

Abstract
Salicylic acid (SA) plays an important role in signaling the activation of plant defense responses against pathogen attack including induction of pathogenesis-related (PR) proteins. To gain further insight into the SA-mediated signal transduction pathway, we have isolated and characterized a tobacco mosaic virus (TMV)-inducible myb oncogene homolog (myb1) from tobacco. The myb1 gene was induced upon TMV infection during both the hypersensitive response and development of systemic acquired resistance in the resistant tobacco cultivar following the rise of endogenous SA, but was not activated in the susceptible cultivar that fails to accumulate SA. The myb1 gene was also induced by incompatible bacterial pathogen Pseudomonas syringae pv. syringae during the hypersensitive response. Exogenous SA treatment rapidly (within 15 min) activated the expression of myb1 in both resistant and susceptible tobacco cultivars with the subsequent induction of PR genes occurring several hours later. Biologically active analogs of SA and 2,6-dichloroisonicotinic acid (a synthetic functional analog of SA), which induce PR genes and enhanced resistance, also activated the myb1 gene. In contrast, biologically inactive analogs were poor inducers of myb1 gene expression. Furthermore, the recombinant Myb1 protein was shown to specifically bind to a Myb-binding consensus sequence found in the promoter of the PR-1a gene. Taken together, these results suggest that the tobacco myb1 gene encodes a signaling component downstream of SA that may participate in transcriptional activation of PR genes and plant disease resistance.
Keywords: salicylic acid, pathogenesis-related proteins, transcription factor, plant disease resistance
Tobacco mosaic virus (TMV) infection of tobacco cultivars (e.g., Nicotiana tabacum cv. Xanthi nc) that carry a dominant disease resistance gene (e.g., N gene) leads to the hypersensitive response and is subsequently accompanied by the induction throughout the plant of systemic acquired resistance. The hypersensitive response is characterized by host cell death and necrosis at the site of infection and restriction of pathogen growth and movement. Establishment of systemic acquired resistance results in enhanced resistance to a secondary challenge by the same or even unrelated pathogens. An increasing body of evidence suggests that salicylic acid (SA) is an important component of the signal transduction pathway(s) leading to local and systemic plant defense responses, including the activation of pathogenesis-related (PR) proteins (1–5). Studies in our laboratory suggest that one mechanism of SA action is to inhibit catalase and ascorbate peroxidase, thereby elevating endogenous H2O2 levels and generating salicylate free radicals (refs. 6–8; Z. Chen and D.F.K., unpublished data). The increased levels of H2O2 and/or products of reactions induced by salicylate free radicals such as lipid peroxide may act as signals for the activation of plant defense genes such as the PR-1 gene. However, little is known about the molecular mechanisms beyond these signals. To better understand the SA-mediated signal transduction pathway, therefore, it is essential to identify other signaling components.
Oncogenes such as myb play an important role in animal pathogenesis and the immune response; by analogy their plant homologs may also function in plant pathogenesis and defense responses. Animal myb genes encode transcription factors involved in cell cycling, proliferation and differentiation. The c-myb gene, the cellular counterpart of the transforming gene (v-myb) of the avian myeloblastosis virus, is essential for the development of hematopoietic tissue, including the formation of lymphocytes and other leukocytes important for the immune response. The Myb protein not only regulates the expression of cell proliferation genes (e.g., c-myc, DNA-polymerase-α, and p34cdc2), but also activates genes related to the immune response and pathogenesis (e.g., lysozyme, mim-1, CD4, and T-cell receptors) (9, 10).
During the past several years plant homologs of myb oncogenes have been isolated from maize, barley, petunia, potato, Antirrhinum, and Arabidopsis (11–19). They were shown to regulate flavonoid biosynthesis in maize (11) and Antirrhinum (16) and to determine epidermal cell (trichome) differentiation in Arabidopsis (17). Recently, myb genes have also been shown to be involved in dehydration and salt stress of Arabidopsis (18) and the gibberellin-response pathway of barley (19). However, their involvement in plant-microbe interactions has not been demonstrated. Here we report the isolation and characterization of a tobacco myb oncogene homolog that is induced by TMV infection. The encoded tobacco protein, designated Myb1, appears to function as a signaling component downstream of SA where it may participate in transcriptional activation of PR genes.
MATERIALS AND METHODS
Chemicals and Plant Materials.
SA and its analogs were purchased from Sigma or Aldrich. The N. tabacum cvs. Xanthi nc (NN genotype) and Xanthi (nn genotype), N. sylvestris, and N. tomentosiformis were grown in growth rooms at 22°C in a 14-h light cycle for 6–8 weeks before being used in experimentation.
Pathogen Inoculations and Chemical Treatments.
Two leaves on each tobacco plant were inoculated with TMV strain U1 at a concentration of 1 μg/ml in 50 mM phosphate buffer (pH 7.0) by rubbing with carborundum. Mock-inoculated plants were rubbed with buffer and carborundum. In temperature-shift experiments, plants were moved to 32°C after TMV inoculation and incubated for 48 h before shifting to 22°C. For bacterial infection, suspensions of Pseudomonas syringae pv. syringae strain Pss61 was adjusted in 5 mM MgSO4 solution to 2 × 108 cells per ml and infiltrated into tobacco leaves. For chemical treatments, tobacco leaves were pricked with a needle and then infiltrated with 1 mM solutions of SA, 2,6-dichloroisonicotinic acid (INA), or their analogs (adjusted to pH 6.5 with KOH). In cycloheximide (CHX) tests, leaves were pretreated with CHX (0.3 mM) for 30 min as described (20) before injection of SA (1 mM) plus CHX (0.3 mM). All experiments were performed at least twice.
Isolation and Sequencing of the myb1 cDNA.
Two degenerate oligonucleotides corresponding to highly conserved regions of Myb proteins were synthesized: forward, 5′-AAGTCNTG(C/T)(A/C)GN(C/T)TI(A/C)GITGG-3′; reverse, 5′-AT(C/T)TCGTTGTCNGTNC(G/T)NCC-3′. DNA purified from a tobacco cDNA library (made from TMV-infected leaf mRNA, A. Guo and D.F.K., unpublished data) was used as the template for PCR reactions. The amplified PCR fragments were purified on a 2.5% agarose gel and cloned into pGEM-T vector (Promega). After one of the PCR fragments was identified to be part of a TMV-inducible myb gene by RNA blot analysis, it was used as a probe to screen ≈106 plaques of the TMV-induced tobacco cDNA library. Positive plaques were purified by a second round of plaque hybridization. The pBK-CMV phagemid containing the myb1 cDNA was excised in vivo from the ZAP Express vector using the ExAssist helper phage (Stratagene). The sequence of myb1 was determined for both strands by the dideoxy sequencing method and analyzed using the lasergene software (DNAstar, Madison, WI) and GCG package (version 7.0, Genetics Computer Group, Madison, WI).
DNA and RNA Analysis.
DNA and RNA were purified from leaf tissues by phenol/chloroform extraction. Fifteen micrograms of genomic DNA was digested with restriction enzymes, fractionated on a 1% agarose gel, and blotted onto a nylon membrane (Nytran Plus, Schleicher & Shuell). For detection of myb1 mRNA, 25 μg of total RNA was fractionated on a 1.5% agarose gel containing formaldehyde and blotted onto a nylon membrane. The [α-32P]dCTP-labeled probe corresponding to the gene specific 3′ region (from 724-1344 bp of myb1) was made by the random-priming method with an oligolabeling kit (Pharmacia). Hybridization and washing were performed according to the method of Church and Gilbert (21).
Production and Purification of the Recombinant Myb1 Protein.
The myb1 cDNA was recloned into the NdeI/XhoI site of the pET28a(+) vector (Novagen) and the in-frame fusion was verified by DNA sequencing. Cells of Escherichia coli strain BL21 (DE3) transformed with the recombinant plasmid were grown at 37°C in Luria–Bertani medium (22) containing 50 μg/ml kanamycin. The production of the polyhistidine–Myb1 fusion protein was induced in mid-logarithmic E. coli cultures by addition of isopropyl β-d-thiogalactopyranoside (1 mM). After 3 h of growth at 30°C, bacterial cells were harvested and proteins were purified on nickel chelate affinity columns as described by the manufacturer (Novagen). The purified Myb1 protein was eluted from the column using the elution buffer (20 mM Tris, pH 7.9/500 mM NaCl/500 mM imidazole) and stored at −80°C after addition of glycerol to 20%. Protein concentrations were determined with the Bradford reagent (Bio-Rad).
Gel Mobility-Shift Assay.
The PR-1a promoter fragments were amplified by PCR and labeled by filling the 5′ overhangs with [α-32P]dATP and the Klenow fragment. Myb-binding site type I (MBSI) (5′-CGGAATTCCCTAACTGACGCATCGATGGGA-3′) and MBSII (5′-CGGAATTCTGTTTGGTATGCATCGATGGGA-3′) oligonucleotides and their mutated forms (see Fig. 8B) were annealed with a primer (5′-TCCCATCGATGC-3′) and then extended with the Klenow fragment in presence of [α-32P]dCTP to make double-stranded probes for the assay. DNA binding reactions were performed in 20 μl of binding buffer (10 mM Tris, pH 8.0/50 mM NaCl/1 mM dithiothreitol/1 mM EDTA/1 μg/μl BSA/10% glycerol) that contained 2 μg of double-stranded poly(dI-dC), 200 ng of the purified recombinant Myb1 protein, and 0.5–2 ng of 32P-labeled probe (10,000–40,000 cpm). After incubation at 4°C for 30 min, the reaction mixtures were electrophoresed on a 5% polyacrylamide gel in 0.5× Tris·borate·EDTA buffer (22) at 100 V. The gels were then dried and autoradiographed.
Figure 8.
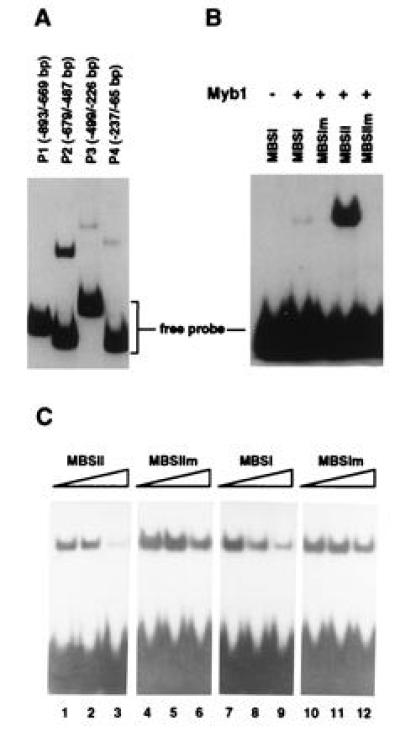
Gel mobility-shift assays with purified recombinant Myb1 protein. (A) DNA-binding activity of Myb1 to several PR-1a promoter fragments. The locations of the fragments in the PR-1a promoter relative to the transcriptional start site are given in parenthesis. (B) The Myb1 protein binds preferentially to MBSII (GTTTGGT) found in PR-1a promoter fragment P2. The binding reaction fractionated in the first lane did not contain Myb1 while those in other lanes contained 200 ng of Myb1. The following double-strand oligonucleotides were used as probes: MBSI (5′-CGGAATTCCCTAACTGACGCATCGATGGGA-3′), MBSIm (5′-CGGAATTCCCTCCCTGACGCATCGATGGGA-3′), MBSII (5′-CGGAATTCTGTTTGGTATGCATCGATGGGA-3′), and MBSIIm (5′-CGGAATTCTGTTGCCTATGCATCGATGGGA-3′). (C) Competition for binding of Myb1 to the MBSII probe. The binding assay was performed by preincubating unlabeled competitor oligonucleotides for 15 min with Myb1 before addition of 1 ng of 32P-labeled MBSII probe. Reactions fractionated in lanes 1, 4, 7, and 10 contained no competitors, while those in lanes 2, 5, 8, and 11 contained 20 ng competitors and those in lanes 3, 6, 9, and 12 contained 200 ng competitors.
RESULTS
Cloning and Sequence Analysis of a TMV-inducible myb Homolog.
To study the potential role of myb homologs in the tobacco-TMV interaction, PCR fragments (160 bp) of tobacco myb genes were amplified and cloned from TMV-induced tobacco cDNAs using degenerate oligonucleotide primers corresponding to highly conserved regions of Myb proteins (underlined in Fig. 1A). Sequence analysis of 20 myb PCR fragments indicated the existence of at least three groups of tobacco myb genes with DNA sequence identity among them ranging from 56–72%. Based on Northern blot analysis, one group was found to be induced upon TMV infection. The corresponding TMV-inducible tobacco myb gene, designated myb1, was isolated by screening a cDNA library made from the resistant N. tabacum cv. Xanthi nc after TMV infection. This cDNA is 1344 bp in length and contains a 194-bp 5′ untranslated region and a 363-bp 3′ untranslated region with a typical polyadenylation signal. The ORF encodes a full-length Myb1 protein of 278 amino acid residues with a predicted molecular mass of 32 kDa (Fig. 1A). The hydrophilic Myb1 protein has a calculated isoelectric point of 5.62. Among Myb proteins, the basic N-terminal region is conserved. In the tobacco Myb1, it is composed of two imperfect tryptophan repeats of 53 (residues 12–64) and 51 (residues 65–115) amino acids, while the mouse c-Myb contains three tryptophan repeats (R1-R3, Fig. 1B). Secondary structure analyses of Myb1 (and mouse c-Myb) predict that the tryptophan repeats form helix–turn–helix DNA-binding motifs. Myb1 and c-Myb also share a potential ATP/GTP-binding site (P-loop), a redox sensitive cysteine, and a nuclear localization sequence within their N-terminal DNA-binding domains. The acidic C-terminal region of Myb1 (residues 179–278, pI 3.6) forms amphiphathic α-helices, which are characteristic of transcriptional activation domains. The corresponding activation domain of the mouse c-Myb does not share sequence homology with Myb1 and is bounded on its C-terminal ends by a large phosphorylatible negative regulatory domain that controls the activity of c-Myb (Fig. 1B). This negative regulatory domain is absent from Myb1.
Figure 1.
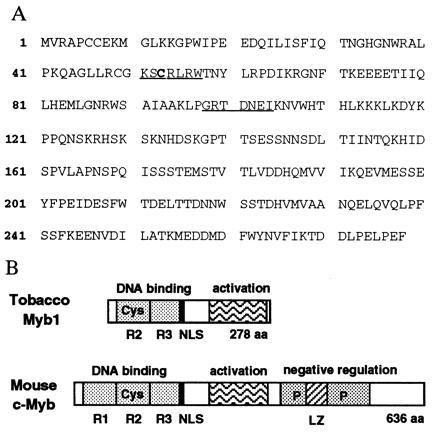
The tobacco Myb1 protein. (A) The deduced amino acid sequence of the Myb1 protein. The underlined sequences were used to design degenerate primers for the PCR amplification of the conserved regions of myb genes. The conserved redox sensitive cysteine residue at position 53 is indicated in boldface. (B) Schematic comparison of structural features between Myb1 and the mouse c-Myb. R, tryptophan repeat; Cys, conserved redox sensitive cysteine (Cys-53 in Myb1 and Cys-130 in c-Myb); P, phosphorylation sites; NLS, nuclear localization sequences; LZ, leucine zipper; aa, amino acid residues.
The putative DNA-binding domain of the tobacco Myb1 is highly homologous with that of animal and plant Myb proteins. Amino acid sequence comparisons of the DNA-binding domains reveal that Myb1 is 90% identical to Myb.Ph2 of petunia (12), 52% identical to Atmyb2 of Arabidopsis (18), and 45% identical to mouse c-Myb (9). Outside of the DNA-binding domain, only petunia Myb.Ph2 shows limited homology with the C-terminal region of Myb1. In contrast to tobacco Myb1, Myb.Ph2 is constitutively expressed in petunia leaf tissues (12).
Southern blot analysis of tobacco (cvs. Xanthi nc and Xanthi) genomic DNAs digested with EcoRI and HindIII identified two bands when probed with the gene-specific 3′ region (from position 724 bp to 1344 bp) of myb1 cDNA (Fig. 2). The two copies of myb1 found in the amphidiploid N. tabacum cultivars are probably derived from the diploid parental species N. sylvestris and N. tomentosiformis as one of the two bands was present in N. sylvestris and the other in N. tomentosiformis. Two different size RNAs (1.4 kb and 1.2 kb) that hybridized with the gene-specific myb1 probe were found in N. tabacum (see Fig. 3). Since both RNAs were also detected in N. sylvestris and N. tomentosiformis, which contain only one myb1 gene each (data not shown), they may result from alternative splicing or polyadenylation of the myb1 transcript.
Figure 2.
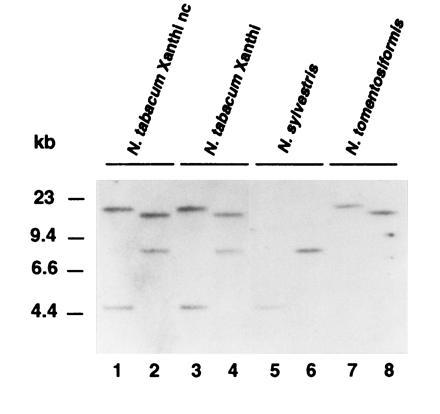
Southern analysis of the myb1 gene with genomic DNAs from several Nicotiana species. The genomic DNAs (15 μg) of the amphidiploid N. tabacum (cvs. Xanthi nc and Xanthi) and its parental diploid species, N. sylvestris and N. tomentosiformis were digested with EcoRI (lanes 1, 3, 5, and 7) or HindIII (lanes 2, 4, 6, and 8). After blotting, the DNAs were hybridized with a gene-specific myb1 probe.
Figure 3.
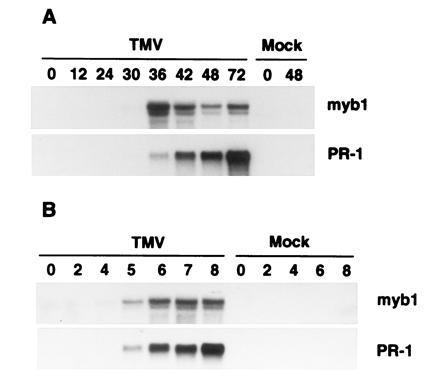
Induction of myb1 by TMV infections during the hypersensitive response. (A) RNA blot analysis of myb1 and PR-1 gene expression following TMV infection at 22°C in the resistant tobacco cultivar Xanthi nc. Time is indicated in h postinfection. (B) Accumulation of myb1 and PR-1 mRNA following shift of Xanthi nc plants from a temperature where resistance to TMV cannot develop (32°C) to a temperature at which resistance is established (22°C). RNAs were isolated from inoculated leaves. Time is indicated in h posttemperature shift. TMV- and mock-inoculated plants were maintained at 32°C for 48 h after inoculation before being transferred to 22°C.
Induction of myb1 Expression by TMV and Bacterial Infections.
To examine the expression of myb1 in response to TMV infection, RNA blots prepared from mock- and TMV-infected tobacco were probed with the 3′ gene-specific myb1 cDNA. The blots were also probed with a cDNA for the PR-1 genes, which served as a molecular marker for activation of plant defense responses. The myb1 genes were not detected in healthy tobacco leaves, but were induced to low levels by 30 h and to high levels by 36 h after TMV infection at 22°C in Xanthi nc, which carries the N resistance gene (Fig. 3A). The kinetics of myb1 expression closely paralleled the rise of endogenous SA levels that occurs 24–36 h after TMV inoculation (1). Expression of myb1 peaked at 36 h after infection, preceding activation of the PR-1 genes whose mRNA continued to rise through at least 72 h post inoculation. In contrast, there was no activation of myb1 in the TMV-infected susceptible cultivar Xanthi (data not shown), which lacks the N gene and does not accumulate SA or express PR genes (1). The myb1 genes also were not induced by wounding with a hemostat (data not shown) or by rubbing with carborundum (i.e., mock inoculation in Fig. 3).
In cultivars containing the N gene, resistance to TMV is reversibly blocked at high temperatures (>28°C) and there is neither elevation of SA levels nor PR-1 gene activation. Shifting infected plants to a lower temperature (e.g., 22°C) results in (i) a rapid and dramatic rise in SA between 4–6 h, (ii) activation of PR-1 genes, and (iii) development of resistance (ref. 23; A. Guo and D.F.K., unpublished data). At the high temperature (32°C), TMV-infected Xanthi nc also failed to express myb1 genes while transfer to a lower temperature (22°C) resulted in activation of myb1 and PR-1 within 5 h (Fig. 3B).
Systemic expression of myb1 was also analyzed in TMV-infected tobacco (Fig. 4). The myb1 gene, as well as PR-1 genes, was induced in upper, uninoculated leaves within 6 days after TMV inoculation of lower leaves. Therefore, activation of myb1 is associated with not only the hypersensitive response in local, infected leaves, but also with the development of systemic acquired resistance in distal, uninfected leaves.
Figure 4.
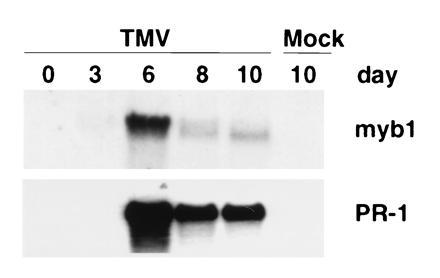
Induction of myb1 by TMV infections during development of systemic acquired resistance. The RNAs were isolated from upper, uninoculated leaves of the resistant tobacco cultivar Xanthi nc following TMV infection of lower leaves. Time is indicated in days postinfection.
To provide further evidence for the importance of myb1 expression in plant–pathogen interactions, the bacterial pathogen P. syringae pv. syringae strain Pss61 was tested for its ability to induce myb1 expression. Pss61 is an incompatible pathogen of tobacco and elicits a rapid hypersensitive response. Infection of tobacco with Pss61 induced myb1 expression within 16 h, closely followed by the continuous accumulation of PR-1 mRNAs (Fig. 5).
Figure 5.
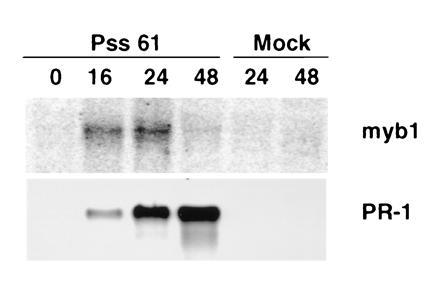
Induction of myb1 by an incompatible bacterial pathogen. RNAs were isolated from tobacco leaves infected with P. syringae pv. syringae strain Pss61 or mock-inoculated with 5 mM MgSO4 solution. Time is indicated in h postinfection.
Rapid Activation of myb1 Expression by SA, INA, and Their Biologically Active Analogs.
Since the induction of myb1 appears to follow the rise of endogenous SA levels in TMV-infected tobacco leaves, we tested whether SA treatments would activate expression of myb1. Exogenous application of 1 mM SA induced expression of myb1 within 15 min in both the resistant cultivar Xanthi nc (Fig. 6A) and the susceptible cultivar Xanthi (data not shown). Activation of myb1 by SA was transient, with myb1 mRNA levels peaking at 30 min and then rapidly declining. This may be due to rapid inactivation of SA via glycosylation in tobacco leaves (24). In contrast, PR-1 induction did not occur until 6–12 h after treatment.
Figure 6.
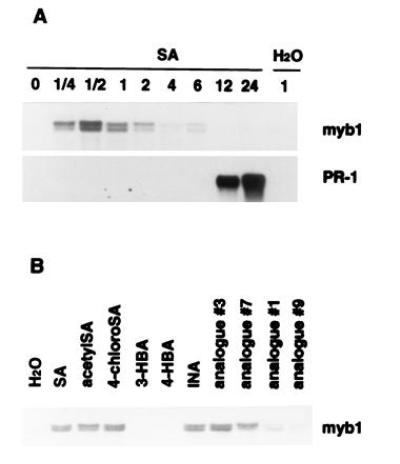
Effect of SA, INA and their analogs on myb1 gene expression. (A) Rapid activation of myb1 by SA (1 mM) preceded induction of PR-1. Time is indicated in h postinjection of SA (or H2O as a control) into tobacco leaves. (B) Induction of myb1 by SA, INA, and their biologically active analogs (acetylSA, 4-chloroSA, analogs 3 and 7). Inactive analogs [3- and 4-hydroxybenzoic acid (HBA), analogs 1 and 9] were poor inducers. Expression of myb1 was analyzed 30 min after injection with 1 mM of the various compounds. The chemical structures of the INA analogs were presented in a previous report (25).
The synthetic compound INA is a potent inducer of PR genes and enhanced disease resistance and appears to act as functional analog of SA (7, 25). It also induced myb1 expression (Fig. 6B). To further assess the functional relevance of SA and INA in the activation of myb1, several analogs of SA and INA were compared for their ability to activate myb1 expression vs their biological activity—i.e., the ability to induce PR genes and enhanced disease resistance. Expression of myb1 was effectively activated by the biologically active analogs of both SA (acetylSA and 4-chloroSA) and INA (analogs 3 and 7). Biologically inactive analogs (3-hydroxybenzoic acid, 4-hydroxybenzoic acid, analogs 1 and 9) were much poorer inducers (Fig. 6B). Thus, there is a good correlation between biological activity and the ability to induce myb1 gene expression.
Effect of Cycloheximide on myb1 Expression.
The rapid activation of myb1 by SA suggested that this induction did not require synthesis of a new protein(s). To test this possibility, protein synthesis was blocked by pretreatment for 30 min with CHX before addition of SA and CHX. As anticipated, CHX did not inhibit SA-mediated activation of myb1 (Fig. 7). However, CHX treatment alone induced the myb1 genes and greatly enhanced their expression when combined with SA. This is similar to barley GAmyb expression in response to CHX and gibberellin treatments (19). In contrast, SA induction of PR-1 genes was blocked by CHX, in agreement with a previous report (20).
Figure 7.
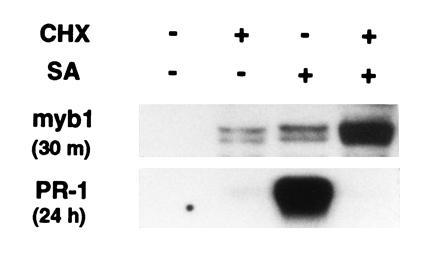
Effect of CHX on myb1 and PR-1 gene expression. RNA blot analysis of tobacco leaves pretreated with CHX (0.3 mM) for 30 min followed by treatment with SA (1 mM) plus CHX (0.3 mM) for an additional 30 min or 24 h.
Specific Binding of the Myb1 Protein to the PR-1a Promoter.
Induction of PR-1 genes by SA required de novo protein synthesis as demonstrated above. The tobacco Myb1 protein may be one of these newly synthesized factors since activation of myb1 by SA preceded the expression of PR-1 genes (Fig. 6). In fact, sequence analysis uncovered multiple MBSs in the promoters of several PR genes including PR-1 and PR-2 (encoding β-1,3-glucanase). Myb proteins are known to bind to two types of MBS. Type I (MBSI) has the consensus sequence (T/C)AAC(T/G)G and is bound by animal and some plant Myb proteins (10, 18). Type II (MBSII) has the consensus sequence G(G/T)T(A/T)G(G/T)T and is bound by several plant Myb proteins (11, 13, 16). The PR-1a promoter (26) contains both types of binding sites: MBSI (TAACTG) located at −169 to −174 and MBSII (GTTTGGT) at −520 to −514. In addition, four MBSI-like sites, with 5 of 6 nt matching the consensus sequence, are positioned at −643 to −639 (AACTG), −526 to −522 (AACTG), −332 to −328 (TAACG), and −241 to −237 (TAACT).
To examine the interaction between the Myb1 protein and the PR-1a promoter, the histidine-tagged recombinant Myb1 protein was produced in E. coli and purified by nickel affinity chromatography. Using the gel mobility-shift assay, purified Myb1 was found to strongly bind to the P2 fragment (−679 to −487) of the PR-1a promoter that contains a MBSII (GTTTGGT) and two MBSI-like sites. (Fig. 8A). Myb1 also weakly bound to the P4 fragment (−237 to −65), which contains a MBSI (TAACTG) and to the P3 fragment (−499 to −226), which contains two type I-like sites. Myb1 did not bind to the P1 fragment (−893 to −669), which does not contain MBS.
To prove that Myb1 indeed binds to MBS in the PR-1a promoter, 30 bp oligonucleotides containing MBSI (TAACTG), MBSII (GTTTGGT) or their mutated forms were synthesized, labeled, and used in the gel mobility-shift assay. Myb1 preferentially bound to MBSII and only very weakly bound to MBSI (Fig. 8B). It did not bind the mutated forms of MBSI and MBSII. Therefore, the tobacco Myb1 protein indeed binds to MBS in the PR-1a promoter. The much higher binding affinity for MBSII is in agreement with its stronger binding of the P2 fragment (which contains MBSII) than the P4 fragment (which contains MBSI).
To further demonstrate the specific binding of Myb1 to MBSII, competition experiments were performed with MBSII as the labeled probe. Binding of Myb1 to MBSII was effectively reduced by the addition of unlabeled MBSII (Fig. 8C). In contrast, competition by MBSI was less effective, while the mutated forms of MBSI and MBSII had marginal effects on binding. In summary, Myb1 preferentially bound to MBSII in the PR-1a promoter, suggesting that Myb1 may be involved in the transcriptional activation of this gene during plant defense responses.
DISCUSSION
A major objective in studying plant disease resistance is to identify and characterize plant components crucial for signal transduction processes involved in activation of host defenses after pathogen attack. In this study, we have identified a TMV-inducible myb oncogene homolog from tobacco and demonstrated its potential role in SA-mediated induction of plant defense responses. Activation of myb1 by TMV infection, which occurred during both the hypersensitive response and development of systemic acquired resistance, appears to depend on the N resistance gene-mediated signaling process and is closely associated with the rise of endogenous SA levels (ref. 1, Figs. 3 and 4). In addition to TMV, the incompatible bacterial pathogen P. syringae pv. syringae also induced myb1 expression (Fig. 5). Exogenous treatment with SA rapidly activated expression of myb1, which preceded the induction of PR-1 genes (Fig. 6A). Furthermore, analysis of analogs of SA and INA showed a good correlation between biological activity of the analogs and their ability to induce myb1 gene expression (Fig. 6B). Together, these results suggest that myb1 encodes a signaling component downstream of SA. Although myb homologs have been isolated from several plant species, to our knowledge, this is the first time that a myb gene has been shown to be involved in plant defense responses against microbial pathogens.
Currently, it is not known which transcription factor or factors control PR gene expression. Activation of the myb1 gene by SA did not require de novo protein synthesis and preceded induction of PR-1 genes that did require protein synthesis (Fig. 7). These results suggest that myb1 may encode a transcription factor involved in PR gene expression. The discovery of multiple MBS in the promoters of PR-1 and PR-2 genes and the specific binding of purified recombinant Myb1 protein to these sites in the PR-1a promoter (Fig. 8) adds further support for this model. However, it should be noted that a role for myb1 in PR-1 gene expression has yet to be rigorously established.
The tobacco Myb1 protein had different affinities for the two types of MBS (Fig. 8B) as reported for several other plant Myb proteins (11, 18). It preferentially bound to the MBSII (GTTTGGT) found in the PR-1a promoter (−520 to −514 bp). MBSII is a H-box-like sequence present in promoters of many genes involved in phenylpropanoid biosynthesis (e.g., pal, chs, and dfr) and is implicated in the response to UV light and other environmental stresses (16). Indeed, the maize, petunia, and Antirrhinum Myb transcription factors are known to control phenylpropanoid biosynthesis (11, 13, 16). Since many phytoalexins (compounds with antibacterial and antifungal activities) are derived from phenylpropanoids (27), tobacco Myb1 may not only regulate PR gene expression, but also transactivate phytoalexin biosynthetic genes that might be induced by TMV or SA. Similar scenarios may extend to other plant species, especially those with well characterized flavonoid phytoalexins.
There was a significant delay between SA activation of myb1 (within 15 min) and induction of PR-1 genes (between 6–12 h; Fig. 6). If Myb1 acts directly on the PR-1 promoters, this delay suggests that while myb1 activation by SA may be necessary, it is not sufficient for induction of PR-1 genes. Myb proteins are known to activate gene expression in combination with other transcription factors. The yeast Myb protein, BAS1, activates HIS4 transcription only in combination with the homeodomain protein, BAS2 (28). The chicken c-Myb and v-Myb proteins require bZIP factors such as C/EBP to synergistically activate mim-1 gene expression (29), while maize Myb and Myc-like factors often act together to activate expression of flavonoid biosynthetic genes (11). Therefore, the tobacco Myb1 protein may need additional factor(s) such as bZIP- or Myc-related proteins for efficient activation of PR genes. In fact, the PR-1a promoter contains three Myc-binding consensus sequences (CANNTG) that are similar to the G-box core sequence (CACGTG) and may serve as binding sites for a Myc-related protein and/or a member of the bZIP class of G-box-binding proteins.
Rapid induction of myb1 gene expression by SA was insensitive to cycloheximide treatment (Fig. 7) and thus does not require de novo protein synthesis. In mammalian cells, members of the NF-κB family have been shown to transactivate the c-myb gene by binding to its first intron that contains NF-κB-binding sites (30). The transcription factor NF-κB is posttranscriptionally activated by reactive oxygen species such as H2O2 and mediates expression of many genes involved in acute phase, immune, and inflammatory responses (31). In plants, elevation of H2O2 levels, due to inhibition of catalase and ascorbate peroxidase, appears to be one mode of SA action for induction of PR genes (6–8). By analogy, a posttranslationally activated NF-κB-like factor might be involved in the rapid activation of the tobacco myb1 gene by SA.
There are other intriguing parallels between animal and plant defense responses as demonstrated by the structural and functional conservation of some of their signal transduction processes. The N gene of tobacco, which confers resistance to TMV, shows considerable homology with the gene encoding the human interleukin 1 receptor, which participates in interleukin 1 activation of NF-κB (32). SA (and aspirin), H2O2, and Myb transcription factors appear to affect or participate in not only animal immune responses, but also plant defense responses. Interestingly, one of the major plant PR proteins, PR-1, is conserved throughout eukaryotes including mammals, insects, fungi, and plants (5, 33–35). The tobacco PR-1 has antifungal activity but its mechanism of action is unknown (36, 37). A recent report has shown that one of the human PR-1 homologs is associated with brain tumors and is induced by viral infection or phorbal ester treatment (35). Furthermore, the plant hypersensitive response triggered by pathogen attack shares many cytological and biochemical characteristics of animal programmed cell death (a key mechanism for eliminating transformed or infected cells; ref. 38). Therefore, characterization of plant signaling components such as myb1 will not only help elucidate plant defense mechanisms and develop new strategies for plant disease control, but also enhance our understanding of host-pathogen interactions in general.
Acknowledgments
We thank members of the laboratory for their critical reading of the manuscript. Helmut Kessmann, Theo Staub, and John Ryals are acknowledged for generously providing INA and its analogs. Sheng Yang He is acknowledged for kindly providing the P. syringae strain. This work was supported in part by a Charles Johanna Busch postdoctoral fellowship awarded to Y.Y. from the Waksman Institute and National Science Foundation Grant MCB-9310371 to D.F.K.
Footnotes
Abbreviations: SA, salicylic acid; CHX, cycloheximide; INA, 2,6-dichloroisonicotinic acid; MBS, Myb-binding site; MBSI and MBSII, MBS type I and type II; PR, pathogenesis-related; TMV, tobacco mosaic virus.
Data deposition: The sequence reported in this paper has been deposited in the GenBank data base (accession no. U72762U72762).
References
- 1.Malamy J, Carr J P, Klessig D F, Raskin I. Science. 1990;250:1002–1004. doi: 10.1126/science.250.4983.1002. [DOI] [PubMed] [Google Scholar]
- 2.Métraux J P, Signer H, Ryals J, Ward E, Wyss-Benz M, Gaudin J, Raschdorf K, Schmid E, Blum W, Inverardi B. Science. 1990;250:1004–1006. doi: 10.1126/science.250.4983.1004. [DOI] [PubMed] [Google Scholar]
- 3.Gaffney T, Friedrich L, Vernooij B, Negrotto D, Nye G, Uknes S, Ward E, Kessmann H, Ryals J. Science. 1993;261:754–756. doi: 10.1126/science.261.5122.754. [DOI] [PubMed] [Google Scholar]
- 4.Delaney T, Uknes S, Vernooij B, Friedrich L, Weymann K, Negrotto D, Gaffney T, Gut-Rella M, Kessmann H, Ward E, Ryals J. Science. 1994;266:1247–1250. doi: 10.1126/science.266.5188.1247. [DOI] [PubMed] [Google Scholar]
- 5.Klessig D F, Malamy J. Plant Mol Biol. 1994;26:1439–1458. doi: 10.1007/BF00016484. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Silva H, Klessig D F. Science. 1993;262:1883–1886. doi: 10.1126/science.8266079. [DOI] [PubMed] [Google Scholar]
- 7.Durner J, Klessig D F. Proc Natl Acad Sci USA. 1995;92:11312–11316. doi: 10.1073/pnas.92.24.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durner, J. & Klessig, D. F. (1996) J. Biol. Chem. 271, in press. [DOI] [PubMed]
- 9.Graf T. Cur Opin Genet Dev. 1992;2:249–255. doi: 10.1016/s0959-437x(05)80281-3. [DOI] [PubMed] [Google Scholar]
- 10.Thompson M A, Ramsay R G. BioEssay. 1995;17:341–350. doi: 10.1002/bies.950170410. [DOI] [PubMed] [Google Scholar]
- 11.Grotewold E, Drummond B J, Bowen B, Peterson T. Cell. 1994;76:543–53. doi: 10.1016/0092-8674(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 12.Avila J, Nieto C, Canas L, Benito M J, Paz-Ares J. Plant J. 1993;3:553–562. doi: 10.1046/j.1365-313x.1993.03040553.x. [DOI] [PubMed] [Google Scholar]
- 13.Solano R, Nieto C, Paz-Ares J. EMBO J. 1995;14:1773–1784. doi: 10.1002/j.1460-2075.1995.tb07166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baranowskij N, Frohberg C, Willmitzer L. EMBO J. 1994;13:5383–5392. doi: 10.1002/j.1460-2075.1994.tb06873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson D, Culianez-Macia F A, Prescott A G, Roberts K, Martin C. Plant Cell. 1991;3:115–125. doi: 10.1105/tpc.3.2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sablowski R W, Moyano E, Culianez-Macia F A, Schuch W, Martin C, Bevan M A. EMBO J. 1994;13:128–137. doi: 10.1002/j.1460-2075.1994.tb06242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oppenheimer D G, Herman P L, Sivakumaran S, Esch J, Marks M D. Cell. 1991;67:483–493. doi: 10.1016/0092-8674(91)90523-2. [DOI] [PubMed] [Google Scholar]
- 18.Urao T, Yamaguchi-Shinozaki K, Urao S, Shinozaki K. Plant Cell. 1993;5:1529–1539. doi: 10.1105/tpc.5.11.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gubler F, Kalla R, Roberts J K, Jacobsen J V. Plant Cell. 1995;7:1879–1891. doi: 10.1105/tpc.7.11.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uknes S, Dincher S, Friedrich L, Negrotto D, Williams S, Thompson-Tayler H, Potter S, Ward E, Ryals J. Plant Cell. 1993;5:159–169. doi: 10.1105/tpc.5.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Church G M, Gilbert W. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 23.Malamy J, Hennig J, Klessig D F. Plant Cell. 1992;4:359–366. doi: 10.1105/tpc.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hennig J, Malamy J, Grynkiewicz G, Indulski J, Klessig D F. Plant J. 1993;4:593–600. doi: 10.1046/j.1365-313x.1993.04040593.x. [DOI] [PubMed] [Google Scholar]
- 25.Conrath U, Chen Z, Ricigliano J W, Klessig D F. Proc Natl Acad Sci USA. 1995;92:7143–7147. doi: 10.1073/pnas.92.16.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Payne G, Parks T D, Burkhart W, Dincher S, Ahl P, Métraux J P, Ryals J. Plant Mol Biol. 1988;11:89–94. doi: 10.1007/BF00015662. [DOI] [PubMed] [Google Scholar]
- 27.Dixon R A, Paiva N L. Plant Cell. 1995;7:1085–1097. doi: 10.1105/tpc.7.7.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tice-Baldwin K, Fink G R, Arndt K T. Science. 1989;246:931–935. doi: 10.1126/science.2683089. [DOI] [PubMed] [Google Scholar]
- 29.Burk O, Mink S, Ringwald M, Klempnauer K H. EMBO J. 1993;12:2027–2038. doi: 10.1002/j.1460-2075.1993.tb05852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toth C R, Hostutler R F, Baldwin A S, Bender T P. J Biol Chem. 1995;270:7661–7671. doi: 10.1074/jbc.270.13.7661. [DOI] [PubMed] [Google Scholar]
- 31.Shreck R, Rieber P, Baeuerle P A. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Whitham S, Dinesh-Kumar S P, Choi D, Hehl R, Corr C, Baker B. Cell. 1994;78:1101–1115. doi: 10.1016/0092-8674(94)90283-6. [DOI] [PubMed] [Google Scholar]
- 33.Fang K S F, Vitale M, Fehlner P, King T P. Proc Natl Acad Sci USA. 1988;85:895–899. doi: 10.1073/pnas.85.3.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuren F H J, Asgeirsdöttir S A, Kothe E M, Scheer J M J, Wessels J G H. J Gen Microbiol. 1993;139:2083–2090. doi: 10.1099/00221287-139-9-2083. [DOI] [PubMed] [Google Scholar]
- 35.Murphy E V, Zhang Y, Zhu W, Biggs J. Gene. 1995;159:131–135. doi: 10.1016/0378-1119(95)00061-a. [DOI] [PubMed] [Google Scholar]
- 36.Alexander D, Goodman R M, Gut-Rella M, Glascock C, Weymann K, Friedrich L, Maddox D, Ahl-Goy P, Luntz T, Ward E, Ryals J. Proc Natl Acad Sci USA. 1993;90:7327–7331. doi: 10.1073/pnas.90.15.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niderman T, Genetet I, Bruyère T, Gees R, Stintzi A, Legrand M, Fritig B, Mösinger E. Plant Physiol. 1995;108:17–27. doi: 10.1104/pp.108.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mittler R, Lam E. Trends Microbiol. 1996;4:10–15. doi: 10.1016/0966-842x(96)81499-5. [DOI] [PubMed] [Google Scholar]