

Abstract
We describe a novel plant transformation technique, termed “agrolistic,” that combines the advantages of the Agrobacterium transformation system with the high efficiency of biolistic DNA delivery. Agrolistic transformation allows integration of the gene of interest without undesired vector sequence. The virulence genes virD1 and virD2 from Agrobacterium tumefaciens that are required in bacteria for excision of T-strands from the tumor-inducing plasmid were placed under the control of the CaMV35S promoter and codelivered with a target plasmid containing border sequences flanking the gene of interest. Transient expression assays in tobacco and in maize cells indicated that vir gene products caused strand-specific nicking in planta at the right border sequence, similar to VirD1/VirD2-catalyzed T-strand excision observed in Agrobacterium. Agrolistically transformed tobacco calli were obtained after codelivery of virD1 and virD2 genes together with a selectable marker flanked by border sequences. Some inserts exhibited right junctions with plant DNA that corresponded precisely to the sequence expected for T-DNA (portion of the tumor-inducing plasmid that is transferred to plant cells) insertion events. We designate these as “agrolistic” inserts, as distinguished from “biolistic” inserts. Both types of inserts were found in some transformed lines. The frequency of agrolistic inserts was 20% that of biolistic inserts.
Gene delivery by particle bombardment has become a widely used technique with broad applications in plant transformation (for review, see ref. 1). For example, maize resistant to European corn borer has been developed by this technique (2). In the course of product development, the structure and copy number of the transgenes as well as their stability must be established. The most desirable product would be one with a single simple insert and no extraneous plasmid vector DNA. However, transformation by particle bombardment often leads to integration at one locus of complex arrays of multiple copies of the introduced genes including plasmid vector, often fragmented and rearranged (3–8). Because the multiple copies inserted during biolistic transformation are usually genetically linked, they cannot be segregated during subsequent breeding.
Multiple copies of transgenes can lead to instability of their expression by several mechanisms (for review, see ref. 9); multiple copies of transgenes can interact to inactivate each other and related host genes by epigenetic mechanisms variously labeled “cosuppression” or “gene silencing.” In addition, homologous recombination may cause genetic instability of multiple copies. For these reasons, reduction of the copy number of inserted transgenes and simplification of their arrangement should prove beneficial for maintaining the fidelity and expression of introduced genes.
The integration pattern for foreign genes introduced via Agrobacterium-mediated transformation is in general strikingly different from the pattern resulting from particle bombardment of plant cells (for review, see ref. 10). The number of copies of intact and rearranged transgenes resulting from biolistic delivery exceeds, often greatly, the copy number of transgenes introduced into plants by the Agrobacterium system.
Agrobacterium has evolved a natural plant transformation mechanism in which the transferred genes are located on plasmids, called tumor-inducing (Ti) or root-inducing (Ri) plasmids (for review, see ref. 11). A specific segment of Ti or Ri plasmids, called T-DNA, is flanked by 25-bp directly repeated border sequences. T-DNA travels by a conjugation-like process from the bacterium to the plant cell nucleus and becomes integrated into the plant’s chromosomal DNA. An elaborate mechanism for DNA transfer is encoded by a series of virulence (vir) genes (for review, see ref. 12). Activation of the vir genes results in the generation of site-specific nicks within the T-DNA border repeats and produces a linear single-stranded DNA molecule (T-strand) corresponding to the bottom strand of the T-DNA.
T-strand nicking requires two polypeptides encoded by the virD operon: VirD1 and VirD2 (13–20). VirD2 has a site-specific endonuclease activity that cleaves the lower strand of the border sequence (21–24). A type I topoisomerase activity has been described for VirD1-containing extracts (25). It has been proposed that this activity is required for relaxing the DNA to prepare it for cleavage by VirD2 (25). However, purified VirD1 protein does not exhibit such topoisomerase activity in vitro, leaving the exact role of VirD1 unclear (26). In vitro experiments have demonstrated that purified VirD2 can specifically cleave single-stranded oligonucleotides at the expected position in the 25-bp border sequence (27, 28). Neither supercoiled nor relaxed double-stranded DNA acts as substrate for cleavage by VirD2 alone in vitro (26), but the combination of VirD1 and VirD2 has been reported to be sufficient to catalyze this cleavage in vitro (27). When it cleaves the 25-bp border sequence, VirD2 becomes covalently attached to the 5′ end of the nicked DNA (18, 29, 30) via tyrosine residue 29 (17, 27, 31). Similar VirD2-catalyzed cleavage at the left border sequence leads to the liberation of the T-strand perhaps by repair replication and displacement.
T-strand is believed to be coated along its length by a single-strand binding protein, VirE2, at some point in the transfer process. Both VirE2 and VirD2 contain nuclear localization signals that are believed to pilot the T-strand into the plant cell nucleus (32–36). The nuclear localization signals of VirD2 and VirE2 are recognized in tobacco and in maize (37), but their efficiency is dependent on the developmental stage of the tissue. Recent indirect evidence supports the view that VirD2 may participate in the ligation of the 5′ end of the T-strand to the plant DNA (38), accounting for the precise joining of T-DNA to plant DNA at the terminal nucleotide of the T-strand.
In the present study, we have developed a novel plant transformation technique that combines some of the advantages of the Agrobacterium system with the proven high efficiency of the biolistic delivery system for a wide range of crop plants. It is designed to integrate the gene of interest with no vector sequence, as in T-DNA inserts, and to control the copy number. The approach uses plant expression cassettes for virD1 and virD2 genes codelivered with a vector containing T-DNA border sequences flanking a gene of interest. We have demonstrated that virD1 and virD2 gene products can cleave T-DNA border sequences in planta and produce transformants with T-DNA-type insertion events (“agrolistic” events) after biolistic delivery.
MATERIALS AND METHODS
Plasmids.
The structures of all constructs used in this study are presented in Figs. 1 and 2.
Figure 1.
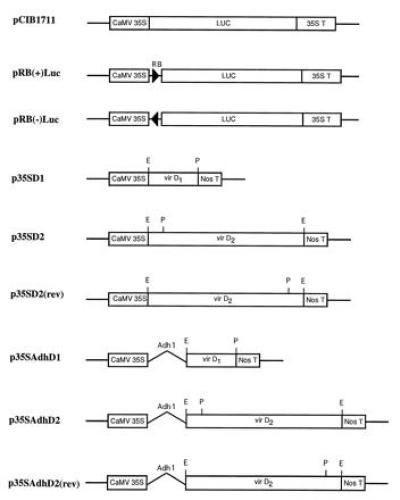
Constructs. Components of the plasmids are as described in Materials and Methods. RB, the 25-bp right border sequence. Restriction sites are indicated as: E, EcoRI; P, PstI,
Figure 2.
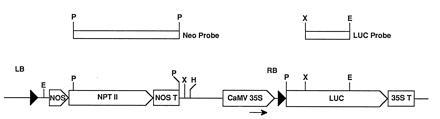
Schematic diagram of pNeoRBLuc. LB, left border; RB, right border. The arrow indicates the location of the primer used to sequence the junction. The top boxes above the diagram indicate the probes used for Southern blot analysis of transformants. Restriction sites are indicated as follows: E, EcoRI; H, HindIII; P, PstI; X, XbaI.
Construction of pRB(+)Luc and pRB(−)Luc.
The starting plasmid for these constructs was pCIB1711, containing a 35S-promoter, 35S-leader, and 35S-terminator flanking the firefly luciferase coding region (Luc) (39) as shown in Fig. 1. Plasmid pRB(+)Luc was formed by introducing the Ti plasmid right border sequence between the promoter and leader of pCIB1711 using the following pair of BamHI–PstI-ended synthetic oligonucleotides corresponding to the right border sequence of LBA5269 (40) (5′-GATCCGGCAGGATATATACCGTTGTAATTCTGCA-3′ and 5′-GAATTACAACGGTATATATCCTGCCG-3′). The plasmid was assembled by four-way ligation of (i) HindIII–BamHI 0.9-kb fragment of pCIB1711; (ii) BamHI–PstI oligonucleotide pair with right border sequence; (iii) PstI–ClaI 1.7-kb fragment; and (iv) ClaI–HindIII 3-kb fragment of pCIB1711. The construction of pRB(−)Luc was identical except that a different pair of oligonucleotides was used, in which the border sequence was in the reverse orientation with respect to BamHI and PstI overhanging ends.
Construction of pNeoRBLuc and pNeoLuc.
pNeoRBLuc was designed for stable transformation of tobacco suspension cells and contains a left border sequence, the neomycin phosphotransferase gene (nptII) and the luciferase gene with the right border inserted between the promoter and the leader [excised from pRB(+)Luc] (Fig. 2). Plasmid pNeoRBLuc was constructed by four-way ligation of the isolated fragments as follows. The 1.2-kb BglII–EcoRI left border fragment from pBin19 (41) was subcloned into pUC21 and then excised as an XbaI–SacII fragment. The nos/nptII/nos gene was excised from pBin19 as a 2.2-kb SacII–HindIII fragment. A ClaI–XbaI fragment of pCIB1711 (3.2 kb) and the HindIII–ClaI fragment of pRB(+)Luc were excised.
As a control, plasmid pNeoLuc (not shown) was constructed identically, except that the 2.4-kb HindIII–ClaI fragment was derived from pCIB1711 and lacked the T-DNA border sequence.
Construction of virD1 and virD2-Derived Plasmids.
virD1 and virD2 genes from pTiA6 were subcloned into expression vector pMF6 (42), consisting of the CaMV35S promoter (0.5 kb), the Adh1 intron 1 (0.5 kb), and the nopaline synthase (nos) polyadenylylation region (0.25 kb) (Fig. 1). A fragment of 0.6 kb (EcoRI–PstI) from pAD1187 (20) corresponding to the virD1 coding sequence was cloned into pMF6, yielding p35SAdhD1. The virD2 coding sequence was excised as a 1.8-kb EcoRI fragment from pAD1190 (20) and cloned in pMF6. The plasmids obtained, p35SAdhD2 and p35SAdhD2(rev), carried the virD2 coding region in either the sense or the antisense orientation. The Adh1 intron sequence was deleted from p35SAdhD1, p35SAdhD2, and p35SAdhD2(rev) to create p35SD1, p35SD2, and p35SD2(rev), respectively, for experiments designed for tobacco tissues.
pGUS.
pGUS (43) is a pUC derivative containing the β-glucuronidase (GUS) coding sequence under the control of the CaMV35S promoter and the castor bean catalase gene intron.
Plant Material.
Maize suspension cells. Suspension cultures of maize (Zea mays L.) were initiated from cryopreserved embryogenic type II callus selected from immature embryos of an elite genotype related to B73. To initiate cultures, about 1 g of callus (44) was added to 50 ml N6 liquid medium (45) supplemented with 30 g/liter sucrose and 2 mg/liter 2,4-dichlorophenoxy acetic acid (2,4-D) (2N63S). Maize cell suspensions used for bombardment experiments were taken from 3-day-old rapidly growing cultures. Before bombarding, ≈0.5 ml of packed volume cells was vacuum-filtered onto 7-cm filters (Whatman no. 4). Prior to bombardment, the filters were placed on gelrite-solidified N6 medium containing 120 g/liter of sucrose and incubated for 4 hr at 25°C.
Tobacco suspension cells.
The Nicotiana tabacum cell line NT-1 (46) was grown in Murashige and Skoog (MS) medium (47) supplemented with 2 mg/liter of 2,4-D and sucrose (30 g/liter) (MS3S). Aliquots of 0.5 ml from 4-day-old cultures were spread onto sterile filters (Whatman no. 4), which were then transferred onto MS medium supplemented with 12% sucrose and kept at room temperature for 4 hr before bombardment.
Bombardment of Plant Cells.
Tissues were bombarded with gold microprojectiles onto which was precipitated a mixture of plasmids. pGUS plasmid DNA was used as internal standard for transformation efficiency in all maize and tobacco experiments. For cotransformation experiments, the gold particles carried either an equal mass of all plasmid DNAs (0.5 μg of each plasmid DNA per target plate) or 2:1 weight ratio of plasmids carrying virD1 and virD2 genes to substrate plasmid. For stable transformation experiments, cotransformation mixtures contained a 5:1 weight ratio of plasmids carrying virD1 and virD2 genes to nptII selection plasmid. Each aliquot of plasmid mixture bombarded per target plate consisted of 0.1 μg of the target plasmid with selectable marker and 0.5 μg each of p35SD1 and p35SD2 plasmid DNAs. Appropriate quantities of each DNA were mixed in a total volume of 10 μl and precipitated with 50 μl of 2.5 M CaCl2 and 20 μl of 0.1 M spermidine-free base to effect precipitation onto 50 μl of 1.0 μm gold microcarriers (60 mg/ml). Microprojectile bombarment was performed with the PDS-1000 He biolistic device (DuPont) using 1500 psi rupture discs with the sample positioned 8 cm below the stopping screen shelf.
Stable Transformation of Tobacco Suspension Cells.
Twenty-four hours after bombardment, tobacco cells were transferred onto MS3S plates with 300 μg/ml kanamycin. Independent microcalli that appeared about 3 weeks after bombardment were transferred onto fresh plates supplemented with 300 μg/ml kanamycin. After two subcultures on the same medium, suspension cultures were initiated by inoculating about 100 mg of tobacco cells into 25 ml liquid medium supplemented with 300 μg/ml kanamycin.
Transient Expression Assays.
Luciferase was assayed in tissue extracts according to the recommendation of the supplier (Luciferase assay system, Promega). GUS activity was determined by a chemoluminescent assay with the GUS-Light kit (Tropix, Bedford, MA). Luciferase and GUS activities are expressed as light units detected by an Analytical Luminescence Laboratory (San Diego) model 2001 Luminometer integrated over 10 sec at 25°C.
DNA Extraction and Southern Blot Hybridization.
Cell cultures were harvested by filtration 10 days after inoculation and were frozen in liquid nitrogen. DNA was isolated as described (48). Approximately 5 μg of genomic DNA was used for digestion with EcoRI. Following separation on a 0.7% agarose gel, the DNA was transferred to Genescreen Plus membrane and hybridization performed according to the conditions described by the manufacturer (DuPont/NEN). DNA probes were labeled with [α-32P]dCTP using the oligo labeling kit of Pharmacia. The neo probe corresponded to a 2-kb PstI fragment of the nptII gene (Fig. 2). The luc probe corresponded to a 0.7-kb XbaI–EcoRI fragment of the luciferase gene (Fig. 2). For removal of probes, membranes were stripped with a solution of 0.1% SDS at 100°C for 5 min.
Cloning of T-DNA/Plant DNA Junctions.
DNA (30 μg) from transgenic tobacco calli was digested with EcoRI and subjected to preparative electrophoresis on a 1% SeaPlaque agarose gel (FMC). Slices of agarose corresponding to the size of fragments to be cloned were cut out of the gel, and DNA was extracted from agarose with QIAquick Gel Extraction Kit (Qiagen, Chatsworth, CA). Fragments were then cloned into the dephosphorylated EcoRI site of pUC19. Ligation mixes were used to transform Escherichia coli HB101 cells by electroporation. Colonies containing the plasmid with the correct insert were identified by colony filter hybridization, using a 0.5-kb CaMV35S promoter fragment as probe. Sequence of the junction of donor plasmid DNA with plant DNA was obtained using the primer (CCACTATCCTTCGCAAGACC) located in the CaMV35S promoter at a distance of 106 bp from the right border sequence.
RESULTS
Experimental Design.
To investigate whether virD1 and virD2 gene products can nick a T-DNA border sequence when expressed in plant cells, a test plasmid pRB(+)Luc was constructed. It contained a substrate T-DNA border sequence between the promoter and the leader of the luciferase gene. For this (+) orientation of the T-DNA border, a site-specific nick introduced by virD1 and virD2 gene products in the bottom strand of the border sequence would interrupt the DNA strand that is the template for luciferase mRNA, and thus decrease the production of luciferase transcript and enzyme. After cobombardment of plant cells with pRB(+)Luc and plasmids carrying the vir D genes, any nicking at the border sequence should be measurable quantitatively by assaying luciferase activity. However, any decrease of luciferase activity could also be explained by the binding of virD1 and virD2 gene products at the border sequence located between the promoter and the coding sequence, binding that might inhibit the transcription of the luciferase gene. pRB(−)Luc, a plasmid that contains the border sequence in reverse orientation with respect to the promoter, was therefore tested to distinguish between these two possibilities. If a decrease of luciferase activity is the result of the binding of the virD gene product(s) to the border sequence, then this decrease would probably be observed even with the border sequence in reverse orientation.
Since virD1 and virD2 gene products must be produced transiently in the bombarded plant cells before they can nick the border sequence, any such nicking would occur after transcripton of the luciferase gene has already started. Therefore, luciferase activity measurements probably underestimate the VirD1 and VirD2 activity in plant cells.
To express virD1 and virD2 genes in plant cells, their respective open reading frames (ORFs) were placed under the control of the CaMV35S promoter. The virD2 ORF was also introduced in antisense orientation with respect to the promoter to serve as control. As the presence of the maize Adh1 intron 1 has been found to increase the expression of genes in maize (42), p35SAdhD1 and p35SAdhD2 (containing the intron inserted in the leader region just ahead of the coding region of virD1 and virD2, respectively) were constructed for use in maize transient expression experiments. A plasmid expressing the GUS gene, pGUS, was included in each bombardment as an internal standard to control for the efficiency of DNA transfer. In all cases, the activity of reporter is expressed as a ratio of luciferase to GUS activity, to correct for variability in efficiency of DNA delivery.
Transient Expression Assays to Test for Cleavage of the Border Sequence by virD1 and virD2 Gene Products in Planta.
Maize and tobacco cells were first transiently transformed with the test plasmid codelivered with virD1 and virD2 genes separately to determine their ability individually to affect transcription through the T-DNA border sequence. Following codelivery of either p35SD1 DNA (tobacco) or p35SAdhD1 DNA (maize) with pRB(+)LUC DNA, 80% of the control level of luciferase to GUS activity was observed in both tobacco and maize cells (Tables 1 and 2). Codelivery of p35SD2 DNA (tobacco) or p35SAdhD2 DNA (maize) with pRB(+)Luc DNA resulted in 50% and 80% of the control level of luciferase to GUS activity, respectively (Tables 1 and 2).
Table 1.
Activity of virD1 and virD2 in tobacco suspension cells
| Plasmids + pGUS | Mean ± SD | % of control |
|---|---|---|
| pRB(+)Luc | 1.36 ± 0.06 | — |
| pRB(+)Luc + p35SD1 | 0.98 ± 0.03 | 72 |
| pRB(+)Luc + p35SD2 | 0.69 ± 0.07 | 50 |
| pRB(+)Luc + p35SD1 + p35SD2 (1:1:1) | 0.27 ± 0.05 | 20 |
| pRB(+)Luc + p35SD1 + p35SD2 (1:2:2) | 0.14 ± 0.05 | 10 |
| pRB(+)Luc + p35SD1 + p35SD2(rev) (1:1:1) | 1.08 ± 0.13 | 80 |
| pRB(+)Luc + p35SD1 + p35SD2(rev) (1:2:2) | 1.13 ± 0.08 | 83 |
| pRB(−)Luc | 1.40 ± 0.14 | — |
| pRB(−)Luc + p35SD1 | 1.33 ± 0.13 | 95 |
| pRB(−)Luc + p35SD2 | 1.19 ± 0.12 | 85 |
| pRB(−)Luc + p35SD1 + p35SD2 | 1.56 ± 0.38 | 112 |
Plasmid constructs are described in the legend to Fig. 1 and were delivered to tobacco cells by the biolistic device. Numbers in parentheses indicate the molar ratio of plasmids. Following incubation for 24 hr, tissues were homogenized and enzyme activities determined. Activities are expressed as a ratio of luciferase (Luc) to β-glucuronidase (GUS). Independent bombardments were analyzed, and data are presented as mean values of six repetitions ± SD. % control values are determined from the ratio of the luciferase to GUS activities to those activities observed with control plasmid.
Table 2.
Activity of virD1 and virD2 genes in maize suspension cells
| Plasmids + pGUS | Mean ± SD | % of control |
|---|---|---|
| pRB(+)Luc | 1.26 ± 0.27 | — |
| pRB(+)Luc + p35SAdhD1 | 1.32 ± 0.28 | 105 |
| pRB(+)Luc + p35SAdhD2 | 1.02 ± 0.15 | 81 |
| pRB(+)Luc + p35SAdhD1 + p35SAdhD2 (1:1:1) | 0.11 ± 0.03 | 8.7 |
| pRB(+)Luc + p35SAdhD1 + p35SAdhD2 (1:2:2) | 0.006 ± 0.007 | 0.5 |
| pRB(+)Luc + p35SAdhD1 + p35SAdhD2(rev) (1:1:1) | 0.99 ± 0.20 | 78.6 |
| pRB(+)Luc + p35SAdhD1 + p35SAdhD2(rev) (1:2:2) | 0.96 ± 0.26 | 76 |
Activities are expressed as described in Table 1.
The two vir genes together appeared to have a synergistic effect. Codelivery by the biolistic device of equal amounts of pRB(+)LUC DNA with both plasmids carrying virD1 and virD2 genes (ratio of 1:1:1) reduced luciferase activity to ca. 20% of control in tobacco (Table 1) and 10% of control in maize cells (Table 2). At a higher ratio of virD1 and virD2 plasmids to test plasmid (2:2:1), the luciferase activity was reduced further to ca. 10% in tobacco cells (Table 1) and 1% in maize cells (Table 2). Analogous experiments using the control plasmid p35SD2(rev) (tobacco) or p35SAdhD2(rev) (maize) with the virD2 coding sequence in antisense orientation gave results similar to those with virD1 gene alone (Tables 1 and 2) as expected.
Reversal of orientation of the T-DNA border in the test plasmid reduced any influence of virD1 and/or virD2 genes on transient expression of the luciferase gene. When pRB(−)Luc was cobombarded into tobacco cells with p35SD1 and p35SD2 plasmid DNA, no significant decrease in luciferase activity was observed. Codelivery of pRB(−)Luc performed with p35SD1 or p35SD2 separately likewise showed no significant decrease of luciferase activity (Table 1). These observations strongly indicated that the decrease of luciferase activity seen with pRB(+)Luc test plasmid plus virD1 and virD2 genes was the result of a strand-specific nick at the right border sequence by vir gene products similar to that observed in Agrobacterium (11).
Analysis of Stable Transformants.
Stable transformation of tobacco suspension cells was undertaken to assess the effect of virD1 and virD2 gene products on the pattern of DNA integration after codelivery of these genes with their substrate DNA. For these experiments, we used pNeoRBLuc, which contains a left T-DNA border, nptII as selectable marker, and the 35SRB(+)Luc gene with the right T-DNA border inserted between promoter and luciferase coding region. In the Results and Discussion, we designate as “agrolistic events” those DNA insertions into the tobacco genome that would result after VirD1 and VirD2 activity on border sequences, generating a T-strand. In contrast, we designate as “biolistic events” those DNA inserts representing the process normally occurring after gene delivery into plant cells by the biolistic device. The initial screen to distinguish biolistic events and putative agrolistic events was absence of luciferase activity in the transformed clone, arising from exclusion of the Luc coding region by T-DNA excision from pNeoRBLuc. In Southern blot analysis, the transgenes representing an agrolistic event should hybridize with the neo probe and not with the luc probe. Moreover, in an agrolistic event, the sequence of the junction between introduced DNA and plant DNA should correspond precisely to the right border end of a T-strand. Both types of events may occur in the same plant cell, but such clones would be scored genetically as biolistic events based on the presence of luciferase activity.
Tobacco suspension cells were bombarded with microprojectiles coated with pNeoRBLuc plasmid DNA together with p35SD1 and p35SD2 DNAs in a ratio 1:5:5. As controls, pNeoRBLuc plasmid was also bombarded alone and the borderless control plasmid pNeoLuc was cobombarded with p35SD1 and p35SD2. Stable transformants were selected by growth on kanamycin-containing medium. An average of 40 kanamycin-resistant clones appeared per bombarded filter, but only one or two calli per plate were analyzed further. Similar numbers of kanamycin-resistant calli were recovered following bombardment with the control plasmids pNeoRBluc DNA alone or pNeoLuc DNA plus p35SD1 DNA and p35SD2 DNA.
A rough estimate of the frequency of agrolistic events could be made by the ratio of the total number of kanamycin-resistant calli analyzed that do not express luciferase to total kanamycin calli. By this criterion, the frequency of agrolistic events was about 10%; of 32 callus lines analyzed, 3 did not express luciferase activity.
Southern Blot Analysis of Control “Biolistic” Events.
Southern blot hybridzation was performed on DNA from control kanamycin-resistant callus lines obtained after bombardment with (i) pNeoRBLuc alone (Fig. 3, lanes A–F) and (ii) pNeoLuc plasmid cobombarded with p35SD1 and p35SD2 DNAs (Fig. 3, lanes G and H). Genomic DNA was digested with EcoRI, which produces a 3.9-kb fragment from the pNeoRBLuc plasmid that is homologous to both neo and luc probes (Fig. 2). When genomic DNA digests were hybridized with the neo probe, all lanes exhibited a hybridizing band of the predicted size (3.9 kb) (Fig. 3, lanes A–F), and the number of intact fragment copies varied from 1 to more than 10 per nucleus (Fig. 3). Southern blot analysis of transformed lines from cobombardment with p35SD1 DNA and p35SD2 DNA together with the borderless control plasmid pNeoLuc are presented in lanes G and H of Fig. 3. These exhibited both intact and rearranged copies of the nptII gene. Such rearrangements are often observed in transformants obtained by the biolistic device.
Figure 3.
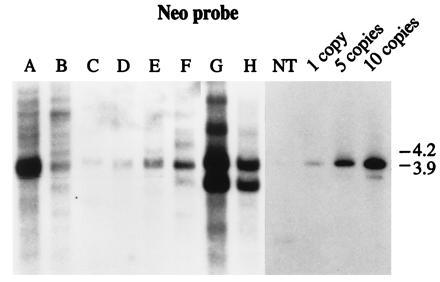
Southern blot hybridization analysis of DNA from kanamycin-resistant calli derived from cells bombarded with control plasmids. pNeoRBLuc plasmid (lanes A–F) and the borderless construct pNeoLuc together with p35SD1 and p35SD2 plasmids (lanes G and H) were delivered to maize cells. The blot was hybridized with the neo probe. NT, DNA from nontransformed tobacco cells. Lanes 1, 5, and 10: genomic DNA mixed with pNeoRBLuc to reconstruct the integration of 1, 5, or 10 copies of the gene per diploid nucleus, respectively.
Southern Blot Analysis of Candidate Agrolistic Events.
Southern blot analysis of DNA from 16 kanamycin-resitant callus lines obtained after cobombardment of pNeoRBLuc with p35SD1 and p35SD2 plasmid DNAs is presented in Fig. 4. For Southern blot analysis, 13 callus lines were chosen randomly from the 32 that tested positively for the luciferase activity. Eight of the Southern blot analyses from these clones are shown in Fig. 4 (lanes 4–11) together with those of the three clones found not to express luciferase (Fig. 4, lanes 1–3). The callus lines in lanes 4 to 11 show a band of the predicted size (3.9 kb) hybridizing with the neo probe and the number of intact nptII gene copies ranges from 1 to 10 per nucleus. The number of copies observed is much lower in calli transformed with pNeoRBLuc DNA and p35SD1 and p35SD2 DNA. The estimated copy number of the nptII gene in callus lines 1, 2, and 3 is, respectively, 1, 2, and 1 (Fig. 4, lanes 1 to 3).
Figure 4.
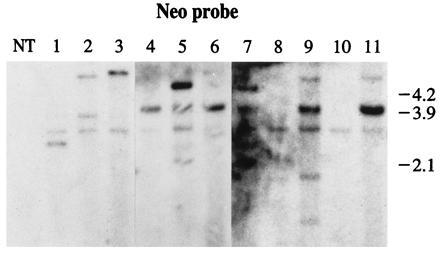
Southern blot analysis of kanamycin-resistant calli derived from cells bombarded with pNeoRBLuc and p35SD1 and p35SD2 plasmid DNAs. NT, DNA from nontransformed tobacco callus. The transformed calli are labeled from lanes 1 to 11. The blot was hybridized with the neo probe.
When blots were hybridized with the luc probe, three groups of transgenic callus lines could be distinguished: (i) callus lines with inserts hybridizing with the neo probe and the luc probe; (ii) callus lines in which some inserts hybridized with only the neo probe and some inserts hybridized with both probes neo and luc; and (iii) callus lines with inserts hybridizing only with the neo probe. The first group of calli probably did not contain agrolistic events. The second group of calli probably contained two types of events: agrolistic events evidenced by the 4.8-, 4.6-, and 5-kb fragments in lanes 5, 7, and 9, respectively, and biolistic events evidenced by the 3.9-kb fragment. The third group of calli exhibited only putative agrolistic events. Lane 1 contains one band of 3.2 kb, lane 2 contains two bands of 3.8 kb and 5 kb, and lane 3 contains one band of 5.5 kb. These three hybridizations patterns were unique, clearly representing independent single cell transformation events.
Among 16 transgenic tobacco lines analyzed by Southern blot hybridization, 10 exhibited biolistic events, 3 exhibited putative agrolistic events, and 3 exhibited both.
DNA Sequence Analysis of Right Border Regions of Agrolistic Events.
The nature of putative agrolistic insertion events was ultimately verified by determining the sequence of the junction between integrated DNA and plant DNA. The 3.2-kb fragment of lane 1, the 3.8-kb and 5-kb fragments of lane 2, the 5-kb fragment of lane 3, and the 4.8-kb fragment of lane 5 were cloned and sequenced outward from inside the T-DNA right border (see Materials and Methods). The nucleotide sequence revealed that each of these fragments contained a right border/plant DNA junction (Fig. 5). The right end point of the T-DNA was identical to the nicking site of the right border sequence of T-DNA.
Figure 5.

Sequence analysis of plant DNA target sites after transformation with pNeoRBLuc, p35SD1, and p35SD2 plasmids. Numbers refer to the target clones as listed in Fig. 4. The right border sequence carried by pNeoRBLuc is underlined (lane RB). The plant target sequence of fragments 1, 2a, 3, and 5 shows 100% homology with PSII of tobacco (GenBank accession no. X62426X62426; nt 908), with NtpII10 of tobacco (GenBank accession no. X70088X70088; nt 573), with ribulose 1,5-bisphosphate carboxylase of tobacco (GenBank accession no. X02353X02353; nt 2174) and 80% homology with a chlorophyll binding protein of petunia (GenBank accession no. M21317M21317; nt 1013), respectively. Numbers between brackets indicate the accession number and the nucleotide coordinate from which the homology starts.
The plant nucleotide sequence in four of five cases perfectly matched tobacco sequences that have been reported previously (see legend to Fig. 5). Interestingly, in each of the four cases, the right border of T-DNA is inserted with the CaMV35S promoter oriented in the antisense direction with respect to the plant gene, and is situated in a high AT region near the polyadenylylation site in the 3′-untranslated region of the gene. No additional nucleotides and no repeated sequences were observed at the right junction sites. It is not possible to conclude whether any deletions of the target sites have occurred because the left border of the insert was not determined.
Fragments of 3.9 kb from lanes 5, 7, and 9 were also cloned, and their nucleotide sequence did not show any right border sequence-plant DNA junction, but rather the full-length right border sequence and the expected luciferase coding sequence beyond (data not shown).
CONCLUSION
The Ti plasmid-encoded virulence proteins VirD1 and VirD2 are required for the formation of T-strands in Agrobacterium. Here we present evidence for T-DNA formation in planta and its integration into the plant genome. Nearly 20% of the transformed tobacco calli exhibited only agrolistic inserts, i.e., DNA integrated after the action of virD1 and virD2 gene products only. A similar fraction of transformed calli contained both agrolistic events and biolistic events. The transgene::plant DNA junctions in agrolistic events demonstrated the occurrence of precise site-specific cleavage within the right border sequence followed by precise insertion into plant DNA, in accordance with data from Agrobacterium-mediated transformants showing the right border T-DNA ends just after the first three nucleotides of the 25-bp repeat (49–52).
The integration sites of the agrolistic events characterized here are in transcribed regions, supporting the observation that T-DNA is preferentially integrated into potentially transcribed genomic loci in different plant species (53–55) with the T-DNA insertions randomly distributed in plant chromosomes (56, 57). Although T-DNA integration is usually not correlated with large rearrangements in the plant DNA, deletions, inversions, and duplications of target plant DNA sequences can occur during (or following) T-DNA insertion. For the agrolistic events examined here, no major rearrangement was noted in the plant target sites, at least on the right border side.
The consistent pattern of agrolistic integrations near the polyadenylylation signal of known tobacco light-inducible and/or photosynthetic genes with the CaMV35S promoter at the right border directed in an antisense orientation to the open reading frame is intriguing. In this orientation, the insertion of the T-DNA structure could potentially generate an antisense transcript that may inactivate expression of the corresponding tobacco gene. Such photosynthetic genes are, however, not required by these non-photosynthetic cultured NT1 cells. It is possible that similar integrations in essential functions lead to nonviable transformants; thus, the estimate of frequency of agrolistic events may be lower than would be found with a T-DNA lacking any promoter at the right border.
The virD1 and virD2 genes that were codelivered with the selectable marker in our stable transformation studies presumably are biolistically integrated into the same transformed lines at high frequency; cotransformation is very efficient with the biolistic device. However, because of their biolistic insertion mechanism, they are unlikely to be linked to the “agrolistic” insert and may be eliminated by subsequent breeding of the transgenic plant. This presumption could not be tested with these transgenic NT1 cells, which are not regenerable.
The agrolistic transformation system offers several distinct advantages: (i) It should be immediately applicable to any plant target tissue susceptible to biolistic transformation methods. (ii) The inserted DNA does not carry extraneous vector DNA. (iii) Fewer copies of the gene of interest are inserted than is the case for DNA delivered by the normal biolistic mechanism. This should minimize regions of homology which may contribute to genetic and/or epigenetic instability. Additional copies inserted as independent T-strand insertions would likely be unlinked and could be separated by segregation after genetic crossing of the transgenic plant.
The agrolistic approach thus combines the advantages of efficient biolistic delivery with the elegance and precision of the Agrobacterium T-DNA insertion mechanism to afford a new, widely applicable technology for producing transgenic crop plants of value to agriculture.
Acknowledgments
We thank Dr. Anath Das for providing pAD1187 and pAD1190.
Footnotes
Abbreviations: Ti, tumor inducing; Ri, root inducing; T-DNA, the portion of the Ti plasmid that is transferred to plants; GUS, β-glucuronidase.
References
- 1.Ahl Goy P, Duesing J H. Bio/Technology. 1995;13:454–458. [Google Scholar]
- 2.Koziel M G, Beland G L, Bowman C, Carozzi N B, Crenshaw R, Crossland L, Dawson J, Desai N, Hill M, Kadwell S, Launis K, Lewis K, Maddox D, McPherson K, Meghji M R, Merlin E, Rhodes R, Warren G W, Wright M, Evola S V. Bio/Technology. 1993;11:194–200. [Google Scholar]
- 3.Klein T M, Harper E C, Svab Z, Sanford J C, Fromm M E, Maliga P. Proc Natl Acad Sci USA. 1988;85:8502–8505. doi: 10.1073/pnas.85.22.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein T M, Kornstein L, Sanford J C, Fromm M E. Plant Physiol. 1989;91:440–444. doi: 10.1104/pp.91.1.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon-Kamm W J, Spencer T M, Mangano M L, Adams T R, Daines R J, Start W G, O’Brien J V, Chambers S A, Adams R A, Willets N G, Rice T B, Mackey C J, Krueger R W, Kausch A P, Lemaux P G. Plant Cell. 1990;2:603–618. doi: 10.1105/tpc.2.7.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vasil V, Castillo A M, Fromm M E, Vasil I K. Bio/Technology. 1992;10:667–674. [Google Scholar]
- 7.Wan Y, Lemaux P G. Plant Physiol. 1994;104:37–48. doi: 10.1104/pp.104.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Register J C, III, Peterson D J, Bell P J, Bullock W P, Evans I J, Frame B, Greenland A J, Higgs N S, Jepson I, Jiao S, Lewnau J L, Sillick J M, Wilson H M. Plant Mol Biol. 1994;25:951–961. doi: 10.1007/BF00014669. [DOI] [PubMed] [Google Scholar]
- 9.Matzke M A, Matzke A J M. Plant Physiol. 1995;107:679–685. doi: 10.1104/pp.107.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chilton M-D. Proc Natl Acad Sci USA. 1993;90:3119–3120. doi: 10.1073/pnas.90.8.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kado C I. In: Bacterial Conjugation. Clewell D B, editor. New York: Plenum; 1993. pp. 243–254. [Google Scholar]
- 12.Zambryski P. Annu Rev Plant Physiol. 1992;43:465–490. [Google Scholar]
- 13.Stachel S E, Nester E W. EMBO J. 1986;5:1445–1454. doi: 10.1002/j.1460-2075.1986.tb04381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stachel S, Timmerman B, Zambryski P. EMBO J. 1987;6:857–863. doi: 10.1002/j.1460-2075.1987.tb04831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrera-Estrella A, Chen Z, Van Montagu M, Wang K. EMBO J. 1988;7:4055–4062. doi: 10.1002/j.1460-2075.1988.tb03299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Vos G, Zambryski P. Mol Plant Microbe Interact. 1989;2:43–52. doi: 10.1094/mpmi-2-043. [DOI] [PubMed] [Google Scholar]
- 17.Durrenberger F, Crameri A, Hohn B, Koukolikova-Nicola Z. Proc Natl Acad Sci USA. 1989;86:9154–9158. doi: 10.1073/pnas.86.23.9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howard E A, Winsor B, A, De Vos G, Zambryski P C. Proc Natl Acad Sci USA. 1989;8:4017–4021. doi: 10.1073/pnas.86.11.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koukolikova-Nicola Z, Raineri D, Stephens K, Ramos C, Tinland B, Nester E, Hohn B. J Bacteriol. 1993;175:723–731. doi: 10.1128/jb.175.3.723-731.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filichkin S A, Gelvin S B. Mol Microbiol. 1993;8:915–926. doi: 10.1111/j.1365-2958.1993.tb01637.x. [DOI] [PubMed] [Google Scholar]
- 21.Stachel S E, Timmerman B, Zambryski P. Nature (London) 1986;322:706–712. [Google Scholar]
- 22.Yanofsky M F, Porter S G, Young C, Albright L M, Gordon M P, Nester E W. Cell. 1986;47:471–477. doi: 10.1016/0092-8674(86)90604-5. [DOI] [PubMed] [Google Scholar]
- 23.Wang K, Stachel S E, Timmerman B, Van Montagu M, Zambryski P. Science. 1987;235:587–591. doi: 10.1126/science.235.4788.587. [DOI] [PubMed] [Google Scholar]
- 24.Albright L M, Yanofsky M F, Leroux B, Ma D, Nester E W. J Bacteriol. 1987;169:1046–1055. doi: 10.1128/jb.169.3.1046-1055.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghai J, Das A. Proc Natl Acad Sci USA. 1989;8:3109–3113. doi: 10.1073/pnas.86.9.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheiffele P, Pansegrau W, Lanka E. J Biol Chem. 1995;270:1269–1276. doi: 10.1074/jbc.270.3.1269. [DOI] [PubMed] [Google Scholar]
- 27.Pansegrau W, Schoumacher F, Hohn B, Lanka E. Proc Natl Acad Sci USA. 1993;90:11538–11542. doi: 10.1073/pnas.90.24.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jasper F, Koncz C, Schell J, Steinbiss H-H. Proc Natl Acad Sci USA. 1994;91:694–698. doi: 10.1073/pnas.91.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward E R, Barnes W M. Science. 1988;242:927–930. [Google Scholar]
- 30.Young C, Nester E W. J Bacteriol. 1988;8:3367–3374. doi: 10.1128/jb.170.8.3367-3374.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogel A M, Das A. J Bacteriol. 1992;174:303–312. doi: 10.1128/jb.174.1.303-308.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herrera-Estrella A, Van Montagu M, Wang K. Proc Natl Acad Sci USA. 1990;87:9534–9537. doi: 10.1073/pnas.87.24.9534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howard E A, Zupan J R, Citovsky V, Zambryski P. Cell. 1992;68:109–118. doi: 10.1016/0092-8674(92)90210-4. [DOI] [PubMed] [Google Scholar]
- 34.Shurvinton C E, Hodges L, Ream W. Proc Natl Acad Sci USA. 1992;89:11837–11841. doi: 10.1073/pnas.89.24.11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tinland B, Koukolikova-Nicola Z, Hall M N, Hohn B. Proc Natl Acad Sci USA. 1992;89:8000–8004. doi: 10.1073/pnas.89.16.7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossi L, Hohn B, Tinland B. Mol Gen Genet. 1993;239:345–353. doi: 10.1007/BF00276932. [DOI] [PubMed] [Google Scholar]
- 37.Citovsky V, Warnick D, Zambryski P. Proc Natl Acad Sci USA. 1994;91:3210–3214. doi: 10.1073/pnas.91.8.3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tinland B, Schoumacher F, Angel A M B, Hohn B. EMBO J. 1995;14:3585–3595. doi: 10.1002/j.1460-2075.1995.tb07364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Wet J R, Wood K V, Deluca M, Helsinki D R, Subramani S. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Haaren M J J, Sedee N J A, De Boer H A, Schilperoort R A, Hooykaas P J J. Plant Mol Biol. 1989;13:523–531. doi: 10.1007/BF00027312. [DOI] [PubMed] [Google Scholar]
- 41.Bevan M. Nucleic Acids Res. 1984;12:8711–8721. doi: 10.1093/nar/12.22.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callis J, Fromm M E, Walbot V. Genes Dev. 1987;1:1183–1200. doi: 10.1101/gad.1.10.1183. [DOI] [PubMed] [Google Scholar]
- 43.Ohta S, Mita S, Hattori T, Nakamura K. Plant Cell Physiol. 1990;31:805–813. [Google Scholar]
- 44.DiMaio J J, Shillito R D. J Tissue Culture Methods. 1992;12:163–169. [Google Scholar]
- 45.Chu C C, Wang C C, Sun C S, Hsu C, Yin K G, Chu C Y, Bi F Y. Sci Sin. 1975;18:659–668. [Google Scholar]
- 46.An G. Plant Physiol. 1985;79:568–570. doi: 10.1104/pp.79.2.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murashige T, Skoog F. Physiol Plant. 1962;15:473–497. [Google Scholar]
- 48.Hall G, Allen G C, Loer D S, Thompson W F, Spiker S. Proc Natl Acad Sci USA. 1991;88:9320–9324. doi: 10.1073/pnas.88.20.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koncz C, Nemeth K, Redei G P, Schell J. In: Homologous Recombination and Gene Silencing in Plants. Paszkowski J, editor. Dordrecht, The Netherlands: Kluwer; 1994. pp. 167–189. [Google Scholar]
- 50.Gheysen G, Villaroel R, Van Montagu M. Genes Dev. 1991;5:287–297. doi: 10.1101/gad.5.2.287. [DOI] [PubMed] [Google Scholar]
- 51.Mayerhofer R, Koncz-Kalman Z, Nawrath C, Bakkeren G, Crameri A, Angelis K, Redei G P, Schell J, Hohn B, Koncz C. EMBO J. 1991;10:697–704. doi: 10.1002/j.1460-2075.1991.tb07999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohba T, Yoshioka Y, Machida C, Machida Y. Plant J. 1995;7:157–164. doi: 10.1046/j.1365-313x.1995.07010157.x. [DOI] [PubMed] [Google Scholar]
- 53.Koncz C, Martini N, Mayerhofer R, Koncz-Kalman Zs, Körber H, Redei G P, Schell J. Proc Natl Acad Sci USA. 1989;86:8467–8471. doi: 10.1073/pnas.86.21.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herman L, Jacobs A, Van Montagu M, Depicker A. Mol Gen Genet. 1990;224:248–256. doi: 10.1007/BF00271558. [DOI] [PubMed] [Google Scholar]
- 55.Kertbundit S, De Greve H, Deboeck F, Van Montagu M, Hernalsteens J-P. Proc Natl Acad Sci USA. 1991;88:5212–5216. doi: 10.1073/pnas.88.12.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chyi Y S, Jorgensen R A, Golstein D, Tanksley S D, Loaiza-Figueroa F. Mol Gen Genet. 1986;204:64–69. [Google Scholar]
- 57.Wallroth M, Gerats A G M, Rogers S G, Fraley R T, Horsch R B. Mol Gen Genet. 1986;202:6–15. [Google Scholar]