

Abstract
Heterobimetallic CuPd and CuPt bis(μ-oxo) complexes have been prepared by the reaction of (PPh3)2MO2 (M = Pd, Pt) with LCu(I) precursors (L = β-diketiminate and di- and triamine ligands) and characterized by low-temperature UV-vis, resonance Raman, and 1H and 31P{1H} NMR spectroscopy in conjunction with DFT calculations. The complexes decompose upon warming to yield OPPh3, and in one case this was shown to occur by an intramolecular process through crossover experiments using double-labeling (oxo and phosphine). The reactivity of one of the complexes, LMe2Cu(μ-O)2Pt(PPh3)2 (LMe2 = β-diketiminate), with a variety of reagents including CO2, 2,4-di-tert-butylphenol, 2,4-di-tert-butylphenolate, [NH4][PF6], and dihydroanthracene, was compared to that of homometallic Pt2 and Cu2 counterparts. Unlike typical [Cu2(μ-O)2]2+ cores which have electrophilic oxo groups, the oxo groups in the [Cu(μ-O)2Pt]+ core behave as bases and nucleophiles, similar to previously described Pt2 compounds. In addition, however, the [Cu(μ-O)2Pt]+ core is capable of oxidatively coupling 2,4-di-tert-butylphenol and 2,4-di-tert-butylphenolate. Theoretical evaluation of the electron affinities, basicities, and H-atom transfer kinetics and thermodynamics of the Cu2 and CuM (M = Pd, Pt) cores showed that the latter are more basic and form stronger O-H bonds.
Introduction
In efforts to gain mechanistic understanding of oxidation catalysis, an important objective is to characterize metal-oxygen complexes relevant to postulated reaction intermediates.1 Homometallic bis(μ-oxo) complexes with [M2(μ-O)2]n+ cores are important examples, having been studied extensively for M = Cu,2 Mn,3 Ni,4 Co,4a,b Fe,2a,5 and Pt,6 in large part because of their possible involvement in biological and/or biomimetic oxidations. For example, the [Cu2(μ-O)2]2+ core has been considered as the active oxidant in phenol hydroxylation by tyrosinase,7 as well as in methane hydroxylations by particulate methane monooxygenase8,9 and a heterogeneous zeolite-based system.10 Typically prepared via the reaction of O2 with mononuclear Cu(I) complexes supported by N-donor ligands, [Cu2(μ-O)2]2+ complexes have been shown to contain Cu(III) ions11 bound to oxo groups that exhibit electrophilic character.2c While the reactivity of these complexes depends in often subtle ways on the nature of their supporting N-donor ligands, counterions, and/or reaction conditions, they generally share a propensity to abstract hydrogen atoms from ligand substituents12 or exogeneous reagents such as phenols.13,14 Oxo transfers to thioethers15 and phosphines16 also have been observed. The oxo groups in [M2(μ-O)2] cores with M = Ni, Co, and Fe also exhibit electrophilic character,2a,4,5 but profoundly different reactivity has been found for M = Pt.6 Countercations (e.g. Li+) associate with oxo groups bridging Pt(II) in the solid state, and these oxo groups act as nucleophiles and Brønsted bases.17
Mixed metal or heterobimetallic oxygen intermediates are less common than homometallic species, yet they are of great interest due to the possible ‘synergistic’ effect of two different metal ions acting together. An illustrative example is the activation of dioxygen by the cytochrome c oxidase Cu-Fe(heme) site and relevant synthetic models.18 Heterometallic species are prevalent in heterogeneous catalysis,19 and have been shown to be useful for homogeneous catalytic oxidations.20 Of particular relevance in the current context are Cu-Pd intermediates postulated in Wacker-type oxidations.21 Examples such as these support the notion that two metals might work together to activate O2 in such a way that the resulting oxo moiety exhibits reactivity different from that observed using either of the metals individually.
Because of the possibility of obtaining intractable reaction mixtures, the procedure typical for preparing homobimetallic metal-oxygen intermediates (e.g. [M2(μ-O)2]n+ species) whereby a monomeric precursor is treated with O2 generally cannot be used to cleanly isolate heterobimetallic products (for exceptions, see ref. 18a). An alternate route involves the reaction of a pre-formed mononuclear metal-dioxygen complex with a second reduced metal compound, in the absence of free O2. This route has been successfully used to prepare dimetal complexes with different supporting ligands on each metal (e.g. with Cu22 and Ni23), as well as heterobimetallic oxygen-containing species with CuNi,24 CuGe,25 PtMo,26 and PtGe27 combinations. Herein we report extension of this strategy for the synthesis of novel heterobimetallic CuPd and CuPt complexes from the reaction of copper(I) complexes supported by N-donor ligands of variable types with the metal peroxo species (PPh3)2MO2 (M = Pd, Pt). The thermally unstable products were shown to contain [M(μ-O)2Cu]1+ cores on the basis of UV-vis and resonance Raman spectroscopic data, with further structural insights provided by NMR spectroscopy and DFT calculations. Comparison of the reactivity of a member of the class, LCu(μ-O)2Pt(PPh3)2 (L = β-diketiminate), with that of a homometallic dicopper counterpart revealed significant differences, particularly with respect to the course of their reactions with phenols, phenolates, and weak acids. These differences have been examined via theoretical evaluation of the thermodynamic and kinetic parameters associated with electron, proton, and/or H-atom transfer reactions of the Cu2 and CuPt cores supported by a range of ligand sets. Preliminary aspects of this work have been communicated.24
Experimental Section
General Considerations
All solvents and reagents were obtained from commercial sources and used as received unless noted otherwise. The solvents tetrahydrofuran (THF), toluene, diethyl ether (Et2O), pentane, and dichloromethane (CH2Cl2) were degassed and passed through a solvent purification system (Glass Contour, Laguna CA) before use. All metal complexes were prepared and stored in a Vacuum Atmospheres inert atmosphere glove box under a dry nitrogen atmosphere or were manipulated using standard inert atmosphere vacuum and Schlenk techniques. Labeled dioxygen (18O2, 99%) was obtained from Cambridge Isotopes, Inc. The complexes Pt(PPh3)4 28, Pd(P(p-tolyl)3)3,29 (PPh3)2PdO2, 24 (PPh3)2PtO2, 30 [(Me4chd)Cu(MeCN)]PF6,31 [(Me4pda)Cu(MeCN)]OTf,32 [LMe2Cu]2,33 and [(iPr3tacn)Cu(MeCN)]SbF634 were synthesized according to published procedures. The NMR solvents CDCl3 and C6D6 were stirred over CaH2 and vacuum distilled prior to use.
Physical Methods
NMR spectra were recorded on a VXR-300 or VI-300 spectrometer. Chemical shifts (δ) for 1H and 13C NMR spectra are reported versus tetramethylsilane and were referenced to residual protium in the deuterated solvent. 31P{1H} NMR spectra are referenced to an external H3PO4 standard (85%). Mass spectra were obtained on a Bruker Biotof II instrument. UV-vis spectra were recorded on an HP8453 (190-1100 nm) diode array spectrophotometer equipped with a Unisoku low-temperature device. X-band EPR spectra were recorded on a Bruker E-500 spectrometer with an Oxford Instruments EPR-10 liquid helium cryostat (4-20K, 9.61 GHz). Quantization of EPR signal intensity was accomplished by comparing the integration with that of H(Me2LiPr2)CuCl.35 Resonance Raman spectra were collected on an Acton AM-506 spectrometer using a Princeton Instruments LN/CCD-11100-PB/UVAR detector and ST-1385 controller interfaced with Winspec software. A Spectra-Physics BeamLok 2065-7S Ar laser provided excitation at 457.9 nm. The spectra were obtained at -196 °C using a backscattering geometry. Samples were frozen in an NMR tube submerged in liquid N2, or frozen in a copper cup attached to a coldfinger Dewar filled with liquid nitrogen. Raman shifts were externally referenced to liquid indene. IR spectra were obtained using a ThermoNicolet Avatar 370 FT-IR equipped with an ATR attachment.
(PPh3)2Pt18O2
A method adapted from the reported procedure for (PPh3)2Pt16O2 was used.30 A 25 mL Schlenk flask was charged with a suspension of Pt(PPh3)4 (200 mg, 0.16 mmol) in 10 mL of Et2O. The suspension was frozen at -196 °C, the headspace evacuated, and 18O2 was transferred from a glass bulb into the flask. Warming to ambient temperature and stirring for 2 h resulted in the deposition of a tan solid, which was collected, washed with Et2O (3 × 5 mL) and dried under reduced pressure (92 mg, 77%). FT-IR (νO-O): 776 cm-1 (Δ18O2 = 48 cm-1).
(P(p-tolyl)3)2Pd16O2
A 50 mL Schlenk flask was charged with a suspension of Pd(P(p-tolyl)3)3 (200 mg, 0.2 mmol) in 20 ml Et2O. Bubbling of dry O2 into the solution for 10 min resulted in the precipitation of the product as a pale blue solid, which was collected, washed with Et2O (3 × 5 mL) and dried under reduced pressure (91 mg, 61%). FT-IR (νO-O): 882 cm-1. 1H NMR (CDCl3): δ 7.22 (m, 12H), 6.97 (d, 12H), 2.31 (s, 18H) ppm. 13C{1H} NMR (CDCl3): δ 140.2, 133.9 (t), 129.0 (t), 21.5 ppm. 31P{1H} NMR (CDCl3): δ 31.7 ppm. Anal. Calcd for C42H42O2P2Pd: C, 67.52; H, 5.67. Found: C, 67.22; H, 5.97.
(P(p-tolyl)3)2Pd18O2
A 25 mL Schlenk flask was charged with a suspension of Pd(P(p-tolyl)3)3 (200 mg, 0.2 mmol) in 10 mL of Et2O. The suspension was frozen at -196 °C, the headspace evacuated, and 18O2 was transferred from a glass bulb into the flask. Warming to ambient temperature and stirring for 20 min resulted in the deposition of a light blue solid, which was collected, washed with Et2O (3 × 5 mL) and dried under reduced pressure (84 mg, 56%). FT-IR (νO-O): 835 cm-1 (Δ18O2 = 47 cm-1).
Characterization of [Cu(μ-O)2M]+ Complexes
(a) UV-vis Spectroscopy
Anaerobic solutions of (PPh3)2MO2 (M = Pd, Pt) in CH2Cl2, toluene, or THF (0.2 mM, 2 mL) in a UV-vis cuvette were cooled to -80 °C in the UV-vis cryostat. One equivalent of the copper(I) reagent in 0.2 mL of solvent was injected by syringe into the stirred cold metal peroxo solution. The progress of intermediate formation and decay was followed by monitoring the main absorption feature at ∼450 nm associated with the [Cu(μ-O)2M]+ cores.
(b) EPR Spectroscopy
Anaerobic solutions of (PPh3)2MO2 (M = Pd, Pt) in CH2Cl2, toluene, or THF (2 mL, 1.0 mM) in a 10 mL Schlenk flask were cooled to -80 °C in a MeOH/liquid N2 bath. One equivalent of the desired copper(I) reagent in 0.2 mL of solvent was then added by syringe, and the resulting solution was transferred via a pre-cooled cannula to a pre-cooled EPR tube, which was immediately frozen at -196 °C.
(c) NMR and resonance Raman Spectroscopy
An NMR tube was purged with argon gas through an 8” needle for approximately 15 min. The tube was charged with an anaerobic solution of (PPh3)2MO2 (M = Pd, Pt) in CH2Cl2, toluene, or THF (0.5 mL, 10 mM) by syringe and cooled to -80 °C by submersion in a MeOH/liquid N2 bath while maintaining the argon purge. One equivalent of the desired copper(I) reagent in 0.1 mL solvent was then added rapidly by syringe. The NMR tubes were maintained at -80 °C for the collection of NMR spectra, and were frozen at -196 °C for the collection of resonance Raman data. For L = iPr3tacn, one equivalent of the copper(I) complex in 0.2 mL CH2Cl2 was added by syringe to anaerobic solutions of (PPh3)2MO2 (M = Pd, Pt) in CH2Cl2 (2 mL, 10 mM) at -80 °C in a 10 mL Schlenk flask. The resulting solution was stirred for 30 min and transferred by micropipet to a copper cup attached to a liquid N2 coldfinger Dewar for collection of the resonance Raman spectra.
Reactivity of (LMe2Cu)2(μ-O)2 with Exogenous Substrates
Anaerobic solutions of [LMe2Cu]2 in THF or toluene (2 mL, 0.1 mM for UV-vis; 3 mL, 5 mM for preparatory scale) were cooled to -80 °C and dry O2 was bubbled for 5 min. The solutions were then stirred at -80 °C for 30 min to ensure complete formation of (LMe2Cu)2(μ-O)2. Excess O2 was removed by purging with argon in the UV-vis experiments and by 6-10 vacuum/purge cycles with argon in the preparatory scale experiments. Reactivity in the UV-vis experiments was followed by monitoring the decay of the absorption feature at 422 nm associated with the [Cu2(μ-O)2]2+ core. In the following, the number of equivalents of substrate are relative to the amount of the [Cu2(μ-O)2]2+ core.
(a) [NH4][PF6], PPh3, Thioanisol, 1-Decene, 2,4-Di-tert-Butylphenol, Sodium 2,4-Di-tert-Butylphenolate, 9,10-Dihydroanthracene, and CO2 at -80 °C by UV-vis
Ten equivalents of the substrates were added in 0.2 mL solvent by syringe (for CO2, the dry gas was bubbled for 10 s). No change in the absorption feature at 422 nm was observed after several h for any of the substrates.
(b) 2,4-Di-tert-Butylphenol at -40 °C. UV-vis
Ten equivalents of 2,4-di-tert-butylphenol were added by syringe in 0.2 mL solvent to a solution of (LMe2Cu)2(μ-O)2 in THF or toluene at -40 °C. The absorption feature at 422 nm decayed gradually over the course of several h (t1/2 ∼ 4 hrs).
Preparatory Scale
Two equivalents of the substrate were added by syringe in 0.2 mL solvent to a solution of (LMe2Cu)2(μ-O)2 in THF or toluene at -80 °C. The solution was then warmed to -40 °C, where it was stirred for 12 h. The solution was warmed to room temperature and the solvent was removed under reduced pressure. The resulting brown residue was dissolved in 5 ml CH2Cl2 and stirred with 5 mL aqueous ammonium hydroxide solution. The organic fraction was collected, and the aqueous solution was extracted with a further 2 × 5 ml CH2Cl2. The organic fractions were combined, dried over Na2SO4, and filtered through a plug of neutral Alumina, which was washed with a further 5 ml of CH2Cl2. The solvent was removed under reduced pressure, and two equivalents of 1,3,5-trimethoxybenzene were added as an internal standard. The solution was dissolved in CDCl3 and characterized by GC/MS and 1H NMR. The yield of the coupled phenolic product was 60% (1.2 equiv phenol coupled, 0.8 equiv recovered unreacted).
Decomposition Reactions of [M(μ-O)2Cu]+ Complexes
Anaerobic solutions of (PPh3)2MO2 (M = Pd, Pt) in CH2Cl2, toluene, or THF (3 mL, 10 mM) were cooled to -80 °C, and one equivalent of the copper(I) complex in 0.2 mL solvent was added by syringe to form the [M(μ-O)2Cu]+ complex. The solutions were allowed to decompose and/or were warmed to room temperature, and the solvent was removed under reduced pressure. The resulting brown residue was dissolved in 5 ml CH2Cl2 and stirred with 5 mL aqueous ammonium hydroxide solution. The organic fraction was collected, and the aqueous solution was extracted with a further 2 × 5 ml CH2Cl2. The organic fractions were combined, dried over Na2SO4 and filtered through a plug of neutral Alumina, which was washed with a further 5 ml of CH2Cl2. The solvent was removed under reduced pressure, and two equivalents of triphenylphosphate per [M(μ-O)2Cu]+ core (31P{1H}, δ = -18 ppm) were added as an internal standard for quantification of the phosphorus containing products. The resulting mixture was dissolved in C6D6 or CDCl3 for characterization by 1H and 31P{1H} NMR spectroscopy.
Crossover Experiments
Anaerobic solutions of (PPh3)2Pd18O2 (3.0 mg, 0.0045 mmol) and (P(p-tolyl)3)2Pd16O2 (3.4 mg, 0.0045 mmol) were added sequentially via syringe to a -80 °C solution of [LMe2Cu]2 (3.4 mg, 0.0045 mmol) in 3 mL toluene. The solution immediately became deep yellow-brown, indicating formation of the [Cu(μ-O)2Pd]+ core.36 The solution was stirred at low temperature for 1 h and then allowed to decompose by warming to room temperature. Analysis of the product mixture by ESI-MS revealed the phosphine oxidation products in the ratios 18OPPh3(∼80%):16OPPh3(∼20%) and 16OP(p-tolyl)3(100%):18OP(p-tolyl)3(0%). An analogous experiment using (PPh3)2Pd16O2 and (P(p-tolyl)3)2Pd18O2 revealed the formation of products in the ratios 18OPPh3(0%):16OPPh3(100%) and 16OP(p-tolyl)3(32%):18OP(p-tolyl)3(68%). Control experiments involving characterization of the decomposition products of LMe2Cu(μ-18O)2Pd(PPh3)2 and LMe2Cu(μ-18O)2Pd(P(p-tolyl3)2 revealed the formation of ∼80% 18OPPh3 and ∼68% 18OP(p-tolyl)3, respectively, with adventitious 16O2 or water possibly resulting in the 16O incorporation in the phosphine oxide products.
Reactivity of LMe2Cu(μ-O)2Pt(PPh3)2 with Exogenous Substrates
Anaerobic solutions of (PPh3)2PtO2 in THF (2 mL, 0.2 mM for UV-vis; 3 mL, 5 mM for preparatory scale) were cooled to -80 °C and 0.5 equiv of [LMe2Cu]2 in 0.2 mL THF was injected by syringe into the cold platinum peroxo solution. The solutions were maintained at -80 °C for 30 min to ensure complete formation of the intermediate before substrate addition. In the following, the number of equivalents of substrate are relative to the amount of the [Cu(μ-O)2Pt]1+ core.
(a) [NH4][PF6], UV-vis Scale
Two equivalents of [NH4][PF6] were added in 0.1 mL THF by syringe to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2, at which time immediate bleaching of the absorption feature at 455 nm in the UV-vis spectrum was observed.
Preparatory Scale
Two equivalents of [NH4][PF6] were added in 0.5 mL THF by syringe to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2, at which time the solution immediately lightened from deep orange-brown to pale green-brown. The solution was stirred for an additional 30 min, warmed to room temperature, and the solvent was removed under reduced pressure to give a brown residue. ESI-MS (m/z): Calculated for [PtCuP2N2C57H57O2]+, 1121.2840; Found, 1121.2883.
(b) 2,4-Di-tert-butylphenol, UV-vis Scale
Two equivalents of 2,4-di-tert-butylphenol were added in 0.1 mL THF by syringe to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2, at which time the immediate bleaching of the absorption feature at 455 nm in the UV-vis spectrum was observed.
Preparatory Scale
One or two equiv. of 2,4-di-tert-butylphenol were added in 0.5 mL THF by syringe to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2, at which time the solution immediately lightened from deep orange-brown to pale yellow-brown. The solution was stirred for an additional 30 min, warmed to room temperature, and the solvent was removed under reduced pressure to give a brown residue. The resulting brown residue was dissolved in 5 ml CH2Cl2 and stirred with 5 mL aqueous ammonium hydroxide solution. The organic fraction was collected, and the aqueous solution was extracted with a further 2 × 5 ml CH2Cl2. The organic fractions were combined, dried over Na2SO4 and filtered through a plug of neutral Alumina, which was washed with a further 5 ml of CH2Cl2. The solvent was removed under reduced pressure, and two equivalents of 1,3,5-trimethoxybenzene were added as an internal standard. The solution was dissolved in CDCl3 and characterized by GC/MS and 1H NMR. The yield of coupled phenolic product was ∼80% for addition of one equiv of phenol and 46% for two (i.e., ∼one equiv substrate coupled in each case, the other left unreacted).
(c) Sodium 2,4-Di-tert-butylphenolate, UV-vis Scale
Two equivalents of sodium 2,4-di-tert-butylphenolate were added in 0.1 mL THF by syringe to a -80 °C solution of [LMe2Cu(μ-O)2Pt(PPh3)2, at which time the immediate bleaching of the absorption feature at 455 nm in the UV-vis spectrum was observed.
Preparatory Scale
Two equivalents of sodium 2,4-di-tert-butylphenolate were added in 0.5 mL THF by syringe to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2, at which time the solution immediately lightened from deep orange-brown to pale yellow-brown. The solution was stirred for an additional 30 min, warmed to room temperature, and the solvent was removed under reduced pressure to give a brown residue. The resulting brown residue was dissolved in 5 ml CH2Cl2 and stirred with 5 mL aqueous ammonium hydroxide solution. The organic fraction was collected, and the aqueous solution was extracted with a further 2 × 5 ml CH2Cl2. The organic fractions were combined, dried over Na2SO4 and filtered through a plug of neutral Alumina, which was washed with a further 5 ml of CH2Cl2. The solvent was removed under reduced pressure, and two equivalents of 1,3,5-trimethoxybenzene were added as an internal standard. The solution was dissolved in CDCl3 and characterized by GC/MS and 1H NMR spectroscopy. The yield of coupled phenolic product was 56% for addition of one equiv of phenolate and 33% for two equiv phenolate added (i.e. ∼0.6 eq substrate coupled, 1.4 eq recovered unreacted).
(d) PPh3, 1-Decene, Thioanisol, and 9,10-Dihydroanthracene
Ten equivalents of the substrates in 0.2 mL THF were added by syringe to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2. No change in the absorption spectrum was observed for any substrate.
(e) With CO2, UV-vis
Dry CO2 gas was bubbled through a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2 in THF for 5 s, upon which time the immediate formation of an absorption feature at 550 nm was observed.
Preparatory Scale
Dry CO2 gas was bubbled for 5 s through a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2, prepared from (PPh3)2PtO2 (20 mg, 0.026 mmol) and [LMe2Cu]2 (10 mg, 0.013 mmol) in THF (5 mL), during which time the solution turned deep purple. Over the course of several min, the solution then lightened to pale brown. The solution was warmed to room temperature, and the solvent was removed under reduced pressure to give a brown residue. The residue was washed on a frit with benzene (5 mL) to give a tan solid (12 mg, 58%). FT-IR (νC=O = 1680 cm-1); 31P{1H} NMR (CDCl3): δ 7.15 ppm (JPtP = 3697 Hz); 1H NMR (CDCl3): δ 7.3 ppm (m); ESI-MS (m/z): Calculated for [PtP2C37H31O3]+, 780.1355; Found, 780.1412.
Theory
Geometry optimizations were performed with Density Functional Theory (DFT) using the B98 functional37 (a ten parameter hybrid functional designed by Becke) as implemented in the Gaussian03 suite of ab initio quantum chemistry programs.38 The 6-31G(d) and 6-311G(d,p) basis sets were used for H and C atoms and N, O and P atoms, respectively. For Cu, Pd, and Pt, relativistic Stuttgart effective core potentials and associated basis functions were used.39 This combination of functional and basis sets gave bond distances and angles consistent with analogous model systems and chemical intuition for all of the compounds described in this work. For a particular case for which an X-ray crystal structure is available, [CuIII(H33m)]2+ (H33m is the macrocyclic ligand 3,7,11-triaza-bicyclo[11.3.1]heptadeca-1(16),13(17),14-triene), bond distances and angles computed at the B98 level show excellent agreement with the experimental structure (supporting information). In addition, a recent comparison of thirty-four density functionals40 found the B98 functional to be one of the two best functionals for the prediction of a variety of molecular properties, including geometries, electron affinities, and hydrogen-atom transfer barriers, all three of which are particularly important to the present study. Partial atomic charges were computed by the methods of Löwdin41 and Mulliken.42 Vibrational frequencies were determined analytically in order to confirm the computed stationary points as either minima or transition-state structures and were also used in the computation of zero-point energy, enthalpy, and entropy under the standard rigid-rotor, harmonic-oscillator, ideal-gas approximation.43
Single-point solvation free energies in chloroform as solvent were calculated using the Polarized Continuum Model (PCM).44 The PCM cavity was constructed from united atom radii (UA0).45
Results and Discussion
Synthesis and Characterization of [LCu(μ-O)2M(PPh3)2]n+ (M = Pd, Pt)
Addition of one equivalent of [LCuI(MeCN)]+ (L = Me4pda, Me4chd, iPr3tacn) or 0.5 equivalent of [LMe2 Cu]2 to a solution (0.2 mM) of (PPh3)2MO2 (M = Pd, Pt) in CH2Cl2, toluene, or THF at -80 °C yielded heterobimetallic bis(μ-oxo) complexes [LCu(μ-O)2M(PPh3)2]n+ (Scheme 1), which were identified on the basis of UV-vis, resonance Raman, and, in selected cases, 1H and/or 31P{1H} NMR spectroscopy. The products are thermally sensitive, as indicated by bleaching of their UV-vis absorption features upon warming to ambient temperature, and attempts to isolate them as crystalline solids have not been successful. For L = LMe2, Me4pda, and Me4chd, addition of the copper(I) reagent resulted in an immediate color change from nearly colorless to deep yellow-to orange-brown, with concurrent formation of a strong absorption feature in the UV-vis spectrum at 450-480 nm (ε ∼ 2500-6000 M-1cm-1) and a weaker shoulder at ∼550-650 nm (Table 1). The reaction proceeded similarly for L = iPr3tacn, but at a significantly decreased rate (∼1 h). A representive spectrum for the case where L = LMe2 and M = Pt is shown in Figure 1, with others provided in the supporting information (Figures S1-S6). In one case, L = LMe2 and M = Pd, a spectrophotometric titration was performed, which showed that the maximum aborbance was reached at a Cu:Pd complex ratio of 1:1 (Figure S7).
Scheme 1.
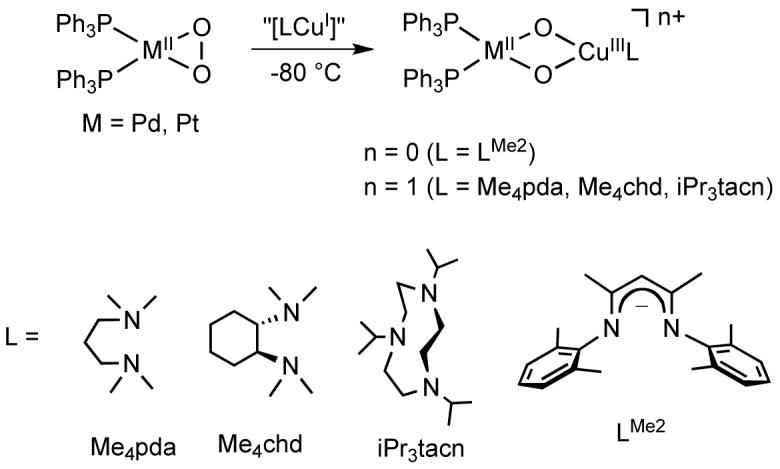
Synthesis of heterobimetallic bis(μ-oxo) complexes.
Table 1.
Selected spectroscopic data for heterobimetallic bis(μ-oxo) complexes
| Complex | UV-Vis λmax (ε)a | Raman Shift (18O)b |
|---|---|---|
| LMe2Cu(μ-O)2Pd(PPh3)2c | 448 (∼5900) | 660 (631) |
| LMe2Cu(μ-O)2Pt(PPh3)2d | 450 (∼5600) | 628,613 (594) |
| [(Me4pda)Cu(μ-O)2Pd(PPh3)2]OTfe | 472 (∼2800) | 610 (580) |
| [(Me4pda)Cu(μ-O)2Pt(PPh3)2]OTfe | 483 (∼2700) | 595 (569) |
| [(Me4chd)Cu(μ-O)2Pd(PPh3)2]PF6e | 463 (∼3500) | 640,616 (600) |
| [(Me4chd)Cu(μ-O)2Pt(PPh3)2]PF6e | 457 (∼3600) | 647,616 (603) |
| [(iPr3tacn)Cu(μ-O)2Pd(PPh3)2]SbF6e | 458 (∼2500) | 630/f |
| [(iPr3tacn)Cu(μ-O)2Pt(PPh3)2]SbF6e | 462 (∼3000) | 628 (601,585) |
Measured in solution at -80 °C. Units: λmax = nm, ε = M-1cm-1 per complex.
Measured as frozen solutions at -196 °C with λex = 457.9 nm. Units: cm-1.
Solvent = THF.
Solvent = 4:1 v/v CH2Cl2/THF.
Solvent = CH2Cl2.
18O-isotope data not collected.
Figure 1.
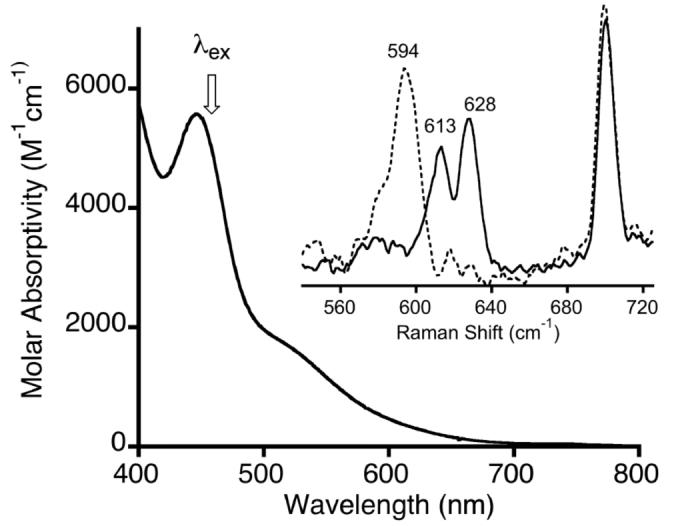
UV-vis spectrum (-80 °C) and resonance Raman spectra (inset, -196 °C, λex = 457.9 nm as noted by arrow in the UV-vis spectrum; solid line = 16O, dashed line = 18O) of [LMe2Cu(μ-O)2Pt(PPh3)2]PF6 in 4:1 v/v THF/CH2Cl2. The peak at ∼700 cm-1 is due to CH2Cl2.
Although we are currently unable to assign the low energy shoulder in the absorption spectra, the similarity of the feature at λmax = 450-480 nm to one typical of [Cu2(μ-O)2]2+ cores2a,b,46 suggests an analogous O → Cu(III) LMCT assignment for a [MCu(μ-O)2]+ core. For the complexes with L = Me4pda, Me4chd, and iPr3tacn, the intensity of this feature (ε ∼ 2500-3600 M-1cm-1) is significantly lower than in the analogous [L2Cu2(μ-O)2]2+ complexes (ε ∼ 5000-10,000 M-1cm-1 per Cu), which may reflect decreased covalency in the Cu-O bonding within the [MCu(μ-O)2]+ units. Consistent with the O → Cu(III) LMCT assignment, laser excitation into this band enables observation of O-isotope sensitive features in Raman spectra (Figure 1, Table 1, and Figures S1-S6 in supporting information). Strong peaks are observed in the ∼600-650 cm-1 region, which shift by ∼25-30 cm-1 upon 18O substitution (using (PPh3)2M18O2 as reagent). We assign these peaks to a [MCu(μ-O)2]+ core vibrational mode(s) on the basis of analogy to the well-studied [Cu2(μ-O)2]2+ systems, for which similar Raman shifts and O-isotope sensitivities were reported.46,46,47 In a few cases (cf. Figure 1), two peaks in the spectrum of the species comprising one O-isotope shifts to a single peak for the other O-isotopomer. Such phenomena were seen previously in [Cu2(μ-O)]2+ 47 and [GeCu(μ-O)2] + 25 complexes and were ascribed to a Fermi doublet that collapses upon isotope substitution, suggesting a similar interpretation of the data for the [MCu(μ-O)2 ]+ cores (M = Pd or Pt). No isotope sensitive vibrations are apparent in the 750-900 cm-1 region, arguing against the presence of peroxo species.48
Solutions of the heterobimetallic reaction products were found to be EPR silent (X-band, 2K), consistent with the diamagnetic ground state expected for the M(II)-Cu(III) core.49 Low temperature (-80 °C, THF) 1H and/or 31P{1H} NMR data were acquired for two representative cases, LMe2Cu(μ-O)2Pd(PPh3)2 and LMe2Cu(μ-O)2Pt(PPh3)2. The spectra support the presence of a single diamagnetic reaction product. For example, in the 31P{1H} NMR spectrum of LMe2Cu(μ-O)2Pd(PPh3)2, a sharp resonance at 26.8 ppm is observed, distinct from that for (PPh3)2PdO2 (34.2 ppm) and the decomposition product OPPh3 (see below; 24.7 ppm). Similarly, the 31P{1H} NMR spectrum of LMe2Cu(μ-O)2Pt(PPh3)2 contains a single resonance at 12.4 ppm (JP-Pt = 3280 Hz, Figure S8) which differs significantly from that of the starting complex (PPh3)2PtO2 (15.6 ppm, JP-Pt = 4061 Hz). Consistent with the structural assignment of a bis(μ-oxo) core, the 31P NMR resonance is similar to that observed in the related complexes [(PPh3)2Pt]2(μ-O)2·LiBF4 (7.8 ppm, JPPt = 3286 Hz) and [(PPh3)2Pt]2(μ-O)2 (8.5 ppm, JPPt = 3183 Hz).17a
Further information on the structures of the heterobimetallic bis(μ-oxo) complexes was obtained from quantum chemical calculations. Systems 1-3, shown in Figure 2, were optimized at the B98 level of density functional theory and their vibrational frequencies were computed for comparison to the experimental Raman data. Selected geometrical data for 1-3 are provided in Figure 2, and the complete set of optimized geometries is provided in the supporting information. Compound 1 has D2h symmetry, with the Cu(III) centers coordinated in a square-planar fashion. Swapping Pd or Pt for Cu, by virtue of the reduced charge on the Group 10 metals, involves replacement of the negatively charged diketiminate ligand with phosphine ligands, as in the experimentally characterized mixed-metal analogs. This introduces asymmetry in the core; compared to Cu-O, the Pd-O and Pt-O bonds in compounds 2 and 3 are about 0.2 Å longer. The CuPd and CuPt distances in 2 and 3 are 0.125 and 0.159 Å longer than is the CuCu distance in 1. Nevertheless, in spite of differences in the second metal and the variation of diketiminate vs. phosphine ligands, there is overall substantial similarity between the various cores.
Figure 2.

Key bond lengths (Å) for structures 1-3 (roman text) optimized at the B98 level (legend inset). The same bond lengths for the one-electron reduced species 4-6 (italic text) are also provided.
Computed vibrational frequencies indicate that the rhomb “breathing” modes46,47 for 1-3 are 635, 597, and 603 cm-1, with 18O isotope shifts of 48, 30, and 30 cm-1, respectively. These data are consistent with those presented in Table 1, where the absolute rhomb breathing frequencies and 18O-isotope shifts of the heterobimetallic compounds are similar to those of characterized homodicopper analogs.46,47 After averaging over Fermi doublet peak positions, the computed isotope shifts for the CuPd and CuPt compounds are in essentially quantitative agreement with experiment, and absolute values of the frequencies are reasonable given the simplicity of the ligands compared to the experimentally characterized systems (which themselves show substantial influence from the various ligands).
Thermal Decomposition Reactions
The [Cu(μ-O)2M]+ complexes decompose upon warming to room temperature as indicated by bleaching of the solution color and decay of the associated UV-vis absorption features. The metal-containing products of the decay process have not been identified, although EPR spectra of the decomposed solutions reveal variable amounts of Cu(II) species (g ∼ 2 signal, ∼10-56% by integration). 31P{1H} NMR spectra of the residue remaining after removal of solvent from the decomposed solutions revealed OPPh3 as the sole observable reaction product in all cases, and in the cases where M = Pd quantitative yields were observed by integration versus an added internal standard (e.g. 2 equiv. OPPh3 per heterobimetallic complex). In the cases where M = Pt, variable yields much less than quantitative (<20%) were observed.
Mechanistic information was obtained from studies of the decomposition of LMe2Cu(μ-O)2Pd(PPh3)2. Monitoring of the reaction by 31P{1H} NMR at -40 °C showed clean conversion of the complex to the product OPPh3, with no other peaks apparent at intermediate reaction times. These data rule out the build-up of measurable quantities of diamagnetic intermediates during the reaction. Decomposition of the 18O-isotopomer derived from (PPh3)2Pd18O2 revealed ∼80% incorporation of the isotopically labeled oxygen in the OPPh3 product by ESI-MS, confirming that the O-atom incorporated into the phosphine is derived from the bis(μ-oxo) core. In order to probe whether the phosphine oxidation involves inter- or intramolecular O-atom transfer, crossover experiments were performed. In these studies, the heterobimetallic bis(μ-oxo) complexes derived from (PPh3)2Pd18O2 and (P(p-tolyl)3)2Pd18O2 were mixed in a 1:1 ratio and then allowed to decompose by warming to ambient temperature. Solvent was removed and the residue was analyzed by ESI-MS, which showed the predominant products to be 18OPPh3 and 16OP(p-tolyl)3; no 18OP(p-tolyl)3 and only a small amount of 16OPPh3 (∼20%, possibly derived from adventitious exchange with air and/or water) were observed. The complementary result was obtained when (PPh3)2Pd16O2 and (P(p-tolyl)3)2Pd18O2 were used. The data clearly show that O-atom transfer occurs via an intramolecular pathway, and rule out mechanisms involving dissociation/association of phosphine ligands.
Monitoring the progress of the decomposition reactions of the various heterobimetallic bis(μ-oxo) complexes by UV-vis spectroscopy revealed irreproducible and/or complicated kinetics that were not amenable to analysis. Despite these difficulties encountered in obtaining and quantitatively interpreting kinetic data, some qualitative observations point to heterometal and ligand effects on the stability of the heterobimetallic [Cu(μ-O)2M]+ cores. For instance, in general, the species with M = Pt are significantly more stable than those with M = Pd, as expected on the basis of their relative metal-ligand bond strengths50 and the kinetic behavior typical for their complexes.51 This stability difference is illustrated by observation of essentially complete decay of [(Me4pda)Cu(μ-O)2Pd(PPh3)2]+ in less than 10 min at -80 °C versus several hours under identical conditions for the Pt analog. The species with L = Me4chd are more stable than those with L = Me4pda, as indicated by a decomposition time for [(Me4chd)Cu(μ-O)2Pd(PPh3)2]+ of ∼1 d at -80 °C and no visible signs of decomposition of the Pt analog after several days under the same conditions.
Reactivity with Exogenous Substrates
In order to assess the impact of metal substitution on the reactivity of the bridging oxo ligands, we compared the reactivity of the known species (LMe2Cu)2(μ-O)252 and [(PPh3)2Pt]2(μ-O)2 17 with the heterobimetallic counterpart LMe2Cu(μ-O)2Pt(PPh3)2 (Figure 3). While comparison to [(PPh3)2Pt]2(μ-O)2 is facilitated by previous reports of aspects of its reactivity,17a,c only observation of the decomposition of (LMe2Cu)2(μ-O)2 to (LMe2Cu)2(μ-OH)2 has been described,53 necessitating parallel studies of its reaction chemistry.
Figure 3.
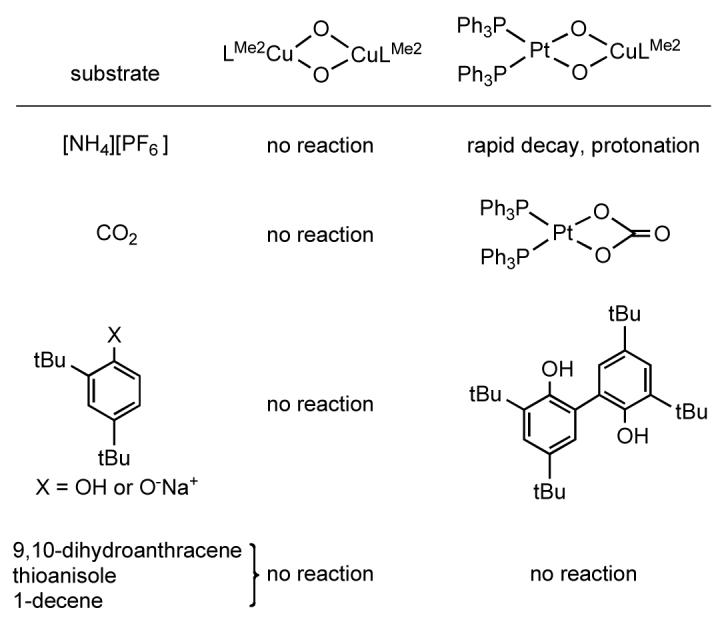
Comparative summary of the reactivity of homo- and heterobimetallic complexes with exogenous substrates. See the text for details of the reaction conditions, yields, and characterization results.
(a) [NH4][PF6]
The oxo groups in [(PPh3)2Pt)]2(μ-O)2 are readily protonated even by weak acids such as H2O (pKa of protonated form approximated as ∼25 in THF),17 but those in [Cu2(μ-O)2]2+ cores are not typically reactive with strong protic acids like HBF4·Et2O and instead typically act as electrophiles.2 Consistent with these previous observations, addition of the weak acid [NH4][PF6] to (LMe2Cu)2(μ-O)2 at -80 °C also resulted in no change in its UV-vis spectral features. In contrast, the similar reaction with LMe2Cu(μ-O)2Pt(PPh3)2 resulted in instantaneous bleaching of the solution color and disappearance of the absorption feature at 455 nm in the UV-vis spectrum. While efforts to isolate and thoroughly characterize the reaction product(s) upon warming have not been successful, high resolution ESI-MS analysis of the crude reaction residue revealed a major peak at m/z = 1121.2883 with the appropriate isotope pattern for the doubly-protonated and 1-electron reduced product [LMe2Cu(μ-OH)2Pt(PPh3)2]+ (predicted m/z = 1121.2838; Figure S9). Thus, the oxo groups in LMe2Cu(μ-O)2Pt(PPh3)2 appear to act as bases similar to [(PPh3)2Pt)]2(μ-O)2.
(b) CO2
Bubbling of CO2 through a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2 resulted in the instantaneous formation of a deep purple solution having a strong absorption feature at 530 nm (ε ∼ 5000 M-1cm-1; Figure S10), which decayed rapidly to give a light brown solution. Removal of the solvent followed by washing with benzene resulted in the isolation of a tan solid (58% yield), the identity of which was confirmed as the known carbonato complex (PPh3)2Pt(CO3) on the basis of FT-IR (νC=O = 1680 cm-1), 1H NMR, 31P NMR (δ = 7.15 ppm, JPtP = 3697 Hz), and high resolution ESI-MS data (m/z = 780.1412 [M+1]+).54 A similar reaction was reported for [(PPh3)2Pt)]2(μ-O)2,17a consistent with analogous nucleophilic attack by the bridging oxo units on the carbon of CO2. No reaction was observed upon treatment of (LMe2Cu)2(μ-O)2 with CO2 at -80 °C, and CO2 fixation has not been seen for other bis(μ-oxo)dicopper systems. Thus, the oxo groups in the heterobimetallic complex can act as nucleophiles, reactivity that is similar to that of the diplatinum analog but is in contrast to that typical of dicopper species.
(c) 2,4-Di-tert-butylphenol, 2,4-di-tert-butylphenolate, and 9,10-dihydroanthracene
Most bis(μ-oxo)dicopper complexes rapidly react at low temperature (-80 °C) with phenols to give products resulting from H-atom abstraction (e.g., to yield a bis(μ-hydroxo)dicopper(II) complex and the coupled diphenol, often in quantitative yields).2 However, at -80 °C no change in the UV-vis spectrum of (LMe2Cu)2(μ-O)2 was observed upon addition of 10 equiv of 2,4-di-tert-butylphenol, even after several hours. After allowing to stir at 24 h at -80 °C followed by warming and analysis by GC/MS and 1H NMR spectroscopy, no coupled phenolic product was seen. When the phenol was added at -40 °C, gradual decay of the bis(μ-oxo) UV-vis feature at 422 nm was observed (t1/2 ∼ 4 h). Analysis of a solution maintained at -40 °C for 12 h and then warmed to room temperature by GC/MS and 1H NMR spectroscopy revealed the main oxidation product to be the coupled diphenol (∼60% yield). No reaction was observed with sodium 2,4-di-tert-butylphenolate or 9,10-dihydroanthracene, which contains relatively weak C-H bonds. The sluggish reactivity with 2,4-di-tert-butylphenol is distinct from what has been observed for other [Cu2(μ-O2)]2+ cores, such as those supported by di- or triamine ligands.2,55 The decreased rate of H-atom abstraction could be a result of inhibition of substrate approach by the steric bulky β-diketiminate, or, perhaps more likely, of decreased oxidizing power due to its strong electron donating properties. Consistent with the latter notion, addition of other electron rich substrates including PPh3, thioanisole, and 1-decene at -80 °C resulted in no observed reaction or oxidation products upon warming.
In contrast to what was observed for (LMe2Cu)2(μ-O)2, addition of one or two equivalents of 2,4-di-tert-butylphenol to a -80 °C solution of LMe2Cu(μ-O)2Pt(PPh3)2 resulted in instantaneous bleaching of the solution color to pale yellow-brown and decay of the 450 nm UV-vis feature. Analysis of the reaction products upon warming revealed the oxidative coupling of 80% of the starting phenol with addition of 1 equiv of phenol, and coupling of 46% of the starting phenol when 2 equiv of phenol were added (i.e. approximately 1 equiv phenol coupled in each case). Similar bleaching of the solution occurred upon addition of sodium 2,4-di-tert-butylphenolate, with formation of the oxidatively coupled diphenol product being observed upon warming, although at a lower yield than for the phenol (56% coupled for 1 equiv phenolate added, 33% coupled for 2 equiv phenolate added). However, no reaction was observed with 9,10-dihydroanthracene, or with the electron rich substrates thioanisole and 1-decene.
Together, these results demonstrate that, unlike (LMe2Cu)2(μ-O)2 and notwithstanding the basic character of its bridging oxo ligands, the [M(μ-O)2 Cu]+ core of LMe2Cu(μ-O)2Pt(PPh3)2 is competent at 1-electron oxidations and/or H-atom abstractions. Considering that (a) LMe2Cu(μ-O)2Pt(PPh3)2 is protonated by NH4+ (vide supra), (b) the oxo groups in LMe2Cu(μ-O)2Pt(PPh3)2 are nucleophilic, as shown by the reaction with CO2 to yield (PPh3)2Pt(CO3), and (c) phenols are weakly acidic similar to NH4+, we believe it is likely that the reaction of 2,4-di-tert-butylphenol occurs via initial proton transfer, rather than H-atom transfer. In this scenario, oxidation to give the coupled diphenol involves subsequent reaction of the protonated complex, which would be expected on the basis of charge considerations alone to be a stronger oxidant than the bis(μ-oxo) precursor.
Theoretical Characterization of Structures and Reactivities
To better understand those factors affecting the reactivity observed for the mixed-metal oxo species, a series of calculations were undertaken on the computational models introduced above. In particular, reduction potentials, proton affinities, and transition-state structures for prototypical H-atom transfer reactions were examined.
(a) Reduction potentials
The potential for complexes 1-3 to accept an electron and form the corresponding anionic complexes 4-6 was investigated in the gas phase and also in solution. Cartesian coordinates for the reduced structures are provided in the supporting information and the most relevant geometrical parameters are displayed in Figure 2 where they may be conveniently compared to those of their oxidized counterparts 1-3. In each case, corresponding structures are similar in the sense that both the [M(μ-O)2Cu]+ core and the β-diketiminate ligand continue to lie in the same plane. The most notable change is the elongation of all Cu-O bonds by roughly 0.1 Å, consistent with Cu being the metal site at which reduction takes place when a choice is available. Thus, in spite of the electron rich character of the anionic β-diketiminate ligand, it appears to be the Cu site that is preferentially reduced in the mixed-metal complexes with Pd(II) and Pt(II), consistent with the rarity of oxidation state +I for these metals in complexes lacking a metal-metal bond.56
Absolute one-electron reduction potentials (Eoabs) in CH2Cl2 solution were calculated from the appropriate thermodynamic cycle,57 adding differential solvation effects to the gas-phase electron affinity including thermal contributions.58,59 Continuum solvation free energies in CH2Cl2 for the different species were calculated using the PCM approach and are included in the supporting information. Absolute reduction potentials were then converted to reduction potentials relative to the standard calomel electrode (SSCE) by the following procedure. The standard reduction potential for [CuIII(H33m)]2+ (where H33m is the macrocyclic ligand 3,7,11-triaza-bicyclo[11.3.1]heptadeca-1(16),13(17),14-triene) has been reported, and its III/II redox couple is well behaved.60 Following the above computational protocol, the absolute reduction potential of this compound was computed (as noted in the Computational Methods section, the choice of the B98 functional was motivated in part by its good performance in reproducing the experimental structure of this compound), and the difference between the absolute value and that relative to the SSCE was used as a constant to place all other computed values on the SSCE scale. Table 2 contains these calculated EoSSCE values for the three redox couples involving complexes 1-3 and their corresponding reduced species 4-6, together with additional thermodynamic data discussed below.
Table 2.
Key computed thermodynamic properties of 1-3
| Compound | EoSSCE, V | pKaa | ΔHoBDE,b kcal/mol | ΔG‡,o,c kcal/mol |
|---|---|---|---|---|
| 1 | -1.99 | 9.5 | 81.7 | 17.4 |
| 2 | -2.19 | 15.7 | 88.1 | 17.8 |
| 3 | -1.86 | 14.0 | 86.2 | 18.9 |
Refers to conjugate acids 4-6.
0 K bond dissociation enthalpy for 7-9.
298 K free energy of activation for H atom abstraction from 1,4-cyclohexadiene.
Table 2 indicates that the reduction potentials for the Pd and Pt complexes are within 200 mV on either side of that for 1, suggesting that the redox active site is indeed Cu (as inferred above on the basis of geometric changes, and as further supported by population analysis which is not discussed here). The distinctly negative reduction potentials are consistent with the strongly electron donating nature of the diketiminate ligand supporting Cu.
(b) Proton affinities
The pKa values for the conjugate acid forms of 1-3 (i.e., 7-9) were computed in the gas phase and also in a hypothetical aqueous solution modeling water as a continuum solvent. Cartesian coordinates for the protonated structures (complexes 7-9 respectively) are provided in the supporting information, while the most relevant geometrical parameters are displayed in Figure 4. In all cases protonation produces a slight bending distortion of the Cu2O2 and MCuO2 cores and also distorts the planarity of the β-diketiminate ligand. The protonation also increases the M-N and M-OH bond distances on the side of the protonated oxygen atom, and the coordination geometry about Cu is distorted from square planar to a D2d-like flattened tetrahedral arrangement. Löwdin partial atomic charges (see supporting information) indicate that the positive charge of the proton is distributed over both metal centers and that a significant portion is delocalized onto the β-diketiminate ligand.
Figure 4.

Key bond lengths (Å) for structures 7-9 (roman text) optimized at the B98 level (see Figure 2 for legend). The same bond lengths for the one-electron reduced species 10-12 (italic text) are also provided.
By combining59 gas phase protonation enthalpies with differential aqueous solvation free energies computed using the PCM continuum solvent model (and the experimental solvation free energy for the proton61), pKa values for 1-3 were computed. Compound 1 is significantly less basic than the mixed-metal cases, consistent with the experimental results of the reactions of (LMe2Cu)2(μ-O)2 and LMe2Cu(μ-O)2Pt(PPh3)2 with [NH4][PF6], as well as the hypothesis that the reaction of the Cu-Pt complex with 2,4-di-tert-butylphenol proceeds by an initial protonation event. This reduced basicity is even larger in the gas-phase than in solution: the gas-phase protonation free energy for 1 is -236.3 kcal/mol compared to about -258 kcal/mol for the Pd and Pt mixed-metal species.
(c) Hydrogen-atom affinities
Having considered individually one-electron reduced and protonated species, we next examined the μ-hydroxo species 10-12 generated by the sum of these two processes applied to 1-3, respectively. Figure 4 contains key geometrical data for these species, where they may be compared to those for their oxidized analogs (full geometries are provided in the supporting information). Inspection of the geometries indicates that M-O and M-N distances for M = Cu are very similar to those in the one-electron reduced species 4-6, while those about M = Pd or Pt are very similar to those in the protonated species 7-9. Thus, transfer of an H atom appears to reduce Cu(III) to Cu(II) and leave the H atom with protonic character.
Interestingly, although cationic 7 was predicted to have Cs symmetry, the reduced species 10 prefers to break symmetry to a mixed valence Cu(III)/Cu(II) species. This C1 radical is about 5 kcal/mol lower in energy than a structure forced to remain in the Cs point group. In every case, then, the Cu-OH bond to the Cu(II) atom is about 2.08 Å (Figure 3).
Gas-phase O-H bond dissociation enthalpies (ΔHoBDE) were calculated for 10-12 and are listed in Table 4. The smallest ΔHoBDE value is associated with the bis-diketiminate homodicopper species, consistent with the experimental finding of significantly reduced reactivity of (LMe2Cu)2(μ-O)2 with 2,4-di-tert-butylphenol, compared to that of LMe2Cu(μ-O)2Pt(PPh3)2. The other data in Table 4 suggest that the dominant cause of the observed reactivity difference between the homometallic and heterobimetallic cores is the higher proton affinity of the latter. The Pd- and Pt-containing species have similar ΔHoBDE values, consistent with their similar net gas-phase electron and proton affinities.
(d) Transition-state structures for C-H bond activation
In order to evaluate the relative abilities of complexes 1-3 to abstract a H atom from an external substrate and to understand the mechanistic character of this process, transition-state (TS) structures were calculated for the reactions of 1-3 with 1,4-cyclohexadiene (C6H8; CHD). The gas phase Cartesian coordinates for these transition-state (TS) structures (species 13-15) are provided in the supporting information and the most relevant geometric parameters are included in Figure 5. The structure of the diamond core in each case is slightly distorted and is intermediate between the initial and final reduced complexes. In the TS structures derived from the mixed metal complexes there is a stereochemical distinction with respect to the orientation of the reacting CHD molecule, which can partially eclipse either the Cu-diketiminate or the Pd or Pt fragments. The energetically preferred structures are the ones shown in Figure 4, with the CHD moiety directed towards the diketiminate; the alternative structures rotated approximately 180 deg about the C - - H - - O axis are 0.6 and 3.8 kcal/mol higher in free energy for 14 and 15, respectively. The most interesting feature of the TS structures is the H atom in flight between an oxo bridging atom and a sp3 carbon atom of CHD. The geometries of the copper-containing portions of 13-15 are remarkably similar to one another, and differences between Pd and Pt are also small. With respect to the H atom in flight, 13 has a slightly later transition state (shorter forming OH bond and longer breaking CH bond) than is found in either of the other two structures, consistent with this reaction being the least exergonic (-14.5 kcal/mol compared to -19.9 and -18.4 for Pd and Pt, respectively), but the bond length differences are not very large.
Figure 5.

Key bond lengths (Å) for structures 13-15 optimized at the B98 level (see Figure 2 for legend).
The 298 K free energies of activation for H-atom abstraction from CHD are included in Table 2. There is only a very small variation (1.5 kcal/mol) over all three systems. The absolute free energies of activation are rather high, considering the very weak nature of the CH bond in CHD (298 K BDE of about 77 kcal/mol)62 and the similarly weak OH bonds in 7-9. For stronger CH bonds, e.g., H-CH2Ph (89.8 Kcal/mol), H-CH2CN (91.0 Kcal/mol), H-C6H11 (95.5 Kcal/mol), H-CH3 (104.9 Kcal/mol) or H-C6H5 (113.1 Kcal/mol),63 one would expect free energies of activation to be substantially increased.
A careful examination of the TS structures in Figure 4, particularly their comparison to structures 7-9 and 10-12 in Figure 3, suggests that the H-atom abstraction process is precisely that, i.e., a process involving roughly simultaneous electron and proton transfer. Thus, the TS geometries in Figure 4 have key bond lengths, particularly about the Cu center that is being reduced, roughly intermediate between protonated and hydrogenated derivatives of 1-3. The case is slightly less clear for 1, because desymmetrization of the two copper centers is not very advanced in the TS structure 13, but is well represented in the structures for 14 and 15.
Karlin et al.64 studied the oxidation of several substrates by a peroxo Cu2O2 complex and found that the mechanism with easily oxidized substrates was a two step electron-transfer (ET)/proton-transfer (PT) process, while with less easily oxidized substrates a concerted ETPT mechanism was observed. The importance of the redox potential for experimental H-atom abstraction by Fe65 and Mn66,67 complexes has also been emphasized. Thus, highly mismatched electron affinities lead to a two-step ET/PT mechanism, highly mismatched proton affinities lead to a two-step PT/ET mechanism, and in the absence of large mismatches a concerted mechanism dominates. The C-H activation cases studied theoretically here appear to fall into the final category, which is consistent with prior work on C-H activation by bis(μ-oxo)dicopper species.12 It is worth noting that the mechanism of phenol oxidation by the [M(μ-O)2Cu]+ cores appears to be rather different from those presumed for the dicopper systems, insofar as proton transfer likely precedes oxidative coupling.
Conclusions
A new class of heterobimetallic CuPd and CuPt complexes has been prepared by the reaction of (PPh3)2MO2 (M = Pd, Pt) with Cu(I) precursors, and the products have been identified as bis(μ-oxo) species on the basis of spectroscopic data and DFT calculations. The thermally unstable complexes decompose to yield OPPh3, and in one instance this was shown to be an intramolecular process from the results of crossover experiments using doubly labeled reagents. Studies of the reactivity of LMe2Cu(μ-O)2Pt(PPh3)2 with CO2 and [NH4][PF6] gave results consistent with basic, nucleophilic oxo groups in the complex, in distinct contrast to what is typical for bis(μ-oxo)dicopper compounds2 but similar to what is known for [M2(μ-O)2] (M = Pd, Pt) cores.6 In addition, oxidative coupling of 2,4-di-tert-butylphenol or 2,4-di-tert-butylphenolate by LMe2Cu(μ-O)2Pt(PPh3)2 demonstrated that the mixed-metal core is also a competent 1-electron oxidant, with significantly increased reactivity compared to the homodicopper analogue (LMe2Cu)2(μ-O)2. Assessment of the electron affinities, basicities, and H-atom transfer kinetics and thermodynamics of Cu2 and CuM bis(μ-oxo) species by theoretical methods provided insight into the bases for the observed reactivity differences. Notably, reduction occurs at the Cu(III) ion in the heterobimetallic systems, which are more basic than the Cu2 counterpart and form stronger O-H bonds. Despite their high proton affinities, all of the systems exhibit low electron affinities which dominate their predicted sluggish C-H bond activation reactivity. In sum, the results reported herein, in conjunction with others reported previously, show that replacement of a Cu(III) ion in a [Cu2(μ-O)2]2+ core with a different metal ion has important electronic structural and reactivity consequences. These results thus motivate further studies of heterobimetallic systems with different metal combinations.
Supplementary Material
Acknowledgements
We thank the NIH (GM47365 to W.B.T.), the NSF (CHE-0610183 to C.J.C.), the MEC (CSD2006-0003 to A.L.) and the University of Minnesota (Doctoral Dissertation fellowship to J.T.Y.) for financial support of this work.
References
- (1)(a).Meunier B. Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations. Springer; Berlin: 2000. Meunier B, editor. Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Imperial College Press; London: 2000. J. Inorg. Biochem. Vol. 100. 2006. pp. 419–880.(issue devoted to “High-valent iron intermediates in biology”).
- (2)(a).Que L, Jr., Tolman WB. Angew. Chem. Int. Ed. 2002;41:1114–1137. doi: 10.1002/1521-3773(20020402)41:7<1114::aid-anie1114>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]; (b) Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; (c) Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (d) Osako T, Terada S, Tosha T, Nagatomo S, Furutachi H, Fujinami S, Kitagawa T, Suzukib M, Itoh S. Dalton Trans. 2005:3514–3521. doi: 10.1039/b500202h. [DOI] [PubMed] [Google Scholar]; (e) Hatcher L, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- (3)(a).Manchanda R, Brudvig GW, Crabtree RH. Coord. Chem. Rev. 1995;144:1–38. [Google Scholar]; (b) Wu AJ, Penner-Hahn JE, Pecoraro VL. Chem. Rev. 2004;104:903–938. doi: 10.1021/cr020627v. [DOI] [PubMed] [Google Scholar]
- (4)(a).Hikichi S, Yoshizawa M, Sasakura Y, Akita M, More-oka Y. J. Am. Chem. Soc. 1998;120:10567–10568. [Google Scholar]; (b) Hikichi S, Yoshizawa M, Sadakura Y, Komatsuzaki H, Moro-oka Y, Akita M. Chem. Eur. J. 2001;7:5012–5028. doi: 10.1002/1521-3765(20011203)7:23<5011::aid-chem5011>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]; (c) Shiren K, Ogo S, Fujinami S, Hayashi H, Suzuki M, Uehara A, Watanabe Y, Moro-oka Y. J. Am. Chem. Soc. 2000;122:254–262. [Google Scholar]; (d) Mandimutsira BS, Yamarik JL, Brunold TC, Gu W, Cramer SP, Riordan CG. J. Am. Chem. Soc. 2001;123:9194–9195. doi: 10.1021/ja016209+. [DOI] [PubMed] [Google Scholar]; (e) Schenker R, Manimutsira BS, Riordan CG, Brunold TC. J. Am. Chem. Soc. 2002;124:13842–13855. doi: 10.1021/ja027049k. [DOI] [PubMed] [Google Scholar]; (f) Itoh S, Bandoh H, Nakagawa M, Nagatomo S, Kitagawa T, Karlin KD, Fukuzumi S. J. Am. Chem. Soc. 2001;123:11168–11178. doi: 10.1021/ja0104094. [DOI] [PubMed] [Google Scholar]
- (5).Skulan AJ, Hanson MA, Hsu H.-f., Que L, Jr., Solomon EI. J. Am. Chem. Soc. 2003;125:7344–7356. doi: 10.1021/ja021137n. [DOI] [PubMed] [Google Scholar]
- (6)(a).Sharp PR. J. Chem. Soc., Dalton Trans. 2000:2647–2657. [Google Scholar]; (b) Li JJ, Li W, Sharp PR. Inorg. Chem. 1996;35:604–613. [Google Scholar]
- (7)(a).Holland PL, Rodgers KR, Tolman WB. Angew. Chem. Int. Ed. 1999;38:1139–1142. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1139::AID-ANIE1139>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; (b) Decker H, Dillinger R, Tuczek F. Angew. Chem. Int. Ed. 2000;39:1591–1595. doi: 10.1002/(sici)1521-3773(20000502)39:9<1591::aid-anie1591>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; (c) Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- (8).Lieberman RL, Rosenzweig AC. Dalton Trans. 2005:3390–3396. doi: 10.1039/b506651d. [DOI] [PubMed] [Google Scholar]
- (9).Chen PPY, Chan SI. J. Inorg. Biochem. 2006;100:801–809. doi: 10.1016/j.jinorgbio.2005.12.014. [DOI] [PubMed] [Google Scholar]
- (10)(a).Smeets PJ, Groothaert MH, Schoonheydt RA. Catal. Today. 2005;110:303–309. [Google Scholar]; (b) Groothaert MH, Smeets PJ, Sels BF, Jacobs PA, Schoonheydt RA. J. Am. Chem. Soc. 2005;127:1394–1395. doi: 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- (11).DuBois JL, Mukherjee P, Stack TDP, Hedman B, Solomon EI, Hodgson KO. J. Am. Chem. Soc. 2000;122:5775–5787. [Google Scholar]
- (12)(a).Mahapatra S, Halfen JA, Tolman WB. J. Am. Chem. Soc. 1996;118:11575–11586. [Google Scholar]; (b) Cole AP, Mahadevan V, Mirica LM, Ottenwaelder X, Stack TDP. Inorg. Chem. 2005;44:7345–7364. doi: 10.1021/ic050331i. [DOI] [PubMed] [Google Scholar]; (c) Cramer CJ, Pak Y. Theor. Chem. Acc. 2001;105:477–480. [Google Scholar]; (d) Cramer CJ, Kinsinger CR, Pak Y. J. Mol. Struct (Theochem) 2003;632:111–120. [Google Scholar]
- (13).Osako T, Ohkubo K, Taki M, Tachi Y, Fukuzumi S, Itoh S. J. Am. Chem. Soc. 2003;125:11027–11033. doi: 10.1021/ja029380+. [DOI] [PubMed] [Google Scholar]
- (14).Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J. Am. Chem. Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- (15).Taki M, Itoh S, Fukuzumi S. J. Am. Chem. Soc. 2002;124:998–1002. doi: 10.1021/ja016023a. [DOI] [PubMed] [Google Scholar]
- (16).Mahadevan V, Henson MJ, Solomon EI, Stack TDP. J. Am. Chem. Soc. 2000;122:10249–10250. [Google Scholar]
- (17)(a).Li W, Barnes CL, Sharp PR. J. Chem. Soc., Chem. Commun. 1990:1634–1636. [Google Scholar]; (b) Li JJ, Li W, Sharp PR. Inorg. Chem. 1996;35:604–613. [Google Scholar]; (c) Sharp PR. J. Chem. Soc., Dalton Trans. 2000:2647–2657. [Google Scholar]; (d) Anandhi U, Sharp PR. Inorg. Chem. 2004;43:6780–6785. doi: 10.1021/ic049463m. [DOI] [PubMed] [Google Scholar]
- (18)(a).Kim E, Chufan EE, Kamaraj K, Karlin KD. Chem. Rev. 2004;104:1077–1134. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]; (b) Collman JP, Boulatov R, Sunderland CJ, Fu L. Chem. Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]; (c) Boulatov R. Pure Appl. Chem. 2004;76:303–319. [Google Scholar]
- (19).Ullmann’s Encyclopedia of Industrial Chemistry. Wiley; New York: 2000. [Google Scholar]
- (20)(a).Hill CL, Prosser-McCartha CM. Coord. Chem. Rev. 1995;143:407–455. [Google Scholar]; (b) Nesterova DS, Volodymyr N. Kokozay;, Dyakonenko VV, Shishkin OV, Jezierska J, Ozarowski A, Kirillov AM, Kopylovich MN, Pombeiro AJL. Chem. Commun. 2006:4605–4607. doi: 10.1039/b608790f. [DOI] [PubMed] [Google Scholar]
- (21)(a).Hosokawa T, Murahashi S-I. Acc. Chem. Res. 1990;23:49–54. [Google Scholar]; (b) Hosokawa T, Takano M, Murahashi S-I. J. Am. Chem. Soc. 1996;118:3990–3991. [Google Scholar]; (c) Hosokawa T, Nomura T, Murahashi S-I. J. Organomet. Chem. 1998;551:387–389. [Google Scholar]
- (22).Aboelella NW, Lewis EA, Reynolds AM, Brennessel WW, Cramer CJ, Tolman WB. J. Am. Chem. Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- (23).Fujita K, Schenker R, Gu W, Brunold TC, Cramer SP, Riordan CG. Inorg. Chem. 2004;43:3324–3326. doi: 10.1021/ic049876n. [DOI] [PubMed] [Google Scholar]
- (24).Aboelella NW, York JT, Reynolds AM, Fujita K, Kinsinger CR, Cramer CJ, Riordan CG, Tolman WB. Chem. Commun. 2004:1716–1717. doi: 10.1039/b404640d. [DOI] [PubMed] [Google Scholar]
- (25).York JT, Young VG, Tolman WB. Inorg. Chem. 2006;45:4191–4198. doi: 10.1021/ic060050q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Alsters PL, Boersma J, van Koten G. Organometallics. 1993;12:1629–1638. [Google Scholar]
- (27)(a).Cygan ZT, Bender JE, Litz KE, Kampf JW, Banaszak Holl MM. Organometallics. 2002;21:5373–5381. [Google Scholar]; (b) Litz KE, Banaszak-Holl MM, Kampf JW, Carpenter GB. Inorg. Chem. 1998;37:6461–6469. doi: 10.1021/ic9808044. [DOI] [PubMed] [Google Scholar]
- (28).Malatesta L, Cariello C. J. Chem. Soc. 1958:2323. [Google Scholar]
- (29).Tolman CA, Seidel WC, Gerlach DH. J. Am. Chem. Soc. 1972;94:2669–2676. [Google Scholar]
- (30)(a).Nyman CJ, Wymore CE, Wilkinson G. J. Chem. Soc. (A) 1968:561–563. [Google Scholar]; (b) Wilke G, Schott H, Heimbach P. Angew. Chem. Int. Ed. 1967;6:92–93. [Google Scholar]
- (31).Brown EC, York JT, Antholine WE, Ruiz E, Alvarez S, Tolman WB. J. Am. Chem. Soc. 2005;127:13752–13753. doi: 10.1021/ja053971t. [DOI] [PubMed] [Google Scholar]
- (32).Mahadevan V, DuBois JL, Hedman B, Hodgson KO, Stack TDP. J. Am. Chem. Soc. 1999;121:5583–5584. [Google Scholar]
- (33).Spencer DJE, Reynolds AM, Holland PL, Jazdzewski BA, Duboc-Toia C, Pape LL, Yokota S, Tachi Y, Itoh S, Tolman WB. Inorg. Chem. 2002;41:6307–6321. doi: 10.1021/ic020369k. [DOI] [PubMed] [Google Scholar]
- (34)(a).Mahapatra S, Halfen JA, Wilkinson EC, Pan G, Cramer CJ, L. Que J, Tolman WB. J. Am. Chem. Soc. 1995;117:8865–8866. [Google Scholar]; (b) Halfen JA, Young VG, Jr., Tolman WB. Inorg. Chem. 1998;37:2102–2103. doi: 10.1021/ic971216d. [DOI] [PubMed] [Google Scholar]
- (35).Holland PL, Tolman WB. J. Am. Chem. Soc. 1999;121:7270–7271. [Google Scholar]
- (36).Addition of 0.5 equiv [LMe2Cu]2 to a THF solution of (P(p-tolyl)3)2PdO2 at -80 °C resulted in the appearance of a UV-vis feature identical to that observed for [LMe2Cu(μ-O)2Pd(PPh3)2], confirming the formation of the analogous species LMe2Cu(μ-O)2Pd(P(p-tolyl)3)2.
- (37)(a).Becke AD. J. Chem. Phys. 1997;107:8554–8560. [Google Scholar]; b) Schmider HL, Becke AD. J. Chem. Phys. 1998;108:9624–9631. [Google Scholar]
- (38).Frisch MJ, et al. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. The complete author list for this references is supplied in the supporting information.
- (39).Dolg M, Wedig U, Stoll H, Preuss H. J. Chem. Phys. 1987;86:866–872. [Google Scholar]
- (40).Riley KE, Op’t Holt BT, Merz KM. J. Chem. Theory Comput. 2007;3:407–433. doi: 10.1021/ct600185a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lowdin P-O. J. Chem.Phys. 1950;18:365–375. [Google Scholar]
- (42).Mulliken RS. J. Chem.Phys. 1955;23:1833–1840. [Google Scholar]
- (43).Cramer CJ. Essentials of Computational Chemistry. Theories and Models. 2nd Ed. John Wiley & Sons; Chichister, UK: 2004. pp. 299–306. [Google Scholar]
- (44).Tomasi J, Mennucci B, Cammi R. Chem. Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- (45).Barone V, Improta R, Rega N. Theor. Chem. Acc. 2004;111:237–245. [Google Scholar]
- (46).Henson MJ, Mukherjee P, Root DE, Stack TDP, Solomon EI. J. Am. Chem. Soc. 1999;121:10332–10345. [Google Scholar]
- (47).Holland PL, Cramer CJ, Wilkinson EC, Mahapatra S, Rodgers KR, Itoh S, Taki M, Fukuzumi S, Que L, Jr., Tolman WB. J. Am. Chem. Soc. 2000;122:792–802. [Google Scholar]
- (48).Additional unassigned peaks in the Raman spectrum of the complexes with L = iPr3tacn were observed (Figure Sx) at 572 cm-1 and 505 cm-1, which shifted upon 18O-substitution to 541 cm-1 and 477 cm-1, respectively (Δ18O = 31 cm-1 and 28 cm-1). With the data currently available, we cannot discern whether these are due to an additional species or to other low frequency modes for the [MCu(μ-O)]+2 core for this case.
- (49).For the system with the ligand Me4pda, significant amounts (∼15%) of Cu(II) decomposition products were observed in the EPR samples due to the extreme thermal sensitivity of the [Cu(μ-O)2M]+ complex.
- (50).Skinner HA, Connor JA. Pure Appl. Chem. 1985;57:79–88. [Google Scholar]
- (51).For example, ligand substitution rates for analogous Pd(II) and Pt(II) complexes vary by ∼105. See:Rund JV. Inorg. Chem. 1974;13:738–740.
- (52).Spencer DJE, Reynolds AM, Holland PL, Jazdzewski BA, Duboc-Toia C, Pape LL, Yokota S, Tachi Y, Itoh S, Tolman WB. Inorg. Chem. 2002;41:6307–6321. doi: 10.1021/ic020369k. [DOI] [PubMed] [Google Scholar]
- (53).Dai X, Warren TH. Chem. Commun. 2001:1998–1999. doi: 10.1039/b105244f. [DOI] [PubMed] [Google Scholar]
- (54).Scherer OJ, Jungmann H, Hussong K. J. Organomet. Chem. 1983;247:c1–c4. [Google Scholar]
- (55).Mahadevan V, DuBois JL, Hedman B, Hodgson KO, Stack TDP. J. Am. Chem. Soc. 1999;121:5583–5584. [Google Scholar]
- (56).Cotton FA, Wilkinson G, Murillo CA, Bochmann M. Advanced Inorganic Chemistry. 6th ed. John Wiley & Sons; New York: 1999. pp. 1063–1084. [Google Scholar]
- (57).Gherman BF, Tolman WB, Cramer CJ. J. Comp. Chem. 2006;27:1950–1961. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]
- (58).Lewis A, Bumpus JA, Truhlar DG, Cramer CJ. J. Chem. Educ. 2004;81:596. [Google Scholar]
- (59).Cramer CJ. Essentials of Computational Chemistry. Theories and Models. 2nd Ed. John Wiley & Sons; Chichister, UK: 2004. pp. 379–381. [Google Scholar]
- (60).Xifra R. l., Ribas X, Llobet A, Poater A, Duran M, Sol M, Stack TDP, Benet-Buchholz J, Donnadieu B, Parella JMT. Chem. Eur. J. 2005;11:5146–5156. doi: 10.1002/chem.200500088. [DOI] [PubMed] [Google Scholar]
- (61)(a).Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Tuttle TR. J. Phys. Chem. A. 1998;102:7787–7794. [Google Scholar]; (b) Kelly CP, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2006;110:16066–16081. doi: 10.1021/jp063552y. [DOI] [PubMed] [Google Scholar]
- (62).McMillen DF, Golden DM. Annu. Rev. Phys. Chem. 1982;33:493–532. [Google Scholar]
- (63)(a).Lide DR. Handbook of Chemistry and Physics. 85th ed. CRC Press; Boca Raton, FL: 20042005. pp. 9–65.pp. 9–68. [Google Scholar]; (b) Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- (64).Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. J. Am. Chem. Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- (65)(a).Jonas RT, Stack TDP. Am. Chem. Soc. 1997;119:8566. [Google Scholar]; (b) Goldsmith CR, Jonas RT, Stack TDP. J. Am. Chem. Soc. 2002;124:83. doi: 10.1021/ja016451g. [DOI] [PubMed] [Google Scholar]
- (66)(a).Baldwin MJ, Pecoraro VL. J. Am. Chem. Soc. 1996;118:11325. [Google Scholar]; b) Caudle MT, Pecoraro VL. J. Am. Chem. Soc. 1997;119:3415. [Google Scholar]
- (67).Goldsmith CR, Cole AP, Stack TDP. J. Am. Chem. Soc. 2005;127:9904. doi: 10.1021/ja039283w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.