

Abstract
There is a grave need for safer antiplatelet therapeutics to prevent heart attack and stroke. Agents targeting the interaction of platelets with the diseased vessel wall could impact vascular disease with minimal effects on normal hemostasis. We targeted integrin α2β1, a collagen receptor, because its overexpression is associated with pathological clot formation whereas its absence does not cause severe bleeding. Structure–activity studies led to highly potent and selective small-molecule inhibitors. Responses of integrin α2β1 mutants to these compounds are consistent with a computational model of their mode of inhibition and shed light on the activation mechanism of I-domain-containing integrins. A potent compound was proven efficacious in an animal model of arterial thrombosis, which demonstrates in vivo efficacy for inhibition of this platelet receptor. These results suggest that targeting integrin α2β1 could be a potentially safe, effective approach to long-term therapy for cardiovascular disease.
Keywords: I-domain, platelet, thrombosis, integrin, alpha(2) beta(1)
Despite recent progress in the development of antithrombotic agents, there remains a need for effective new agents to prevent cardiovascular disease while minimally impairing normal hemostasis. For example, agents that target the interaction of platelets with the diseased blood vessel wall would specifically affect atherosclerotic lesions but with a reduced risk of bleeding side effects. Subendothelial collagen plays an essential role in platelet interaction with diseased blood vessels. Collagen, specifically types I and III, constitutes the major protein in atherosclerotic plaques and strongly contributes to lesion growth and arterial narrowing (1). Platelets express two receptors for this matrix component: integrin α2β1 and glycoprotein VI (2). Integrin α2β1 is a good candidate for antithrombotic therapy because its overexpression is associated with stroke and myocardial infarction (3) and its underexpression results in a mildly prolonged bleeding time, which is quite different from the profound bleeding disorder observed in deficiency of the platelet fibrinogen receptor integrin αIIbβ3 (Glanzmann's thrombasthenia) (4–7). However, previous studies on integrin α2β1 inhibition (including monoclonal antibodies against α2 and murine knockout models) have been controversial, demonstrating equivocal results on the role of the integrin (8). Therefore, a primary goal of this work has been to define the role of the platelet collagen receptor integrin α2β1 in thrombosis as a potential therapeutic target.
Our second goal is to shed light on the activation mechanism of integrins containing I-domains, a less well-studied class of 9 integrins including α2β1 and leukocyte integrins. Integrins are α/β heterodimers whose conformational states are regulated by intracellular signaling pathways (9, 10). I-domain-containing integrins differ from other integrins by virtue of having an inserted domain (I-domain) in their α subunit that is responsible for binding extracellular ligands. No crystallographic or NMR structures have been solved for intact I-domain-containing integrins, although crystal structures for the isolated I-domains are available (11). The mechanism of activation of the ligand-binding I-domain has been inferred through comparison with better-known non-I-domain integrins such as αIIbβ3 and αvβ3, for which more high-resolution structural information is available. The I-domain is homologous to the ligand-binding domain of integrins αIIbβ3 and α4β1, and it directly binds ligand in its “open” conformation, exposing a high-affinity active site (12,13). The β subunit I-like domain regulates the switch of the I-domain from a “closed,” inactive conformation to an open, active conformation (14,15). Thus, the affinity of the I-domain for its ligand is regulated by downward displacement of its C-terminal α7 helix into the β subunit I-like domain (Fig. 1) (16). A class of LFA-1 (integrin αLβ2) antagonists believed to be I-domain inhibitors were proposed to function by an allosteric inhibitory mechanism: by binding the I-like domain, the inhibitors lock the integrin into its inactive conformation (17). A second goal of this work has been to develop I-like domain inhibitors that prevent activation of integrin α2β1 based on this mechanism (Fig. 1).
Fig. 1.

Functional model of integrin α2β1 activation. (Upper Left) Active conformation of integrin α2β1 bound to collagen with the C-terminal α7 helix of the I-domain (red) displaced downward to engage the metal ion (purple) in the I-like domain. (Upper Right) Inactive conformation of integrin α2β1 with stabilizing salt bridges formed by the pair E318 and R288 as well as R310 and E338 (Lower) Inhibitor (orange) docked into integrin α2β1 I-like domain locking the compound into the inactive conformation.
A variety of small molecules that weakly inhibit activation of integrin α2β1 have been found, including naturally occurring collagen-mimetic peptides that bind the I-domain (18–20) and lipophilic compounds that stabilize a hydrophobic patch in the I-domain exposed in the inactive conformation (21). Here, we develop high-affinity small-molecule inhibitors of integrin α2β1 that allosterically regulate the integrin through interaction with the I-like domain. These inhibitors demonstrate efficacy in physiological models of vascular disease, including adhesion to collagen under flow conditions and inhibition of thrombus formation in an animal model of arterial disease. This efficacy in a murine model of arterial disease represents a validation of integrin α2β1 as an antithrombotic therapeutic target in vivo. In addition, the effect of these inhibitors on mutations in different domains of integrin α2β1 has enabled us to propose the mechanism for the activation of I-domain-containing integrins. Clinically, the combination of integrin α2β1 inhibitors with existing antiplatelet agents could prove useful as an improved long-term therapeutic regimen for managing cardiovascular disease.
Results
Increasing the Affinity of Prolyl-2,3-diaminopropionic acid (Pro-Dap) α2β1 Antagonists.
We developed selective small-molecule α2β1 inhibitors based on a 2,3-diaminopropionic acid (Dap) backbone (22). The design of these antagonists was based on α4β1 inhibitors incorporating a benzenesulfonyl-prolyl-phenylalanine (Pro-Phe) scaffold (23–25) and αIIbβ3 inhibitors containing a Dap moiety (26, 27). However, the most potent of the synthesized compounds failed to show sufficient activity in vivo, which prompted us to examine structural modifications to improve the potency. The conformation and physicochemical properties of the Pro residue were systematically varied by using a series of Pro surrogates (Scheme 1). We evaluated the ability of these compounds to inhibit adhesion of human platelets to type I collagen (Table 1 (compound 2). Altering the Pro in the parent compound (6) to a desaturated Pro analog, dehydroproline (9), retained the activity of the parent compound, as did an analog in which the 5-membered ring was expanded to a 6-membered piperidine (11). Contracture of the ring to an azetidine (10) or inclusion of fused rings as in the tetrahydroquinoline derivatives (12 and 13) decreased activity. Various other analogs (7 and 8) led to loss of potency. However, alteration of Pro to thiazolidine (Table 2) (compound 14) significantly increased activity.
Scheme 1.

Synthetic route for the synthesis of integrin α2β1 DAP derivatives 6–27. Reagents and conditions: (a) BnBr, NaHCO3, DMF; (b) (1) TFA, CH2Cl2; (2) i-Pr2EtN, BnNCO, DMF; (c) Et2NH, CH2Cl2; (d) PhSO2Cl, NaHCO3, DMF/H2O; (e) HATU, HOAt, i-Pr2EtN, DMF; (f) for sulfur-containing heterocycles: BCl3, CH2Cl2; for oxygen-containing heterocycles: Pd/C, H2, MeOH.
Table 1.
Proline analogs incorporated into α2β1 inhibitors 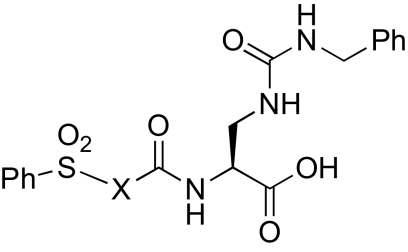
| Compound | X | IC50, nM | Compound | X | IC50, nM |
|---|---|---|---|---|---|
| 6 | 67 | 10 | 215 | ||
| 7 | >1,000 | 11 | 64 | ||
| 8 | >1,000 | 12 | 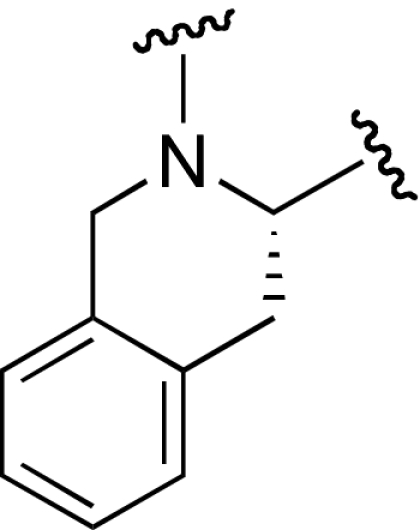 |
>1,000 | |
| 9 | 60 | 13 | 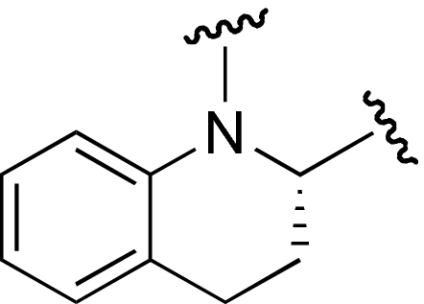 |
127 |
Table 2.
Thiazolidine and oxazolidine inhibitors of α2β1 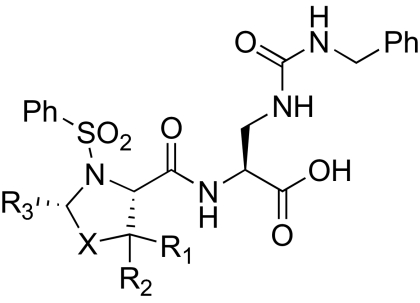
| Compound | X | R1 | R2 | R3 | IC50, nM |
|---|---|---|---|---|---|
| 6 | CH2 | H | H | H | 67 |
| 14 | S | H | H | H | 17 |
| 15 | S | Me | Me | H | 12 |
| 16 | S | H | H | Me | 6 |
| 17 | S | H | H | Et | 49 |
| 18 | S | H | H | i-Pr | 958 |
| 19 | S | H | H | i-Bu | >1,000 |
| 20 | S | H | H | Ph | >1,000 |
| 21 | S | H | H | CH2CH2Ph | 740 |
| 22 | S | Me | Me | Me | 180 |
| 23 | S | Me | Me | Et | 170 |
| 24 | S(O)2 | H | H | H | 227 |
| 25 | O | H | H | H | 82 |
| 26 | O | H | Me | H | 12 |
| 27 | O | H | Me | Me | 25 |
Encouraged by the improved activity of thiazolidine, we investigated other substituted 5-membered heterocycles (Table 2). Addition of a gem dimethyl substitution at the 5-position (15) improved activity slightly over the thiazolidine parent compound (14). Improvement was also observed by addition of a methyl substituent at the 2-position (16). Substitution of larger, sterically demanding groups (17-21) decreased potency. We then combined the potency-enhancing structural features of compounds 15 and 16, which may have been expected to provide an additive benefit; instead, this combination of substituents led to a dramatic loss in activity (22). Replacing the thioether with a sulfone (24) or an ether (25-27) also decreased potency, possibly because of an increase in the polarity of the compounds.
Determining of the Ring Geometry of the Pro Analog in Integrin α2β1 Antagonists.
Substituents in Pro analogs have an important effect on their conformational properties, which in turn strongly affect the energetics of ligand interactions with their receptors (28, 29). Thus, we examined the preferred conformations of the Pro analog in the potent compounds 15 and 16 in contrast to the less potent compound 22, which combined the substitutions in 15 and 16. This set of compounds was chosen because their structures differ by only a single methyl group. The pyrrolidine ring of Pro derivatives can assume two conformations with an UP (Cγ-exo) or a DOWN (Cγ-endo) pucker, based on the relationship between the γ-position ring atom (the thioether in this case) and the carbonyl group (30). In the exo conformation, the γ atom and the carbonyl group are on opposite sides of the plane, whereas these substituents are on the same side of the plane in the endo conformation. A crystal structure of the pyrrolidine ring of 16 showed that the methyl group in the 2-position stabilizes the exo conformation of the thiazolidine ring. This conformation is actually less stable than the endo conformation in the unsubstituted Pro residues found in proteins (28). Supporting the possibility that this difference may be important for activity, quantum mechanical calculations also identified exo as the minimum-energy conformation, shown overlaid with the crystal structure in Fig. 2A. We found the same conformation for the other potent compound 15 (Fig. 2B) and then compared the ring geometry of compounds 15 and 16 with that of the inactive analog 22 (Fig. 2B). We found that the presence of 3 methyl substituents in 22 forces this compound into the endo conformation, whereas the opposite is true for 15 and 16.
Fig. 2.

Quantum mechanical model of inhibitor conformation illustrating prolyl-derived ring structure. (A) Single-crystal X-ray structure for the carboxylic acid derivative of 16 (16_xray, aqua carbon coloring) overlaid with the calculated structure for the amide derivative of 16 (16_frag, green carbon coloring). The energy-minimized model is in excellent agreement with the experimentally derived crystal structure, as illustrated by the similarity in the Ψ2 angles. (B) Comparison of the calculated structure for active inhibitors 15_frag and 16_frag with inactive inhibitor 22_frag. The substituents on compound 22 force the 5-membered ring into the exo conformation (positive Ψ2), whereas 15 and 16 assume the more favorable endo conformation (negative Ψ2). The amide assumes a pseudoequatorial position in 22 and a pseudoaxial position in 15 and 16.
Computational Studies of the α2β1 Antagonist Binding to the β1 I-like Domain.
To shed light on the activation mechanism for I-domain-containing integrins, we generated a computer model of compound 15 docked onto integrin α2β1. Because a high-resolution structure of the intact integrin α2β1 heterodimer is not available, we constructed a model based on the crystal structures of the extracellular portion of integrin αIIbβ3 and the α2β1 I-domain (11,31). Known inhibitors of αIIbβ3 and αvβ3 bind at an allosteric regulatory site between the α and β subunits, invariantly forming an interaction with a divalent metal cation in the β-chain I-like domain (32). In I-domain-containing integrins, a carboxylate-containing side chain (Glu-336 in α2 integrins) binds to this Mg2+ ion, stabilizing the activated conformation of the integrin (15). Antagonists of I-domain-containing integrins have been proposed to lock the integrin in the inactive conformation through binding to this site, the I-like domain (32). Compound 15 docks into the homology model in a geometry appropriate for interaction with the Mg2+ ion at this site in the α/β interface. The I-like domain in α2β1 has a number of notable features (Fig. 3) that help explain the specificity of our antagonists for integrin α2β1 (22). The urea moiety of the Dap side chain projects toward the α2 subunit, explaining the specificity of this class of compounds for α2β1 vs. other β1 integrins (see below) (22). Further, 3 residues in the I-like domain of integrin αIIbβ3 with large side chains change to smaller amino acids in β1, which allows the inhibitors to fit into the binding pocket (Fig. 3).
Fig. 3.

The inhibitor scaffold (thiazolidine-containing dipeptide mimetic) is docked into the binding site on the model of the β1 integrin. Carbon atoms are colored green for the β1 integrin and yellow for the inhibitor. The Mg2+ is coordinated by polar groups from β1 and the terminal carboxylate of the inhibitor. The β1 integrin provides 3 well-positioned backbone polar atoms coming from the end of an α helix (Tyr-122) and a tight turn (Asn-215) that help lock the inhibitor into the binding site. The thiazolidine ring binds into the opening of a hydrophobic pocket with good shape complementarity. The hydrophobic character and the restrictive size of this pocket help explain the allowable substitutions on the thiazolidine ring. There was excellent homology between the β1 integrin and the template β3 integrin around this binding site, except for the hydrophobic pocket that does not exist on the β3 surface. The limited homology between the sequences of α2 and αIIb in this binding region precluded a more detailed analysis of the interactions between the inhibitors and the α subunit. For clarity, some of the surfaces near the Mg2+ have been removed.
We demonstrated that Pro-Dap-based inhibitors do not inhibit binding of isolated α2 I-domains (in vitro) to type I collagen, suggesting that they inhibit adhesion indirectly by binding to the β subunit and preventing activation (22). To confirm this hypothesis, we examined the ability of our inhibitors to block cell adhesion to collagen by using mutants of α2 transfected into rat basophilic leukemia (RBL) hematopoietic cells. RBL cells naturally express β1 integrin but not α2. We first examined a constitutively activating mutant E318A (pictured in Fig. 1) in the α2 chain of the full-length integrin (33). This mutant disrupts a salt bridge in the I-domain that is critical to the stability of the closed conformation of the integrin, bypassing the requirement for an interaction between the α2 I-domain and the β1 I-like domain to achieve the activated conformation (15, 34). Thus, we would predict that our compounds would not inhibit the adhesion of cells bearing this mutation to collagen surfaces (Fig. 4). To test this premise, we first measured the ability of compound 15 to inhibit adhesion of RBL cells bearing wild-type integrin α2β1 to collagen. This compound inhibits adhesion with an IC50 ≈5- to 10-fold higher than for static platelet adhesion, probably reflecting differences in integrin density and/or a greater activation state of α2β1 in RBL cells. By contrast, compound 15 has no effect on adhesion of the constitutively active mutant E318A (IC50 >10 μM), confirming that this mutation functions by inducing a constitutively active conformation in the α2 I-domain.
Fig. 4.

Adhesion of α2β1-expressing RBL cells to type I collagen under static conditions. Compound 15 inhibits adhesion of cells expressing wild-type (WT) and demonstrates no effect on RBL cells expressing an activating I-domain mutation (E318A) but inhibits an activating cytoplasmic domain mutant [Del(GFFKR)] with an IC50 ≈4-fold higher than for wild-type integrin α2β1. The IC50 for WT and Del(GFFKR) varied ≈2-fold on different assay days, but the relative potency of compound for the mutants remained constant.
To test further the activation mechanism and to determine whether these compounds would be effective under conditions that mimic strong activation via inside-out signaling pathways, we examined the effect of these antagonists on an activation mutation in the cytoplasmic tail of α2 (12, 13). Deletion of GFFKR is a well-known way to induce integrin activation through the normal platelet activation pathway (Fig. 1) (35, 36). In this case, the extracellular domains of α2β1 remain intact, but the equilibrium is shifted toward the activated state by mutation of the cytoplasmic domain. Compound 15 should still be able to inhibit this mutant by blocking the I-domain-binding site in the β1 I-like domain [although this inhibitor may not be as potent because Del(GFFKR) indirectly shifts the equilibrium to the activated state]. As expected, compound 15 inhibits Del(GFFKR), and ≈4-fold higher concentrations of the small molecule are required to achieve the same degree of inhibition as for the wild-type integrin α2β1 (Fig. 4). Together, these data suggest that 15 inhibits adhesion by interrupting the interaction of the α2 I-domain with the β1 I-like domain; this compound has no effect on adhesion of cells bearing a mutant (E318A) that bypasses the need for α–β interactions to achieve the activated state [whereas 15 is fully active but has decreased potency when measured with cells bearing Del(GFFKR), which enhances the normal activation via the pathway in Fig. 1].
Pro-Dap-Based Compounds Bind Specifically to α2β1.
Platelets express 5 integrins that bind to ligands in the extracellular matrix: αIIbβ3, αvβ3, α2β1, α5β1, and α6β1 (37). We examined the ability of several compounds (15, 16, and 25) to block the binding of human platelets to the ligands of these other integrins, to assess specificity. To examine α5β1-mediated adhesion, we measured the ability of platelets to adhere to fibronectin-coated surfaces in the presence of abciximab (the human–murine monoclonal antibody to β3 integrins) to control for αIIbβ3- and αvβ3-mediated platelet adhesion to fibronectin (37). As expected, we found that our compounds had no effect on binding at concentrations exceeding 1,000 nM. To assess αvβ3-mediated adhesion, we measured the ability of the compounds to inhibit ADP-stimulated platelet adhesion to osteopontin (38). Again, we observed no effect at compound concentrations >1,000 nM. Similarly, the compounds had no effect on ADP-stimulated platelet aggregation, an integrin αIIbβ3-mediated process, at concentrations as great as 20 μM.
Pro-Dap-Based α2β1 Antagonists Inhibit Arterial Thrombosis in Vivo.
Normal blood flow concentrates platelets near the vessel wall where they can interact with the endothelium and subendothelial matrix proteins. In vascular diseases such as atherosclerosis, the blood vessels are narrowed, which increases the shear stress on platelets and promotes high-shear platelet activation. To replicate this pathological process, we examined the ability of 3 representative compounds to block adhesion of human platelets to fibrillar collagen under flow at 1,000 s−1, a condition chosen to mimic platelet function in vivo. The two most potent compounds, 15 and 16 (12 and 6 nM under static conditions, respectively), remained similar under flow (715 and 698 nM). The IC50 for 6 (67 nM under static conditions) increased to 3.6 μM under flow conditions. Although the relative efficacy of the compounds remained approximately the same, the IC50 of each compound increased compared with static adhesion of gel-filtered platelets, an observation that could explain the difference, in in vivo efficacy, between 15 and 6.
To test the potential of these inhibitors as antithrombotic agents in vivo, we examined their ability to inhibit thrombus formation in a murine model of arterial damage. We initially demonstrated the functional equivalence of human and mouse platelets in the static platelet adhesion assay (data not shown). In this model of arterial thrombosis, we measured the total time to occlusion (TTO) after ferric chloride-induced carotid artery injury after administration of compound 6 or 15 in wild-type mice (Fig. 5) (39, 40). We selected the ferric chloride-mediated model of arterial injury because it recapitulates the physiological pathway of thrombosis in atherosclerotic disease: ferric chloride has been shown to migrate through the endothelium by endocytic–exocytic pathways and cause endothelial denudation, therefore exposing collagen in the subendothelial matrix (41). Mice were given, i.v., either aspirin [10 mg/kg, a dose shown to abolish thromboxane A2 generation (42)] or normal saline, as positive and negative controls. Compound 15 delayed clot formation by 3-fold (TTO = 18.7 ± 12.2 min, P = 0.04 compared with 6.0 ± 1.8 min sham-treated mice), similar to the efficacy of aspirin (TTO = 15.8 ± 11.3 min). By contrast, the less-potent compound 6 had no effect on TTO (6.8 ± 3.0 min, P = 0.62).
Fig. 5.

Wild-type C57BL/6J mice were given i.v. normal saline (NS, 200 μL) (n = 5), aspirin (10 mg/kg) (n = 6), or the integrin α2β1 inhibitor (compound 6 or 15, 60 mg/kg, n = 9 for 15 and n = 6 for 6) 15 min before the assay. The carotid artery was surgically exposed, injured with 10% ferric chloride solution for 2.5 min, and then washed with PBS. Blood flow through the carotid artery was measured for 30 min by a Doppler ultrasound probe. Statistical analysis was performed by using one-way ANOVA analysis relative to sham-treated controls (P = 0.09 for aspirin, P = 0.04 for compound 15, P = 0.62 for compound 6).
Discussion
Interaction between circulating platelets and collagen at sites of vascular injury plays a critical role in pathological thrombus formation. We have used a modified Pro-Dap scaffold to synthesize a series of small-molecule inhibitors that potently and selectively block integrin α2β1 binding to type I collagen. These small-molecule antagonists represent compounds targeting α2β1 that demonstrate in vivo efficacy in a model of pathological thrombosis. The efficacy of our agents corroborates observations of α2-null mice that suggest that integrin α2β1 plays a role in thrombus growth and stabilization (42). The efficacy of the compounds also confirms the link observed in patient populations that first, integrin α2β1 overexpression increases the risk of myocardial infarction and ischemic stroke, and second, that the absence of the integrin still allows fairly normal hemostasis.
These compounds are not only potentially useful as antithrombotic agents but also have helped elucidate the activation pathways of I-domain-containing integrins. The responses of RBL cells expressing different integrin α2β1 mutants to the various compounds shed light on the physiological pathway of integrin activation and subsequent platelet adhesion to collagen. Additionally, our computational model for integrin α2β1 is consistent with binding to the I-like domain and demonstrates the mechanism for selectivity among integrins with related α and β partners. These observations establish the α–β interface as an excellent target for obtaining highly selective inhibitors of the β1 family. Finally, these studies extend and confirm previous suggestions of the mechanism of integrin activation and the mode of binding of this class of inhibitors to the I-domain-containing class of integrins.
Methods
Additional procedures can be found in supporting information (SI) Text.
Blood Collection and Preparation of Gel-Filtered Platelets.
All studies were conducted in accordance with Institutional Review Board-approved protocols at the University of Pennsylvania. Human blood was collected by venipuncture from healthy volunteers using sodium citrate as an anticoagulant. Platelet-rich plasma (PRP) was prepared by centrifuging whole blood (200 × g, 20 min). The PRP was carefully removed and applied to a Sepharose CL-2B column (bed volume, 60 mL; 11 × 3 cm) in GFP buffer [4 mM Hepes (pH 7.4), 135 mM NaCl, 2.7 mM KCl, 3.3 mM PO4, 0.35% BSA, 0.1% glucose, and 2 mM MgCl2] (43). After gel purification, the platelets were counted and diluted to the appropriate final concentration (2–3 × 108 platelets per mL).
Adhesion Assays and Platelet Aggregation.
Immulon 2 flat-bottom 96-well plates (Dynatech Laboratories) were coated with soluble type I collagen (Col1), fibronectin (FN), or osteopontin (OPN) (5 μg/mL) for 48 h at 4 °C. The proteins were dissolved in either 5% aqueous acetic acid or 50 mM NaHCO3 containing 150 mM NaCl (pH 8). The plates were washed and blocked with BSA (5 mg/mL in PBS) for at least 24 h. Adhesion assays using Col1 and FN contained test compound and 1.6 × 107 platelets in GFP buffer in a final volume of 100 μL. OPN binding was assessed in a similar manner, differing in that the platelets were incubated with 10 μM ADP (10 min, 20 °C) before ligand exposure, and assays contained 2.4 × 107 platelets per well. The plates were incubated (37 °C, 30 min) and washed with TBS [10 mM Tris (pH 7.4), 150 mM NaCl] (11, 21). Adherent platelets were determined by staining for acid phosphatase by addition of 5 mM p-nitrophenyl phosphate in 0.1 M sodium citrate (pH 5.4), 0.1% Triton X-100 (100 μL per well) and incubating for 30 min at 37 °C (44). Plates were developed by the addition of 50 μL of 2 N NaOH and read at 405 nm in a microplate reader.
To assess platelet aggregation, purified platelets were incubated with Ca2+ and FN in the presence or absence of antagonist. Next, ADP was added, and aggregation was determined in a Chronolog aggregometer (43, 45).
RBL cells expressing wild-type integrin α2β1 or the constitutively activating mutations E318A or Del(GFFKR) were obtained as a generous gift from Mark L. Kahn (University of Pennsylvania). Collagen adhesion assays were performed as described above for platelet adhesion. EDTA was used as a negative control.
Flow Assays (20).
Glass coverslips were incubated with a suspension of fibrillar collagen (100 μg/mL) overnight at 4 °C. Surfaces were then blocked with denatured BSA (5 mg/mL) for 1 h at room temperature. This was followed by washing with PBS before use in spreading assays. Coverslips were assembled onto a flow chamber (Glyotech) and mounted on the stage of an inverted microscope (Zeiss Axiovert 200M). PPACK (40 μM) anticoagulated whole blood was perfused through the chamber for 3 min at a wall shear rate of 1,000 s−1, and this was followed by washing for 4 min at the same shear rate with modified Tyrode's buffer [imaged by using differential interference contrast microscopy (DIC) microscopy].
Computational Studies.
The molecules 15_frag, 16_frag, and 22_frag were subjected to the conformer distribution routine by using the MMFFaq forcefield, as implemented in PC Spartan 06 V101 (Wavefunction). Geometry optimizations were performed with restricted Hartree Fock SCF (self-consistent field) calculations using the 631G* basis set for the lowest-energy conformers obtained from the conformer distribution routine. For the SCF model, the restricted Hartree Fock calculations were performed by using Pulay DIIS and geometric direct minimization. Aqueous solvation energies were calculated from SM5 models. All 3 molecules studied (15_frag, 16_frag, and 22_frag) were fit to the experimentally derived single-crystal structure for 16_xray by using the Fit Atom routine implemented in Sybyl (Tripos). The atoms from the thiazolidine ring chosen for fitting in the Fit Atom routine were: S-1, C-2, N-3, and C-5).
Structures of the integrin α2β1 were generated through homology modeling from structures of integrin αIIbβ3, including 1AOX (non-collagen-bound) and 1DZI (collagen-bound) (10, 11). Rosetta Dock was used to dock the generated α2 structure onto the β1 homology model. Missing residues 316–338 of helix α2 were completed in the model by using other high-resolution protein structures in the Protein Data Bank.
Thrombosis Assay (39).
All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Wild-type C57BL/6J mice (Jackson Laboratories) were anesthetized with pentobarbital (90 mg/kg) by i.p. injection. The mice were administered vehicle (200 μL of PBS), aspirin (10 mg/kg dissolved in 200 μL of PBS), or the integrin α2β1 inhibitor (60 mg/kg dissolved in 200 μL of PBS) i.v. 15 min before the assay. The anesthetized animals were placed on a 37 °C warming pad throughout the experiment. The right carotid artery was exposed by blunt dissection with minimal blood loss, and a miniature Doppler flow probe was attached to the artery to monitor blood flow continuously. Thrombus formation was subsequently induced by application of filter paper (1 × 1 mm) saturated with 10% FeCl3 solution to the exposed artery in contact with the adventitial surface. After 2.5 min, the filter paper was removed, and the vessel was washed with PBS. The blood flow was monitored continuously for 30 min after injury. Immediately after the experiment, the mice were killed by cervical dislocation while still under deep anesthesia.
Statistics and Data Analysis.
The results obtained from adhesion assays are fitted to: m1+(m2−m1)/1+(M0/m3)⋀m4, where m1 = background binding, m2 = maximal binding, m3 = IC50 (nM), and m4 = cooperativity, using KaleidaGraph (Synergy Software). The results presented are those obtained from a minimum of two independent experiments. This assay gave reproducible results for adhesion. Although we observed differences in the IC50 values from the various donors, the relative potency of the compounds remained the same. For compound 15, which was included as positive control reference in all assays, the calculated IC50 value was 12 nM ± 8 (n = 18).
Supplementary Material
Acknowledgments.
We thank L. Brass, M. Poncz, J. Gewirtz, and T. J. Stalker for technical assistance with the thrombosis model; and David K. Robinson for technical assistance with the platelet adhesion under flow assay. This work was supported by National Center for Research Resources Grant HL62250.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0811622106/DCSupplemental.
References
- 1.Rehkter MD. Collagen synthesis in atherosclerosis: Too much and not enough. Cardiovasc Res. 1999;41:376–384. doi: 10.1016/s0008-6363(98)00321-6. [DOI] [PubMed] [Google Scholar]
- 2.Ruggeri ZM. Platelets in atherothrombosis. Nat Med. 2002;8:1227–1234. doi: 10.1038/nm1102-1227. [DOI] [PubMed] [Google Scholar]
- 3.Kritzik M, et al. Nucleotide polymorphisms in the α2 gene define multiple alleles that are associated with differences in platelet α2β1 density. Blood. 1998;92:2382–2388. [PubMed] [Google Scholar]
- 4.Nieuwenhuis HK, Akkerman JWN, Houdijk WPM, Sixma JJ. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature. 1985;318:470–472. doi: 10.1038/318470a0. [DOI] [PubMed] [Google Scholar]
- 5.Nieuwenhuis HK, Sakariassen KS, Houdijk WP, Nievelstein PF, Sixma JJ. Deficiency of platelet membrane glycoprotein Ia associated with a decreased platelet adhesion to subendothelium: A defect in platelet spreading. Blood. 1986;68:692–695. [PubMed] [Google Scholar]
- 6.Kehrel B, et al. Deficiency of intact thrombospondin and membrane glycoprotein Ia in platelets with defective collagen-induced aggregation and spontaneous loss of disorder. Blood. 1988;71:1074–1078. [PubMed] [Google Scholar]
- 7.Handa M, et al. Platelet unresponsiveness to collagen: Involvement of glycoprotein Ia-IIa (α2β1 integrin) deficiency associated with a myeloproliferative disorder. Thromb Haemostasis. 1995;73:521–528. [PubMed] [Google Scholar]
- 8.Nieswandt B, Watson SP. Platelet–collagen interaction: Is GPVI the central receptor? Blood. 2003;102:449–461. doi: 10.1182/blood-2002-12-3882. [DOI] [PubMed] [Google Scholar]
- 9.Shimaoka M, Springer TA. Therapeutic antagonists and conformational regulation of integrin function. Nat Rev Drug Discov. 2003;2:703–716. doi: 10.1038/nrd1174. [DOI] [PubMed] [Google Scholar]
- 10.Emsley J, Knight CG, Farndale RW, Barnes MJ, Liddington RC. Structural basis of collagen recognition by integrin α2β1. Cell. 2000;101:47–56. doi: 10.1016/S0092-8674(00)80622-4. [DOI] [PubMed] [Google Scholar]
- 11.Emsley J, King SL, Bergelson JM, Liddington RC. Crystal structure of the I-domain from integrin α2β1. J Biol Chem. 1997;272:28512–28517. doi: 10.1074/jbc.272.45.28512. [DOI] [PubMed] [Google Scholar]
- 12.Jung SM, Moroi M. Signal-transducing mechanisms involved in activation of the platelet collagen receptor integrin α2β1. J Biol Chem. 2000;275:8016–8026. doi: 10.1074/jbc.275.11.8016. [DOI] [PubMed] [Google Scholar]
- 13.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 14.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in alternate conformations of the integrin αLβ2 I-domain with disulfide bonds reveals functional relationships among integrin domains. Proc Natl Acad Sci USA. 2001;98:2393–2398. doi: 10.1073/pnas.041618598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connors WL, et al. Two synergistic activation mechanisms of integrin α2β1 integrin-mediated collagen binding. J Biol Chem. 2007;282:14675–14683. doi: 10.1074/jbc.M700759200. [DOI] [PubMed] [Google Scholar]
- 16.Shimaoka M, et al. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimaoka M, Salas A, Yang W, Weitz-Schmidt G, Springer TA. Small-molecule integrin antagonists that bind to the β2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity. 2003;19:391–402. doi: 10.1016/s1074-7613(03)00238-3. [DOI] [PubMed] [Google Scholar]
- 18.Marcinkiewicz C, et al. Isolation and characterization of EMS16, a C-lectin type protein from Echis multisquamatus venom, a potent and selective inhibitor of the α2β1 integrin. Biochemistry. 2000;39:9859–9867. doi: 10.1021/bi000428a. [DOI] [PubMed] [Google Scholar]
- 19.Eble JA, Tuckwell DS. The α2β1 integrin inhibitor rhodocetin binds to the A-domain of the integrin α2 subunit proximal to the collagen-binding site. Biochem J. 2003;376:77–85. doi: 10.1042/BJ20030373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White TC, et al. The leech product saratin is a potent inhibitor of platelet integrin α2β1 and von Willebrand factor binding to collagen. FEBS J. 2007;274:1481–1491. doi: 10.1111/j.1742-4658.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 21.Yin H, et al. Arylamide derivatives as allosteric inhibitors of the integrin α2β1/type I collagen interaction. Bioorg Med Chem Lett. 2006;16:3380–3382. doi: 10.1016/j.bmcl.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 22.Choi S, et al. Small-molecule inhibitors of integrin α2β1. J Med Chem. 2007;50:5457–5462. doi: 10.1021/jm070252b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagmann WK, et al. The discovery of sulfonylated dipeptides as potent VLA-4 antagonists. Bioorg Med Chem Lett. 2001;11:2709–2713. doi: 10.1016/s0960-894x(01)00544-3. [DOI] [PubMed] [Google Scholar]
- 24.Chang LL, et al. The discovery of small-molecule carbamates as potent dual α4β1/α4β7 integrin antagonists. Bioorg Med Chem Lett. 2002;12:159–163. doi: 10.1016/s0960-894x(01)00710-7. [DOI] [PubMed] [Google Scholar]
- 25.Huryn DM, et al. The identification and optimization of orally efficacious, small-molecule VLA-4 antagonists. Curr Top Med Chem. 2004;4:1473–1484. doi: 10.2174/1568026043387467. [DOI] [PubMed] [Google Scholar]
- 26.Xue CB, et al. Discovery of an orally active series of isoxazoline glycoprotein IIb/IIIa antagonists. J Med Chem. 1997;40:2064–2084. doi: 10.1021/jm960799i. [DOI] [PubMed] [Google Scholar]
- 27.Xue C-B, et al. Design, synthesis, and in vitro activities of benzamide-core glycoprotein IIb/IIIa antagonists: 2,3-diaminopropionic acid derivatives as surrogates of aspartic acid. Bioorg Med Chem. 1997;5:693–705. doi: 10.1016/s0968-0896(97)00013-8. [DOI] [PubMed] [Google Scholar]
- 28.Raines RT. 2005 Emil Thomas Kaiser award. Protein Sci. 2006;15:1219–1225. doi: 10.1110/ps.062139406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas KM, Naduthambi D, Zondlo NJ. Electronic control of amide cis-trans isomerism via the aromatic–prolyl interaction. J Am Chem Soc. 2006;128:2216–2217. doi: 10.1021/ja057901y. [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarti P, Chakrabarti S. C–H…O hydrogen bond involving proline residues in α-helices. J Mol Biol. 1998;284:867–873. doi: 10.1006/jmbi.1998.2199. [DOI] [PubMed] [Google Scholar]
- 31.Xiao T, Takagi J, Coller BS, Wang J-H, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo B-H, Carman CV, Springer TA. Structural basis of integrin signaling and regulation. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edelson BT, et al. Novel collectin/C1q receptor mediates mast cell activation and innate immunity. Blood. 2006;107:143–150. doi: 10.1182/blood-2005-06-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang W, Shimaoka M, Salas A, Takagi J, Springer TA. Intersubunit signal transmission in integrins by a receptor-like interaction with a pull spring. Proc Natl Acad Sci. 2004;101:2906–2911. doi: 10.1073/pnas.0307340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Toole TE, Katagiri Y, Faull RJ. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, Leisner TM, Parise LV. Platelet α2β1 integrin activation: Contribution of ligand internalization and the α2 cytoplasmic domain. Blood. 2003;102:1307–1315. doi: 10.1182/blood-2002-09-2753. [DOI] [PubMed] [Google Scholar]
- 37.Shattil SJ, Newman PJ. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 38.Tam SH, Sassoli PM, Jordan RE, Nakada MT. Abciximab (ReoPro, chimeric 7E3 Fab) demonstrates equivalent affinity and functional blockade of glycoprotein IIb/IIIa and αvβ3 integrins. Circulation. 1998;98:1085–1091. doi: 10.1161/01.cir.98.11.1085. [DOI] [PubMed] [Google Scholar]
- 39.Kurz KD, Main BW, Sandusky GE. Rat model of arterial thrombosis induced by ferric chloride. Thromb Res. 1990;60:269–280. doi: 10.1016/0049-3848(90)90106-m. [DOI] [PubMed] [Google Scholar]
- 40.Farrehi PM, Ozaki CK, Carmeliet P, Fay WP. Regulation of arterial thrombosis by plasminogen activator-1 in mice. Circulation. 1998;97:1002–1008. doi: 10.1161/01.cir.97.10.1002. [DOI] [PubMed] [Google Scholar]
- 41.Tseng MT, Dozier A, Haribabu B, Graham UM. Transendothelial migration of ferric chloride in FeCl3-injured murine common carotid artery. Thromb Res. 2006;118:275–280. doi: 10.1016/j.thromres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 42.Kuijpers MJE, et al. Role of murine integrin α2β1 in thrombus stabilization and embolization: Contribution of thromboxane A2. Thromb Haemostasis. 2007;98:1072–1080. [PubMed] [Google Scholar]
- 43.Basani RB, et al. RGD-containing peptides inhibit fibrinogen binding to platelet αIIbβ3 by inducing an allosteric change in the amino-terminal portion of αIIb. J Biol Chem. 2001;276:13975–13981. doi: 10.1074/jbc.M011511200. [DOI] [PubMed] [Google Scholar]
- 44.Bellavite P, et al. A colorimetric method for the measurement of platelet adhesion in microtiter plates. Anal Biochem. 1994;216:444–450. doi: 10.1006/abio.1994.1066. [DOI] [PubMed] [Google Scholar]
- 45.Yin H, et al. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.