 2% (w/v) red blood cells.34 Low-passage L1210 mouse leukemia cells (American Type Culture Collection) were also grown in plasma-supplemented RPMI-1640. Blood products were obtained from Biochemed Services, Virginia. To study inhibition of cell proliferation [3H]-hypoxanthine uptake was measured in drug-treated P. falciparum infected-erythrocytes and L1210 cells described previously.15 Data were fitted to Eq. 3 to determine EC50.
| Eq. 3 |
Curve fitting and error analysis
Enzyme IC50 and parasite EC50 data were determined over a range of inhibitor concentrations using triplicate data points at each concentration. IC50 values were determined using the graphing program Prism (GraphPad). Data reported in Table 1 represents the average of at least 2 independent experiments.
General Chemistry
Unless otherwise indicated, all anhydrous solvents were commercially obtained and stored under nitrogen. Reactions were performed under an atmosphere of dry nitrogen in oven-dried glassware and were monitored for completeness by thin layer chromatography (TLC) using silica gel 60 F-254 (0.25 mm) plates with detection with UV light. 7-Amino substituted [1,2,4]triazolo[1,5-a]pyrimidine compounds (Table 1) were prepared by Scheme 1. Briefly, 3-amino-[1,2,4]triazole 3 was condensed with the substituted ethyl acetoacetates 2a-d to form the substituted 7-hydroxy-[1,2,4]triazolo[1,5-a]pyrimidines 4a-d. Chlorination with phosphorous oxychloride gave the corresponding 7-chloro-[1,2,4]triazolo[1,5-a]pyrimidines 5a-d,35 which upon treatment with substituted aryl amines in ethanol resulted in the desired products (7-20).
Compounds 4a-d
A mixture of 3-amino-1,2,4-triazole 3 (20 mmol) and substituted ethyl acetoacetate 2a-d (20 mmol) was heated under reflux in acetic acid (10 ml) for 3.5-8 h. The product was then cooled to RT, filtered, washed with water, and dried under vacuum to give a white solid with 40-58% yield.
Compounds 5a-d
[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4a-d) (6.5 mmol) was added to 1.82 ml (19.5 mmol) of phosphorus oxychloride and heated under reflux for 30-60 min in a round bottom flask, during which the solid dissolved and hydrogen chloride was evolved. Excess phosphorus oxychloride was removed by distillation at reduced pressure on a steam-bath and the residue triturated with ice water. Product was extracted from the aqueous mixture with methylene chloride, evaporated and purified by column chromatography using 60% EtOAc/Hexane at a yield of 43-65%. Compound 5b was used for the next step without purification.
Compounds 7-20
The appropriate aryl substituted amine (1 mmol) was added to compound 5a-d (1 mmol) in absolute ethanol (10 ml) and stirred at RT for 8-15 h. Products were purified by column chromatography with CH2Cl2/MeOH/NH4OH (23:1:1). Yields ranged from 80-87%.
Analysis
1H NMR spectra were recorded on dilute solutions in CDCl3 or DMSO-d6 at 300 MHz. Chemical shifts are reported in parts per million (δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Electrospray ionization mass spectra were acquired on a Bruker Esquire Liquid Chromatograph-Ion Trap Mass Spectrometer. Flash chromatography was carried out with silica gel (32-63 μm). Melting points were taken in capillary tubes (Mel Temp apparatus) and are uncorrected. Compound 7 was crystallized in CH2Cl2/CH3OH. As a test of purity, compounds were subjected to HPLC analysis on an automated Varian Prep star system using a gradient of 20% MeOH to 100% MeOH (with 0.1% trifluoroacetic acid) over 30 min, using a YMC S5 ODS column (20 × 100 mm, Waters, Inc.). Compounds eluted as a single peak and the activity of the peak fraction was confirmed by demonstrating PfDHODH inhibitory activity. Representative HPLC peaks for the most active compounds, compounds 7 and 20, are provided as Supplemental data.
Physical properties
5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4a)
mp. 287 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.15 (s, 1H), 5.82 (s, 1H), 2.30 (s, 3H). MS m/z 151.1 (M + H+).
5-Trifluoromethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4b)
mp. 263 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.40 (s, 1H), 8.04 (s, 1H, OH), 6.14 (s, 1H). MS m/z 202.9 (M - H+).
5-Ethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4c)
mp. 215 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.18 (s, 1H), 5.82 (s, 1H), 2.60 (m, 2H), 1.21 (m, 3H). MS m/z 162.9 (M - H+).
5,6-Dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4d)
mp. 308 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.15 (s, 1H), 2.29 (s, 3H), 1.92 (s, 3H). MS m/z 163.0 (M - H+).
7-Chloro-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine (5a)
mp. 150 °C. 1H NMR (300 MHz, CDCl3): δ 8.50 (s, 1H), 7.15 (s, 1H), 2.75 (s, 3H). MS m/z 169.1 (M + H+).
7-Chloro-5-ethyl-[1,2,4]triazolo[1,5-a]pyrimidine (5c)
mp. 184 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.52 (s, 1H), 7.13 (s, 1H), 3.04 (m, 2H), 1.40 (m, 3H). MS m/z 183.1 (M + H+).
(7-Chloro-5,6-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidine (5d)
mp. 147 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.59 (s, 1H), 2.63 (s, 3H), 2.40 (s, 3H). MS m/z 183.1 (M + H+).
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine DSM1 (7)
mp. 220 °C (lit.36 216-17 °C). 1H NMR (300 MHz, DMSO-d6): δ 10.35 (brs, NH, exchangeable), 8.50 (s, 1H), 7.85-8.05 (m, 4H), 7.45-7.60 (m, 3H), 6.50 (s, 1H), 2.40 (s, 3H). MS m/z 276.1 (M + H+).
(5-Trifluoromethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine (8)
mp. 239 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.80 (s, 1H), 8.0-8.20 (m, 4H), 7.55-7.70 (m, 3H), 6.70 (s, 1H). MS m/z 330.1 (M + H+).
(5-Ethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine (9)
mp. 238 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.40 (brs, NH, exchangeable), 8.54 (s, 1H), 7.94-8.03 (m, 4H), 7.50-7.64 (m, 3H), 6.50 (s, 1H), 2.70 (m, 2H), 1.20 (m, 3H). MS m/z 290.2 (M + H+).
(5,6-Dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine (10)
mp. 262 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.0 (brs, NH, exchangeable), 8.82 (s, 1H), 7.75-7.95 (m, 3H), 7.35-7.60 (m, 4H), 2.65 (s, 3H), 2.05 (s, 3H). MS m/z 290.1 (M + H+).
Methyl-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine (11)
mp. 158 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.23 (s, 1H), 7.82-7.93 (m, 3H), 7.69 (s, 1H), 7.52 (m, 2H), 7.40 (d, J = 8.7 Hz, 1H), 6.64 (s, 1H), 3.73 (s, 3H), 2.51 (s, 3H). MS m/z 290.2 (M + H+)
Benzyl-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine (12)
mp. 172 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.35 (s, 1H), 7.75-7.90 (m, 4H), 7.40-7.50 (m, 5H), 7.15-7.30 (m, 3H), 6.53 (s, 1H), 5.63 (s, 2H), 2.45 (s, 3H). MS m/z 366.3 (M + H+).
4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ylamino)-benzamide (13)
mp. 294 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.60 (s, 1H), 7.90-8.04 (m, 2H and NH, exchangeable), 7.55 (m, 2H), 7.40 (brs, NH, exchangeable), 6.60 (s, 1H), 2.51 (s, 3H). MS m/z 269.1 (M + H+).
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-quinolin-3-yl-amine (14)
mp. 243 °C. 1H NMR (300 MHz, CDCl3): δ 9.00 (s, 1H), 8.45 (s, 1H), 8.20-8.30 (m, 2H), 8.10 (brs, NH, exchangeable), 7.88 (d, J = 7.5 Hz, 1H), 7.75 (m, 1H), 7.70 (m, 1H), 6.44 (s, 1H), 2.62 (s, 3H). MS m/z 277.1 (M + H+).
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-quinolin-6-yl-amine (15)
mp. 280 °C. 1H NMR (300 MHz, CDCl3): δ 9.00 (s, 1H), 8.45 (s, 1H), 8.20-8.25 (m, 2H), 8.12 (brs, NH, exchangeable), 7.80 (s, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.54-7.60 (m, 1H), 6.54 (s, 1H), 2.55 (s, 3H). MS m/z 277.1 (M + H+).
3-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ylamino)-naphthalen-2-ol (16)
mp. 301 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.51 (s, 1H), 7.95 (s, 1H), 7.87 (m, 1H), 7.78 (m, 1H), 7.41-7.45 (m, 1H), 7.31-7.35 (m, 2H), 6.29 (s, 1H), 2.42 (s, 3H). MS m/z 292.1 (M + H+).
2-Methyl-7-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ylamino)-chromen-4-one (17)
mp. 306 °C. 1H NMR (300 MHz, CDCl3): δ 8.41 (s, 1H), 8.30 (d, J = 8.52 Hz, 1H), 8.15 (brs, NH, exchangeable), 7.44 (s, 1H), 7.40 (d, J = 8.0 Hz, 1H), 6.69 (s, 1H), 6.22 (s, 1H), 2.66 (s, 3H), 2.44 (s, 3H). MS m/z 308.1 (M + H+).
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-1-yl-amine (18)
mp. 192 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.43 (brs, NH, exchangeable), 8.55 (s, 1H), 8.05 (m, 2H), 7.88 (m, 1H), 7.50-7.65 (m, 4H), 5.69 (s, 1H), 2.26 (s, 3H). MS m/z 276.1 (M + H+).
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-phenyl-amine (19)
mp. 187 °C (lit.37 188 °C). 1H NMR (300 MHz, CDCl3): δ 8.32 (s, 1H), 8.01 (brs, NH, exchangeable), 7.48-7.53 (m, 2H), 7.28-7.40 (m, 3H), 6.38 (s, 1H), 2.55 (s, 3H). MS m/z 226.1 (M + H+).
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-anthracen-2-yl-amine (20)
mp. 231 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.45 (brs, NH, exchangeable), 8.61-8.55 (m, 3H), 8.08-8.20 (m, 4H), 7.51-7.65 (m, 3H), 6.63 (s, 1H), 2.45 (s, 3H). MS m/z 326.2 (M + H+).
Supplementary Material
Acknowledgements
The authors would like to gratefully acknowledge Amgen for analytical chemistry support during the structural identification of DSM1 (compound 7). This work was supported by the United States National Institutes of Health grants AI053680 (to MAP and PKR), MG00203 (to NAM), and AI26912 and AI67670 (to PKR). MAP also acknowledges support from the Welch Foundation (I-1257), and PKR support from a Senior Scholar Award in Global Infectious Diseases from the Ellison Medical Foundation and from the UW Keck Center for Microbial Pathogens.
Abbreviations
- PfDHODH
Plasmodium falciparum dihydroorotate dehydrogenase
- CoQ
ubiquinone
- FMN
flavin mononucleotide
- HTS
high-throughput screen
Footnotes
Competing interests statement. The authors declare no competing financial interests.
References
- (1).Breman J, Egan A, Keusch G. The intolerable burden of malaria: a new look at the numbers. Am. J. Trop. Med. Hyg. 2001;64(12 Suppl):iv–vii. doi: 10.4269/ajtmh.2001.64.iv. [DOI] [PubMed] [Google Scholar]
- (2).Snow RW, Craig M, Deichmann U, Marsh K. Estimating mortality, morbidity and disability due to malaria among Africa’s non-pregnant population. Bull World Health Organ. 1999;77:624–40. [PMC free article] [PubMed] [Google Scholar]
- (3).White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–92. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Rathod PK, McErlean T, Lee PC. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA. 1997;94:9389–9393. doi: 10.1073/pnas.94.17.9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415:686–93. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- (6).Pink R, Hudson A, Mouries MA, Bendig M. Opportunities and challenges in antiparasitic drug discovery. Nat Rev Drug Discov. 2005;4:727–40. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- (7).Bray PG, Ward SA, O’Neill PM. Quinolines and artemisinin: chemistry, biology and history. Curr Top Microbiol Immunol. 2005;295:3–38. doi: 10.1007/3-540-29088-5_1. [DOI] [PubMed] [Google Scholar]
- (8).Rosenthal P. Antimalarial chemotherapy: mechanisms of action, resistance, and new directions in drug discovery. Humana Press Inc; Totowa, NJ: 2001. [DOI] [PubMed] [Google Scholar]
- (9).Gero A, O’Sullivan W. Purines and pyrimidines in malarial parasites. Blood Cells. 1990;16:467–484. [PubMed] [Google Scholar]
- (10).Gutteridge W, Trigg P. Incorporation of radioactive precursors into DNA and RNA of Plasmodium knowlesi in vitro. J. Protozool. 1970;17:89–96. doi: 10.1111/j.1550-7408.1970.tb05163.x. [DOI] [PubMed] [Google Scholar]
- (11).Reyes P, Rathod P, Sanchez D, Mrema J, Rieckmann K, Heidrich H. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 1982;5:275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- (12).Sherman I. Biochemistry of Plasmodium. Microbiol. Reviews. 1979;43:453–495. doi: 10.1128/mr.43.4.453-495.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gardner MJ, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).White N. Drug resistance in malaria. British Medical Bulletin. 1998;54:703–715. doi: 10.1093/oxfordjournals.bmb.a011721. [DOI] [PubMed] [Google Scholar]
- (15).Jiang L, Lee P, White J, Rathod P. Potent and selective activity of a combination of thymidine and 1843U89, a folate-based thymidylate synthase inhibitor, against Plasmodium falciparum. Antimicrob. agents and chemotherapy. 2000;44:1047–1050. doi: 10.1128/aac.44.4.1047-1050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rathod PK, Khatri A, Hubbert T, Milhous WK. Selective activity of 5-fluoroorotic acid against Plasmodium falciparum in vitro. Antimicrobial. agents and chemotherapy. 1989;33:1090–1094. doi: 10.1128/aac.33.7.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rathod PK, Reshmi S. Susceptibility of Plasmodium falciparum to a combination of thymidine and ICI D1694, a quinazoline antifolate directed at thymidylate synthase. Antimicrobial. agents and chemotherapy. 1994;38:476–480. doi: 10.1128/aac.38.3.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nagy M, Lacroute F, Thomas D. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl. Acad. Sci. USA. 1992;89:8966–8970. doi: 10.1073/pnas.89.19.8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jones M. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem. 1980;49:253–279. doi: 10.1146/annurev.bi.49.070180.001345. [DOI] [PubMed] [Google Scholar]
- (20).Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- (21).Goldenberg M. Leflunomide, a novel immunomodulator for the treatment of active rheumatoid arthritis. Clinical therapeutics. 1999;21:1837–1852. doi: 10.1016/S0149-2918(00)86732-6. [DOI] [PubMed] [Google Scholar]
- (22).Herrmann M, Schleyerbach R, Kirschbaum B. Leflunomide: an immunomodulatory drug for the treatment of rheumatoid arthritis and other autoimmune diseases. Immunopharmacology. 2000;47:273–289. doi: 10.1016/s0162-3109(00)00191-0. [DOI] [PubMed] [Google Scholar]
- (23).John WP, Paul SC, Michael JE. 3-Carboxy-5-methyl-N-[4-(trifluoromethyl)phenyl]-4-isoxazolecarboxamide, a new prodrug for the antiarthritic agent, 2-Cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide. J. Med. Chem. 1992;35:507–510. doi: 10.1021/jm00081a011. [DOI] [PubMed] [Google Scholar]
- (24).Liu S, Neidhardt E, Grossman T, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 1999;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- (25).Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr D Biol Crystallogr. 2006;62:312–23. doi: 10.1107/S0907444905042642. [DOI] [PubMed] [Google Scholar]
- (26).Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280:21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- (27).Heikkila T, Ramsey C, Davies M, Galtier C, Stead AM, Johnson AP, Fishwick CW, Boa AN, McConkey GA. Design and synthesis of potent inhibitors of the malaria parasite dihydroorotate dehydrogenase. J Med Chem. 2007;50:186–91. doi: 10.1021/jm060687j. [DOI] [PubMed] [Google Scholar]
- (28).Phillips M, Rathod PK, Baldwin J, Gujjar R. WO Patent 2007149211 A1. US Patent 20080027079 A1. Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity. 2008
- (29).Copeland RA. Evaluation of enzyme inhibitors in drug discovery. 2005:185–192. [PubMed] [Google Scholar]
- (30).Malmquist NA, Gujjar R, Rathod PK, Phillips MA. Analysis of Flavin Oxidation and Electron Transfer Inhibition in Plasmodium falciparum Dihydroorotate Dehydrogenase. Biochemistry. 2008;47:2466–2475. doi: 10.1021/bi702218c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–49. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- (32).Baldwin J, Farajallah A, Malmquist N, Rathod P, Phillips M. Malarial dihydroorotate dehydrogenase: substrate and inhibitor specificity. J. Biol. Chem. 2002;277:41827–41834. doi: 10.1074/jbc.M206854200. [DOI] [PubMed] [Google Scholar]
- (33).Malmquist NA, Baldwin J, Phillips MA. Detergent-dependent kinetics of truncated Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2007;282:12678–86. doi: 10.1074/jbc.M609893200. [DOI] [PubMed] [Google Scholar]
- (34).Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16:710–8. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chambers VC. The Structure and Reactivity of 5-Hydroxy-7-methyl-s-triazolo[2,3-a]pyrimidine. J. Am. Chem. Soc. 1960;82:605–609. [Google Scholar]
- (36).Levin YA, Sergeeva EM, Kukhtin VA. Condensed heterocycles. V. Reaction of 4-chloro-6-methyl-1,2,4-triazolo[2,3-α]pyrimidine with some nitrogenous bases. Zhurnal Obshchei Khimii. 1964;34:205–209. [Google Scholar]
- (37).Reynolds GA, VanAllan JA. Structure of certain polyazaindenes. VII. 4-Amino-6-methyl-1,3,3a,7-tetraazaindene and its derivatives. J. Org. Chem. 1961;26:115–117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.