

Abstract
MUC1, a membrane tethered mucin glycoprotein, is overexpressed and aberrantly glycosylated in >80% of human ductal pancreatic adenocarcinoma. However, the role of MUC1 in pancreatic cancer has been elusive, partly due to the lack of an appropriate model. We report the characterization of a novel mouse model that expresses human MUC1 as a self molecule (PDA.MUC1 mice). Pancreatic tumors arise in an appropriate MUC1-tolerant background within an immune competent host. Significant enhancement in the development of pancreatic intraepithelial pre-neoplastic lesions (PanINs) and progression to adenocarcinoma is observed in PDA.MUC1 mice, possibly due to increased proliferation. Tumors from PDA.MUC1 mice express higher levels of cyclooxygenase-2 and indoleamine 2,3, dioxygenase compared to PDA mice lacking MUC1, especially during early stages of tumor development. The increased pro-inflammatory milieu correlates with an increased percentage of regulatory T cells and myeloid suppressor cells in the pancreatic tumor and tumor draining lymph nodes. Data shows that during pancreatic cancer progression, MUC1-mediated mechanisms enhance the onset and progression of the disease which in turn regulate the immune responses. Thus, the mouse model is ideally-suited for testing novel chemopreventive and therapeutic strategies against pancreatic cancer.
Introduction
Approximately 30,000 Americans develop pancreatic cancer each year and nearly as many die from the disease annually (1). Surgical resection remains the only potentially curative intervention for pancreatic cancer, but is contraindicated in most patients because their disease is either locally inoperable or metastatic at presentation (2). Among the minority of patients who undergo surgical resection, the median survival is only 20 months, with a 5-year survival rate of 8-20% (3). Despite some improvements in outcome, pancreas cancer remains a lethal diagnosis for the vast majority of patients. Greater understanding of the disease and development of new strategies to improve patient outcome are in dire need, but progress in these areas has been limited by the lack of an appropriate model that recapitulates the human disease.
Recently, a mouse model of pre-invasive and invasive ductal pancreatic cancer has been developed that recapitulates the full spectrum of human PanINs, putative precursors to pancreatic cancer (4). These mice, designated PDA, were generated using P48-Cre (5) to drive the KRASG12D mutation in pancreatic ductal precursor cells (4). We have further crossed the PDA mice to the human MUC1 transgenic (MUC1.Tg) (6) which express MUC1 in a pattern and level consistent with that in humans. These mice are called PDA.MUC1.
MUC1 is a highly glycosylated type I transmembrane glycoprotein (7) which is overexpressed in ∼70-80% PDA and elevated in the pancreatic juice of pancreatic cancer patients (8-11). MUC1 can function as an enhancer of tumor progression (12, 13), as an oncogene (14), and as a target for therapeutic intervention (7). The antigenic profile of MUC1 on malignant cells is different from normal cells due to changes in its glycosylation and expression levels, making MUC1 immunogenic in tumor-bearing hosts. Patients with pancreatic, breast, and ovarian tumors exhibit increased serum MUC1 levels and spontaneous immune responses including development of antibodies and T cells specific for MUC1 (15-19). Generation of the PDA.MUC1 mouse model that expresses human MUC1 as a self molecule enables examination of MUC1 function during pancreatic cancer progression and evaluation of novel MUC1-targeted immune therapies.
Immune-based therapies, though promising, have not been as successful as hoped, in part due to the immune evasion tactics employed by tumors to escape immune recognition and/or killing. One such evasion mechanism activated in pancreatic cancer is the arachidonic acid / cyclooxygenase 2 (COX-2) pathway (20). COX-2 is an enzyme that is induced during various pathologic conditions including inflammation and cancer; it converts arachidonic acid to prostaglandins. It is now well recognized that tumor-associated COX-2 and its product prostaglandin E2 (PGE2) are highly immunosuppressive. PGE2 directly downregulates cytotoxic T lymphocyte (CTL) and helper T lymphocyte (Th) functions (21, 22). In addition, PGE2 reverses the ability of dendritic cells (DCs) within tumors to effectively present antigens to T cells, inducing the generation of T regulatory cells (Tregs) and myeloid suppressor cells (MSCs) (23, 24).
We have recently shown that inhibiting COX-2 significantly enhances cancer vaccine efficacy by reducing the activity of another enzyme, indoleamine 2,3-dioxygenase (IDO), a major player in inducing immune tolerance (25). IDO catabolizes tryptophan to kynurenine (26) to create a tumor microenvironment that is dangerously low in tryptophan. Immune effector cells, in particular CTLs and Th cells, are highly sensitive to low tryptophan levels and fail to proliferate and function effectively (27-29); however, little is known about IDO function in pancreatic tumors. Herein, we use the PDA.MUC1 model to assess the role of MUC1 in immune modulation in the context of COX-2 and IDO activity in pancreatic tumorigenesis.
Materials and Methods
Generation of PDA.MUC1 mice
PDA mice were generated by breeding P48Cre-expressing mice obtained from Dr. Chris Wright (Vanderbilt University, Nashville, TN) to the LSL-KRASG12D mice obtained from Dr. Tyler Jacks (MIT, Boston, MA) (4); PDA mice were then mated to heterozygous human MUC1.Tg mice (6) (Figure 1). All mice used were congenic on the C57BL/6 background: MUC1.Tg and LSL-KRASG12D mice (30) were maintained as C57BL/6; the P48-Cre mice (5) were obtained on the FVB background and were backcrossed onto C57BL/6 for 10 generations. Genomic DNA was used to genotype the triple transgenic PDA.MUC1 mice by polymerase chain reaction. For KRASG12D, primers were: 5′ wildtype: GTCGACAAGCTCATGCGGGTG; 5′ mutant: AGCTAGCCACCATGGCTTGAGTAAGT CTGCG; and 3′ universal: CCTTTACAAGCGCACGCAGACTGTAGA, creating amplification products of 500 bp for the wildtype and 550 bp for the mutant allele. For P48Cre, the primers were: 5′: TGCTGTTTCACTGGTTATGCGG and 3′: TTGCCCCT GTTTCACTATCCAG with a 700 bp amplification product. For MUC1.Tg the primers were 5′-CTTGCCAGCCATAGCACCAAG-3′ and 5′-CTCCACGTCGTGGACATTGATG-3′ with a 341bp amplification product. Amplified products were confirmed on 1% agarose gels. All mouse protocols were conducted in accordance with stringent regulations laid out by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC).
Figure 1. Schematic representation of PDA and PDA.MUC1 mice.

PDA mice were mated to the human MUC1.Tg mice that were maintained as heterozygotes.
Evaluation and scoring of PanIN lesions
The entire pancreas was dissected free of fat and surrounding tissue, fixed in 10% neutral buffered formalin (pH 6.8-7.2), and embedded in paraffin. Serial sections (4 μm) were cut for histological and immunohistochemical studies. Mouse pancreatic tissue sections stained with hematoxylin and eosin were used for counting the PanINs. PanINs were scored in 5 consecutive sections per pancreas and 10 fields per section. To determine mucus accumulation, sections were stained with periodic acid–Schiff (PAS).
MUC1, COX-2, IDO, and Proliferating Cell Nuclear Antigen immunohistochemistry
For nonenzymatic antigen retrieval, sections were heated to 95°C in Dako antigen retrieval solution 20-40 min and cooled 20 min; all subsequent steps occurred at room temperature. To quench endogenous peroxidase, slides were rinsed and incubated in methanol / 2% H2O2 for 10 min. Sections were then washed, blocked in PBS / 0.1% BSA / 5% normal bovine serum for 45 min, and incubated overnight with primary antibodies. Sections were incubated 1 h with secondary antibody, developed with a diaminobenzidine (DAB) substrate (Vector, Inc.), counterstained with hematoxylin, and mounted with Permount. Primary antibodies used were: mouse anti-MUC1 tandem repeat (TR), BC2 (1:1000 dilution, kind gift from Dr. McGuckin, Queensland, Australia); Armenian hamster anti-MUC1 cytoplasmic tail (CT), CT2 (1:50, own), goat anti-COX-2 (1:100; Santa Cruz Inc.); goat anti-IDO (1:50; Santa Cruz Inc.), mouse anti-PCNA (5 ug/ml at 4°C, BD Biosciences, USA). Secondary antibodies were anti-mouse (1:100, Dako), anti-hamster (1:250, Jackson Labs), and anti-goat (1:100, Dako) IgGs conjugated to horseradish peroxidase. Immunopositivity was assessed using light microscopy and images taken at 100× or 200× magnification.
Cell Culture, Retroviral Infection, and siRNA Transfection
Human pancreatic cancer cell lines, BxPC3 and MiaPaCa2 cells (American Type Culture Collection) were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA, USA) plus 10% fetal calf serum, 1% Glutamax (Invitrogen) and 1% penicillin/streptomycin. The retroviral infection protocol was previously described (31). Briefly, GP2-293 packaging cells (stably expressing the gag and pol proteins) were cotransfected with full-length MUC1 and vector expressing the VSV-G envelope protein (BxPC3.MUC1), or vector alone (BxPC3.Neo). Stable cell lines were selected with 0.5 mg/ml G418 beginning 48 hours post infection, then sorted by flow cytometry. Two independent infections were done, with similar results. Transient siRNA transfection was performed with Lipofectamine2000 (Invitrogen) and 100 nM siRNA oligonucleotides as previously described (32-34). Briefly, MiaPaCa2 (high endogenous MUC1) cells were plated at 225,000 cells/well in 6-well plates and grown to 50% confluence. Cells were transfected with MUC1-specific (siGENOME smartpool), or scrambled or luciferase non-targeting (siCONTROL) siRNA (all from Dharmacon, Lafayette, CO, USA) according to manufacturer's instructions. Post-transfection, MUC1 protein expression was determined at 24, 48, 72, and 96h by flow cytometry and western blotting; MUC1 knockdown was maintained for at least 96h. Data are reported for 48h post siRNA treatment. FITC-conjugated anti-MUC1 (Pharmingen, San Diego, CA, clone HPMV) was used at 1ug/106 cells for flow cytometry.
Serum PGEM, MUC1 and anti-MUC1 antibody ELISA
PGE2 levels in the sera were assessed using a specific ELISA (Cayman Pharmaceuticals, Ann Arbor, MI) for the PGE2 metabolite (PGEM; 13,14-dihydro 15-keto prostaglandin A2), according to the manufacturer's recommended protocol. Results are expressed as pg PGEM / ml serum. Serum MUC1 levels were determined using the CA15-3 ELISA (Genway Biotech Inc. San Diego, CA (35)). Detection of antibody to MUC1 was carried out by a specific ELISA using BC2 (IgG) antibody that recognizes the extracellular MUC1 TR as the standard. The plate was coated with the 24-mer TAPPAHGVTSAPDTRPAPGSTAPP peptide as the capture antigen (6, 36).
Measurement of IDO activity by HPLC analysis of kynurenine and tryptophan
Using a published HPLC assay for IDO enzymatic activity measurement (37) as a starting point, we have optimized and validated a sensitive HPLC assay with UV and fluorescence detection that allows effective chromatographic separation of tryptophan and its metabolite, kynurenine, in serum and cell lysate. Briefly, 50 μl sample diluted in 150 μl PBS was added to 50 μl of the internal standard, 3-nitro-tyrosine (3-NT). Proteins were precipitated with 50 μl 30% trichloroacetic acid (TCA); samples were then spun at 14,000g for 5 min and 200 μl of supernatant transferred to glass tubes for HPLC analysis. All samples were run in duplicate. Calibrators were prepared and frozen in the same fashion as the samples to control for any errors in handling and/or metabolite degradation.
IFN-gamma ELISPOT
Cells from tumor draining lymph nodes (TDLNs) were isolated during tumor development and used as responders in an IFN-γ ELISPOT assay. The stimulators were irradiated autologous DCs prepared as previously described (38), and pulsed with either a human MUC1 peptide (for PDA.MUC1 mice), or a mouse Muc1 peptide (for PDA mice). The peptides used were: human MUC1 TR, STAPPAHGVTSAPDTRPAPGSTAPP; mouse Muc1 CT, SSLSYTNPAVAATSANL. A responder to stimulator ratio of 10:1 was used. Negative control wells contained T cells stimulated with DCs pulsed with an irrelevant peptide (vesicular stomatitis virus peptide, RGYKYQGL). Spot numbers were determined using computer assisted video image analysis by Zellnet Consulting, Inc. (Fort Lee, NJ). Splenocytes from C57BL/6 mice stimulated with concavalin A were used as a positive control.
Cytotoxic T - lymphocyte assay: 51Chromium-release assay
CTL activity was determined by a standard 51Cr-release method using T cells from TDLNs as effector cells and autologous irradiated DCs pulsed with MUC1 TR peptide (same as the ELISPOT) as stimulator cells. Effector and stimulator cells were co-incubated at a 10:1 ratio for 48 h; effectors were then recovered and incubated with 51Cr-labelled tumor target cells at a 50:1 ratio for 6 h. Target cells included the MUC1-negative melanoma cell line B16, transfected with either full-length MUC1 (B16.MUC1) or vector alone (B16.neo)(36). Target cells were treated with 5 ng/ml IFN-γ (Amersham, Piscataway, NJ) one day prior to the assay to up-regulate MHC class I surface expression and loaded with 100μCi 51Cr (Amersham) per 106 target cells for 3h prior to incubation with effectors. Radioactive 51Cr release was determined using the Topcount Micro-scintillation Counter (Packard Biosciences, Shelton, CT) and specific lysis was calculated: (experimental cpms - spontaneous cpms / complete cpms - spontaneous cpms) × 100. Spontaneous 51Cr release in all experiments was 10-15% of complete 51Cr release.
Isolation of tumor infiltrating lymphocytes and flow cytometry
The pancreas was dissected free of fat in DMEM complete medium, rinsed, and cut in small pieces in serum-free DMEM with 1 mg/ml collagenase. Tumor chunks were incubated at 37°C for 30 min, then mashed using frosted glass, filtered through a sieve, and collected. The tumor cells were washed twice in PBS and resuspended in 2 ml of serum-free DMEM. A percoll (Pharmacia, Sweden) gradient was prepared in a glass tube by layering 3 ml of 80% percoll, 3 ml of 40% Percoll, and finally 2 ml of cell suspension; this was spun at 2000 rpm for 20 min ambient without brakes. The cells suspended between 40% and 80% percoll were collected, washed twice with cold PBS, and resuspended in cold FACs buffer (PBS + 1% fetal bovine serum).
Flow cytometry antibodies included: for Tregs, APC-labeled anti-FoxP3 (ebiosciences, San Diego CA, clone FJK-16s), PE-labeled anti-CD25 (Pharmingen, San Diego, CA, clone pc-61), and FITC-labeled anti-CD4 (Pharmingen, clone GK1.5); for MSCs, FITC-labeled anti-CD11b (Pharmingen, clone M1/70) and PE-labeled anti-Gr1 (Pharmingen, clone RB6-8c5). Cells were acquired on a BD Pharmingen Cyan flow cytometer and analyzed with BD Biosciences FlowJo version 8. For Treg and MSC analyses, the lymphocyte and granulocyte/macrophage populations were gated, respectively.
Statistical Analysis
All statistical analyses were performed by the Mayo Clinic Biostatistics Core Facility. A two-factor ANOVA was used to determine significant differences between experimental groups. For the ELISPOT analysis, data were adjusted for operator (different days at which assays were conducted).
Results
Generation of the PDA.MUC1 mouse
To create the PDA.MUC1 line, PDA mice were mated to heterozygous human MUC1.Tg mice (6)(Figure 1), which express human MUC1 in addition to the endogenous mouse Muc1. MUC1.Tg exhibit B- and T-cell compartment tolerance and are refractory to immunization with MUC1(6). Since the human transgene is driven by its own promoter, MUC1 expression levels are tissue-specific and appropriate.
Significantly enhanced tumor development in PDA.MUC1
Both PDA and PDA.MUC1 mice developed progressive PanINs ranging from PanIN-IA to PanIN-3, eventually resulting in adenocarcinoma (Figure 2). However, the kinetics of PanIN development and progression differed significantly between the two lines. In PDA.MUC1 mice, PanIN lesions appeared as early as 6 weeks, though none were detected in age-matched PDA mice (Figure 3A). Larger numbers and higher grade of PanINs continued throughout tumor progression in PDA.MUC1 mice (Figures 3A-E). At 26 weeks, invasive adenocarcinomas were observed in 8 out of 10 PDA.MUC1 mice, whereas only 1 out of 10 PDA mice had invasive disease at this age (Figure 3F). In agreement with this, PDA.MUC1 mice had significantly greater pancreas weights compared to PDA mice at all timepoints (Figure 3G). Tumor metastasis was also more prevalent in the PDA.MUC1: at 34 weeks 4 of 10 PDA.MUC1 mice had lung and liver metastases as compared to 0 of 10 PDA mice; by 48 weeks, 6 of 10 PDA.MUC1 had metastases compared to only 1 of 10 PDA (Figure 4C and data not shown).
Figure 2. Histology of normal pancreas and histopathology of PanIN lesions and adenocarcinoma in pancreas from PDA.MUC1 mice.
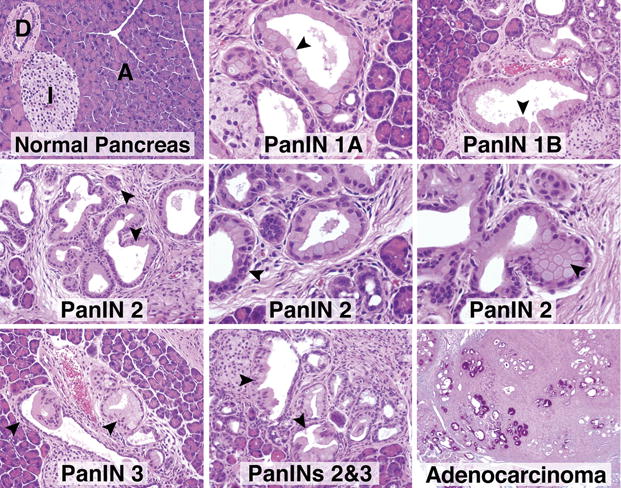
Hematoxylin and eosin (H&E) staining of pancreas tissues from PDA.MUC1 mice. Images are captured at 200× magnification. Representative images are shown depicting the various stages of PanIN lesions and adenocarcinoma. D: duct; I: Islet; A: acinar. Arrows indicate PanINs lesions. Normal pancreas is from a 24-week old MUC1.Tg mouse.
Figure 3. Enhanced progression of tumors in PDA.MUC1 mice.

A-E) Panels showing average number of PanINs in 5 consecutive sections and 10 fields per section from each mouse. Average of 10 mice per time point is shown. F) Numbers of mice (out of 10 per timepoint) with invasive adenocarcinoma. G) Wet weight of pancreas at each timepoint. *p<0.01; **p<0.0001 as compared to PDA mice.
Figure 4. Enhanced PanIN and adenocarcinoma development with liver and lung metastasis in PDA.MUC1.
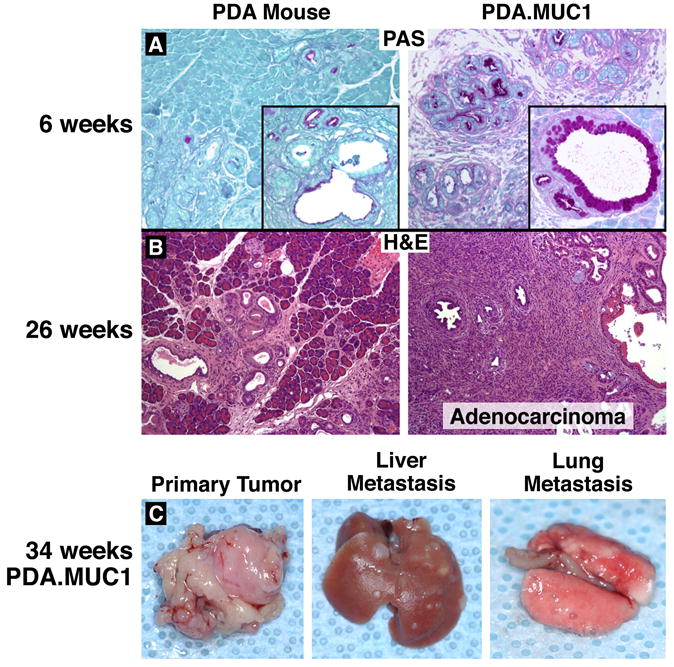
A) Mucus accumulation in PanIN lesions as determined by periodic acid Schiff reaction (purple staining) in pancreas tissues from 6-week-old mice. B) At 26 weeks, PDA.MUC1 mice develop adenocarcinoma (H&E) at which time no adenocarcinoma is observed in the PDA mice. C) Representative 200× images of a primary pancreas tumor, liver, and lung metastases in PDA.MUC1 mice. N=15 mice per time point have been evaluated with similar results.
Greater MUC1 expression and mucus accumulation in PDA.MUC1 mice
Analysis of PanIN lesions using the periodic acid-Schiff (PAS) mucus stain revealed much more mucus accumulation in the PDA.MUC1 pancreas compared to PDA pancreas (Figure 4A). PAS-positive PanINs are generally more aggressive with increased proliferation; (4)these were detected as early as 6 weeks of age in the PDA.MUC1 mice. Similarly, invasive adenocarcinoma was clearly visible in the PDA.MUC1 pancreas, whereas age-matched PDA pancreas did not display the same degree of invasiveness (Figure 4B).
It is known that high MUC1 expression correlates with a highly proliferative and metastatic tumor phenotype in human pancreatic cancer(9, 39). As illustrated in Figure 5, both mouse Muc1 and human MUC1 levels increase with tumor progression in PDA and PDA.MUC1 mice, recapitulating the human disease. However, the kinetics of Muc1/MUC1 expression differ greatly between the lines: Muc1/MUC1 expression is detected much earlier (6 weeks) in PDA.MUC1, correlating with the earlier appearance of PanIN lesions (Figures 5B and C, Figure 3). In general, pancreas sections from PDA.MUC1 mice express higher levels of Muc1/MUC1 as detected by the antibody CT2 that recognizes both the mouse and human forms (Figure 5A and B). This is especially notable at early time points (≤26 weeks). Human MUC1 expression is also increased in the tumors, as compared to the non-tumorigenic MUC1.Tg mice whose pancreas tissues are histologically normal with low-level MUC1 staining (Figure 5D).
Figure 5. Muc/MUC1 expression increases with tumor progression.
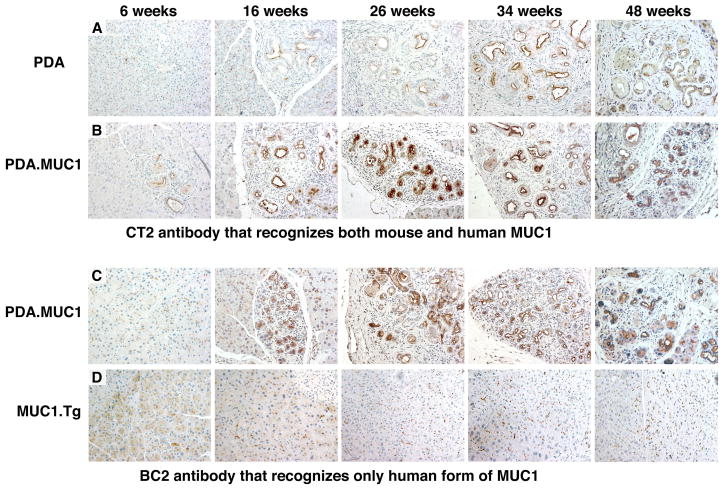
Staining was done using A) CT2, a monoclonal antibody that recognizes the cytoplasmic tail of both mouse Muc1 and human MUC1, and B) BC2, a monoclonal antibody directed against the tandem repeat of only human MUC1. Representative images were captured at 200× magnification. N=15 mice per time point have been evaluated with similar results. For comparison, pancreas sections from age-matched MUC1.Tg mice are shown.
Increased proliferation in PDA.MUC1 tumors
High mucus accumulation and high MUC1 expression correlate with a highly proliferative tumor cell phenotype (4, 9, 39). We therefore examined the expression of proliferating cell nuclear antigen (PCNA), a marker of proliferative cells. Pancreas sections from PDA.MUC1 mice showed considerably higher levels of PCNA-positive cells than do PDA tissues (Figures 6A and B). Pancreas sections from age-matched MUC1.Tg mice are shown for comparison (Figure 6C).
Figure 6. Greater PCNA positivity in the PDA.MUC1 pancreas.
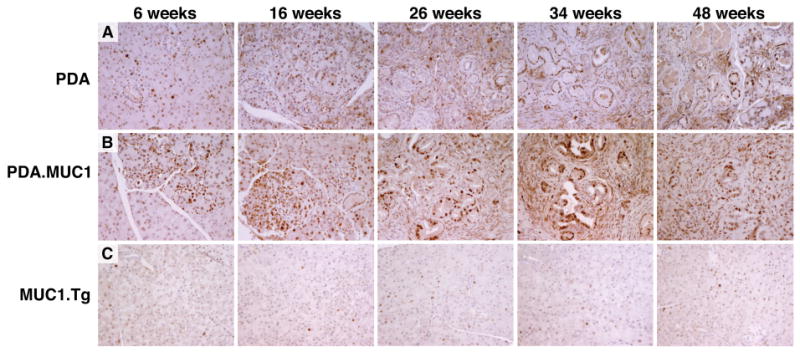
Representative sections were stained with PCNA to assess level of proliferative tumor cells in A) PDA mice, B) PDA.MUC1 mice, and C) MUC1.Tg mice. Dark brown nuclear staining represents proliferating cells. N=15 mice have been assessed giving similar results. Images were captured at 200× magnification.
Increased circulating MUC1 but stable anti-MUC1 levels with tumor progression
A considerable rise in circulating MUC1 levels is observed at each timepoint in PDA.MUC1 mice as PanINs progress to adenocarcinomas (Figure 7A). In contrast, MUC1 levels were undetectable in the non-tumor-bearing MUC1.Tg mice (data not shown). Similarly, circulating anti-MUC1 IgG levels were significantly higher in PDA.MUC1 mice than that found in MUC1.Tg mice (161 ± 45 ug/ml versus <20 ± 10 ug/ml, respectively). However, the antibody levels did not increase significantly with tumor progression (data not shown).
Figure 7. Circulating MUC1 levels and MUC1-specific immune responses.

Circulating MUC1 levels were determined in serum of PDA.MUC1 mice by ELISA Average of n=10 mice are shown. *p<0.001 and **p<0.0001 compared to levels in 6 and 16 week old mice. B) Cells from TDLNs were assessed for IFN-γ production in response to human MUC1 or mouse Muc1 peptides by ELISPOT Average of n=10 mice are shown. *p<0.05 at 6 weeks; **p<0.001 at 16 weeks compared to PDA mice. C) Determination of CTL activity was performed using a standard 51Cr-release method. T cells from TDLNs served as effector cells, autologous irradiated DCs pulsed with MUC1 TR peptide were used as stimulator cells and targets were B16.MUC1 cells (o) or B16.neo cells (x). Data from individual mice are shown. *p<0.001 and **p<0.0001 as compared to previous timepoint.
Early detection of naturally occurring MUC1-specific T cell responses
T cells from TDLNs of PDA and PDA.MUC1 mice were analyzed for MUC1-specific immune responses, namely producing IFN-γ in response to MUC1 antigen and killing MUC1-expressing tumor cells. In both lines, high levels of MUC1-specific IFN-γ-spot producing cells were present at early stages (6-16 weeks), but decreased progressively at later timepoints (Figure 7B). Note that the IFN-γ production is more dramatic in PDA mice; this may reflect differential response between the mouse Muc1 peptide use as a stimulant in the PDA assay as compared to the human MUC1 peptide used for the PDA.MUC1 assay. Similar to the decline in IFN-γ, specific CTL-mediated lysis of MUC1-expressing tumor cells (B16.MUC1) was seen at early times during PanIN development (6-16 weeks) but disappeared by 26 weeks of age (Figure 7C). PDA.MUC1 CTLs did not kill MUC1-negative B16.neo cells; CTL activity was not analyzed for the PDA mice due to lack of a relevant tumor target.
MUC1-associated augmentation of COX-2/PGE2 and IDO/kynurenine pathways
Studies indicate that certain forms of tumor-associated MUC1 are immunosuppressive to T cell and DC function(40, 41). To determine the mechanism of immune regulation by MUC1, we assessed some of the known immune-regulatory pathways activated during carcinogenesis. In PDA.MUC1 mice, expression of both COX-2 and IDO increases with age; the highest expression is observed at 48 weeks when almost all mice have developed adenocarcinoma (Figures 8A and B). COX-2 and IDO expression levels were comparatively lower in PDA pancreatic tumors (data not shown) and absent in the MUC1.Tg normal pancreas (top left panels, Figures 8A and B).
Figure 8. COX-2 and IDO activity is enhanced in PDA.MUC1 mice.

A) COX-2 staining and B) IDO staining of pancreas from various ages of PDA.MUC1 mice. For comparison, MUC1.Tg pancreas from a 36-week old mouse is shown. Images were captured at 200× magnification. Brown staining represents COX-2 or IDO positivity. N=15 mice per time point have been evaluated with similar results. Serum levels of C) PGEM; D) kynurenine; and E) tryptophan in PDA, PDA.MUC1 and MUC1.Tg mice at various ages. An average of ten mice is shown with p-values calculated in comparison with MUC1.Tg mice (control). *p<0.01;**p<0.0001 compared to MUC1.Tg control values.
To confirm the effect of MUC1 on COX-2 and IDO activities, we analyzed three measures of these tumor-enhancing pathways. The first, PGE2 metabolite (PGEM), is a marker of the COX-2 product PGE2 and was significantly higher in the serum of PDA.MUC1 mice as compared to PDA mice, especially at later timepoints (Figure 8C, p=0.05 for 26 weeks, and p<0.001 for 34 and 48 weeks). IDO catabolizes tryptophan to kynurenine, thus high kynurenine and low tryptophan levels are indicative of IDO activity. Significantly higher levels of serum kynurenine were detected in PDA.MUC1 mice compared to PDA mice (Figure 8D, p=0.005 for 26 weeks and p=0.0001 for 34 and 48 weeks). Circulating kynurenine was significantly higher in PDA.MUC1 mice than in non-tumor-bearing MUC1.Tg by 16 weeks (p<0.005 for 16 weeks, p<0.0001 for ≥26 weeks). In contrast, the levels of kynurenine in the PDA mice were not significantly altered compared to MUC1.Tg (Figure 8D). In agreement with the kynurenine results, circulating tryptophan levels were significantly decreased in PDA.MUC1 mice at all time points as compared to MUC1.Tg (Figure 8E, p<0.0001 for all), whereas in PDA mice, the decrease in tryptophan levels was significant only at later stages (p<0.05 for 26 weeks, and p<0.0001 for 34 and 48 weeks). Importantly, PDA.MUC1 mice had significantly lower levels of tryptophan compared to PDA mice (p<0.01 for 26 weeks and p<0.001 for 34, and 48 weeks), suggesting increased IDO activity in the MUC1-expressing tumors.
To further analyze if MUC1 regulates COX-2 and IDO function, we stably infected a MUC1-negative human pancreatic cancer cell line, BxPC3, with full-length MUC1. Two separate stable clones were generated; both culture supernatants and cell lysates were analyzed for PGEM, kynurenine, and tryptophan levels. To complement these studies, MUC1 was depleted from a strongly MUC1-positive human pancreatic cancer cell line, MiaPaCa2, using MUC1-specific siRNA (33). Supernatant and lysates from these cells were analyzed 48h after MUC1 knockdown. BxPC3 cells expressing MUC1 had significantly increased PGEM and kynurenine levels and decreased tryptophan levels compared to wildtype or vector-infected BxPC3 (Table 1). In line with this, knockdown of MUC1 from MiaPaCa2 cells resulted in significantly lower PGEM and kynurenine, with increased tryptophan (Table 1).
Table 1. Levels of PGEM, kynurenine, and tryptophan in BxPC3 and MiaPaCa2 human pancreatic cancer tumor cell lines.
Results are shown for uninfected (wildtype), vector- (neo) or MUC1-infected BxPC3 cells, and for untransfected (wildtype), control siRNA- (luciferase) or MUC1 siRNA-transfected MiaPaCa2 cells. Average values from n=3 samples in 2 separate experiments are reported. For MiaPaCa2 cells, all data are reported for 48h post siRNA treatment. Similar results were obtained with 72h post treatment. * Represents significant difference between MUC1-expressing and MUC1-depleted cells from wildtype and control cells.
| Pancreatic Cancer Cell lines | Percent MUC1+ cells by flow cytometry | PGEM levels in supernatant
(ngs/ml) |
PGEM levels in lysate
(ngs/ml) |
Tryptophan levels in lysate
(ugs/ml) |
Kynurenine levels in lysate
(ugs/ml) |
|---|---|---|---|---|---|
| BxPC3
(wildtype) |
<0.5 | 255 ± 30 | 1025 ± 112 | 18.3 ± 5.5 | 312 ± 35 |
| BxPC3
(neo) |
<1 | 312 ± 55 | 1242 ± 96 | 15.6 ± 6.8 | 437 ± 52 |
| BxPC3
(MUC1) |
85* | 623 ± 73* | 4037 ± 134* | 6.2 ± 2.6* | 865 ± 105* |
| MiaPaCa
(wildtype) |
95 | 1025 ± 213 | 2456 ± 421 | 29.1 ± 6.3 | 1022 ± 112 |
| MiaPaCa
(luciferase siRNA) |
93
(48h post siRNA) |
1546 ± 178 | 3021 ± 265 | 33.8 ± 12.1 | 1189 ± 216 |
| MiaPaCa
(MUC1siRNA) |
<10*
(48h post siRNA) |
735 ± 98* | 964 ± 123* | 10.3 ± 4.8* | 3045 ± 534* |
Increased expression of Treg and MSC in the PDA.MUC1 pancreas
PGE2 is known to induce the generation of Tregs (23) and recruitment of MSCs within the tumor (24). Therefore, we tested the levels of these immune regulatory cells within the tumor microenvironment and in TDLNs. The PDA.MUC1 mice exhibit significantly higher numbers of Tregs (CD4+/CD25+/FoxP3+ cells) in the tumor and in the TDLNs compared to PDA mice at all ages (Figure 9A, p<0.03, p<0.01 and p<0.05 for 6, 26, and 34-48 weeks, respectively). Correspondingly, MSCs (CD11b+/Gr1+ cells, also defined as Mac1+/Ly6G+/PDL1+) were significantly higher in the tumors of PDA.MUC1 mice compared to tumors from PDA mice (Figure 9B, p<0.0001 for 26-34 weeks).
Figure 9. Increased percent of Tregs and MSCs in pancreas and TDLNs of PDA.MUC1 mice.

Flow cytometric analysis evaluating percentages of CD4+/FOXP3+/CD25+ Tregs in A) tumor and B) TDLNs of the PDA.MUC1 and PDA mice as a function of age. N=10 mice per time were evaluated. Significant increase (*p<0.05; **p<0.01; and ***p<0.001) in Tregs are noted. Representative images are shown of i) forward and side scatter; box represents the gate around the lymphocyte population; ii) CD4+ T cells on the gated lymphocyte population, and iii) FL1 and FL2 scatter plot; box represents the CD25+ and FoxP3+ double positive cells gated on the CD4+ T cell population. C) Flow cytometric analysis evaluating percentages of CD11b+/Gr1+ MSCs in tumors of the PDA.MUC1 and PDA mice as a function of age. N=10 mice per time are evaluated. **p<0.0001 at 26 weeks; *p<0.01 at 34 and 48 weeks. Representative images are shown for i) forward and side scatter of the entire pancreas; box represents the gate around the granulocyte population; ii) CD11b+/Gr1+ double positive cells gated on the granulocyte population.
Discussion
Many studies have attempted to elucidate the role of MUC1 in pancreatic cancer progression and explore MUC1 as a target for therapeutic intervention, but lack of appropriate models have made this challenging. We describe a model of spontaneous pancreatic adenocarcinoma that expresses human MUC1 as a self molecule. This mouse model is unique in that the pancreatic tumor arises spontaneously in an appropriate tissue background, within a suitable stromal and hormonal milieu, and in the context of MUC1 tolerance and a viable immune system.
We report that the presence of human MUC1 in the PDA mice significantly enhances the development of PanINs and progression to adenocarcinoma in the presence of KRAS mutation. Muc1/MUC1 expression and mucus accumulation in the PDA.MUC1 pancreas was significantly higher than in PDA mice, a clinically significant observation as higher expression of MUC1 has been associated with greater aggressiveness of PanINs and poorer overall survival in pancreatic cancer (4, 10, 42-45). These findings correlated with the severity of the disease: 80% of PDA.MUC1 mice developed invasive adenocarcinoma by 26 weeks with greater proliferation in situ; in contrast, only 10% of PDA mice developed adenocarcinoma. The results strongly implicate MUC1 as an enhancer of PanIN progression and development of invasive adenocarcinoma in the setting of KRAS mutation.
Circulating MUC1 levels in the PDA.MUC1 mice increased with tumor progression, supporting the ability of the model to recapitulate the human disease. This suggests that the PDA.MUC1 model may be an appropriate setting for exploring the use of serum MUC1 as a prognostic and diagnostic marker for pancreatic cancer. In the past, antibodies to MUC1 have not been specific enough to differentiate aberrantly-glycosylated, tumor-derived MUC1 from other sources of elevated MUC1 such as pancreatitis. However, some success has been shown recently using a PAM4-based immunoassay for circulating MUC1 in diagnosis of pancreatic cancer (46); such assays warrant further investigation in preclinical models.
The PDA.MUC1 model offers an appropriate system to study anti-MUC1 immune responses and MUC1-associated immunosuppression during progression to invasive adenocarcinoma. Robust MUC1-specific T cell responses were detected at early time points. This ties in well with previous studies showing that, though MUC1.Tg non-tumorigenic animals are tolerant to MUC1, very early changes in submicroscopic lesions drive MUC1-specific immune responses, likely through aberrant glycosylation of MUC1. However, anti-MUC1 responses diminished over time, suggesting the existence of immunosuppression with tumor progression. This is supported by a different model of spontaneous pancreatic cancer of acinar origin (36) in which MUC1-specific T cell responses were observed early but not late in oncogenesis. MUC1-specific CTLs from the acinar model were subsequently cloned and used successfully in adoptive transfer experiments (36, 47). The high levels of Tregs and MSCs in the PDA.MUC1 tumors may contribute to the reduction in MUC1-specific immune responses at later times. In humans, MUC1-specific responses have been detected in early-stage cancer patients (15-17, 48), but as in the mouse models, anti-MUC1 immunity in humans does not result in anti-tumor immunity, providing evidence of immunosuppression (49, 50). These immunological characteristics lend credence to the PDA.MUC1 model and create an opportunity to study mechanisms of enhancing pre-existing anti-MUC1 immune responses against the growing tumor in a MUC1-tolerant host.
In addition, mucins produced by cancer cells play a critical role in the induction of COX-2 in the tumor microenvironment (51, 52). Tumor-associated carbohydrate antigens and simple mucin-type O-glycans such as Tn and sialyl-Tn antigens (which may be found on MUC1) correlated with COX-2 overexpression and low CD8+ T cell infiltration in endometrial cancer; strong expression of sialyl-Tn was associated with poor prognosis (52-54). However, few reports address MUC1 as an immune modulator within the pancreatic tumor microenvironment. We show that PDA.MUC1 tumors have higher COX-2 and IDO activity than PDA tumors, possibly a result of MUC1 enhancing tumorigenicity and/or accumulation of acidic mucins. COX-2 and IDO are major players not only in immune tolerance but also in tumor progression, metastasis, and angiogenesis. Thus, it is feasible that MUC1 expression may contribute towards a highly tolerogenic tumor microenvironment by influencing the COX-2/PGE2 and the IDO/tryptophan pathways. We recognize that the effect of MUC1 may not be direct and that increased COX-2 and IDO activities may themselves enhance MUC1 expression.
Taken together, the data suggest that MUC1-specific immune responses are present early in tumor development and are eventually suppressed, possibly by the growing tumor. Immune suppression during tumor progression is a major impediment to successful immune therapies, but the importance of MUC1 remains unclear. It is known that as tumors progress, neutral mucins are replaced by acidic mucins which negatively regulate T cells and DCs to favor tumor cell proliferation (40, 52, 55-59). We have preliminary evidence for the increased accumulation of acidic mucins in PDA.MUC1 tumors compared to PDA, though further analysis is needed. Thus tumor-associated MUC1 may suppress immunity in part through accumulation of acidic mucins.
These studies demonstrate that MUC1 speeds development of PanIN lesions and fosters progression to adenocarcinoma in the presence of oncogenic KRAS. It is clear that MUC1 by itself (in the MUC1.Tg mice) does not initiate PanIN lesions or adenocarcinoma, suggesting that MUC1 may play a role in immune modulation during existing oncogenesis. This makes MUC1 an even more attractive target for immune-based therapies for pancreatic cancer. To further elucidate the role of MUC1 in this setting, we are currently creating the PDA model in a Muc1 null background. These models will advance our understanding of the importance of MUC1 in immune regulation in pancreatic cancer, and may foster use of MUC1 in diagnosis and therapy for this devastating disease.
Acknowledgments
We thank Dr. Sandra Gendler for gifting the MUC1.Tg mice and the MUC1-specific antibodies as well as for her critical comments; Dr. Tyler Jacks and Chris Wright for their willingness to discuss and share the LSL-KRasG12D and P24-Cre mice; our pathologists, Dr. Giovanni De Petris and Dr. R. Marler, for their critical comments on the histology slides; August J. Klug for his contributions to genotyping; all members in the animal, histology, and biostatistics cores; Dr. Christine Hattrup for her help with editing the manuscript; Nichole Boruff in the visual communications core for her help with the preparation of the figures; and Frances Deutschmann for her help in submitting this manuscript.
R01 CA118944; P50 CA102701; AACR/PanCan-Pilot Award, and The Mayo Foundation.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985-1995, using the National Cancer Database. J Am Coll Surg. 1999;189:1–7. doi: 10.1016/s1072-7515(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 3.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, Falconi M, Pederzoli P, Pap A, Spooner D, Kerr DJ, Buchler MW. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–1210. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 4.Hingorani SR, Petricoin EF, III, Maitra A, Rajapaske V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt MA, Jacks T, Wright CV, Hruban R, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 5.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 6.Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998;58:315–321. [PubMed] [Google Scholar]
- 7.Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, Pemberton L, Lalani EN, Wilson D. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem. 1990;265:15286–15293. [PubMed] [Google Scholar]
- 8.Gendler SJ. MUC1, the renaissance molecule. J Mammary Gland Biol Neoplasia. 2001;6:339–353. doi: 10.1023/a:1011379725811. [DOI] [PubMed] [Google Scholar]
- 9.Levi E, Klimstra DS, Andea A, Basturk O, Adsay NV. MUC1 and MUC2 in pancreatic neoplasia. J Clin Pathol. 2004;57:456–462. doi: 10.1136/jcp.2003.013292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chhieng DC, Benson E, Eltoum I, Eloubeidi MA, Jhala N, Jhala D, Siegal GP, Grizzle WE, Manne U. MUC1 and MUC2 expression in pancreatic ductal carcinoma obtained by fine-needle aspiration. Cancer. 2003;99:365–371. doi: 10.1002/cncr.11857. [DOI] [PubMed] [Google Scholar]
- 11.Gronborg M, Bunkenborg J, Kristiansen TZ, Jensen ON, Yeo CJ, Hruban RH, Maitra A, Goggins MG, Pandey A. Comprehensive Proteomic Analysis of Human Pancreatic Juice. J Proteome Res. 2004;3:1042–1055. doi: 10.1021/pr0499085. [DOI] [PubMed] [Google Scholar]
- 12.Kohlgraf KG, Gawron AJ, Higashi M, Meza JL, Burdick MD, Kitajima S, Kelly DL, Caffrey TC, Hollingsworth MA. Contribution of the MUC1 tandem repeat and cytoplasmic tail to invasive and metastatic properties of a pancreatic cancer cell line. Cancer Res. 2003;63:5011–5020. [PubMed] [Google Scholar]
- 13.Spicer AP, Rowse GJ, Lidner TK, Gendler SJ. Delayed mammary tumor progression in Muc-1 null mice. J Biol Chem. 1995;270:30093–30101. doi: 10.1074/jbc.270.50.30093. [DOI] [PubMed] [Google Scholar]
- 14.Schroeder JA, Al Masri A, Adriance MC, Tessier JC, Kotlarczyk KL, Thompson MC, Gendler SJ. MUC1 overexpression results in mammary gland tumorigenesis and prolonged alveolar differentiation. Oncogene. 2004;23:5739–5747. doi: 10.1038/sj.onc.1207713. [DOI] [PubMed] [Google Scholar]
- 15.Barnd DL, Lan MS, Metzgar RS, Finn OJ. Specific, major histocompatibility complex-unrestricted recognition of tumor-associated mucins by human cytotoxic T cells. Proc Natl Acad Sci U S A. 1989;86:7159–7163. doi: 10.1073/pnas.86.18.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ioannides CG, Fisk B, Jerome KR, Irimura T, Wharton JT, Finn OJ. Cytotoxic T cells from ovarian malignant tumors can recognize polymorphic epithelial mucin core peptides. J Immunol. 1993;151:3693–3703. [PubMed] [Google Scholar]
- 17.Jerome KR, Domenech N, Finn OJ. Tumor-specific cytotoxic T cell clones from patients with breast and pancreatic adenocarcinoma recognize EBV-immortalized B cells transfected with polymorphic epithelial mucin complementary DNA. J Immunol. 1993;151:1654–1662. [PubMed] [Google Scholar]
- 18.Rughetti A, Turchi V, Ghetti CA, Scambia G, Panici PB, Roncucci G, Mancuso S, Frati L, Nuti M. Human B-cell immune response to the polymorphic epithelial mucin. Cancer Res. 1993;53:2457–2459. [PubMed] [Google Scholar]
- 19.Kotera Y, Fontenot JD, Pecher G, Metzgar RS, Finn OJ. Humoral immunity against a tandem repeat epitope of human mucin MUC-1 in sera from breast, pancreatic, and colon cancer patients. Cancer Res. 1994;54:2856–2860. [PubMed] [Google Scholar]
- 20.Juuti A, Louhimo J, Nordling S, Ristimaki A, Haglund C. Cyclooxygenase-2 expression correlates with poor prognosis in pancreatic cancer. J Clin Pathol. 2006;59:382–386. doi: 10.1136/jcp.2005.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okuno K, Jinnai H, Lee YS, Nakamura K, Hirohata T, Shigeoka H, Yasutomi M. A high level of prostaglandin E2 (PGE2) in the portal vein suppresses liver-associated immunity and promotes liver metastases. Surg Today. 1995;25:954–958. doi: 10.1007/BF00312380. [DOI] [PubMed] [Google Scholar]
- 22.Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA, Jr, Libby P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem. 2002;277:44147–44154. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- 23.Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006;16:38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–625. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 25.Basu GD, Tinder TL, Bradley JM, Tu T, Hattrup CL, Pockaj BA, Mukherjee P. Cyclooxygenase-2 Inhibitor Enhances the Efficacy of a Breast Cancer Vaccine: Role of IDO. J Immunol. 2006;177:2391–2402. doi: 10.4049/jimmunol.177.4.2391. [DOI] [PubMed] [Google Scholar]
- 26.Kai S, Goto S, Tahara K, Sasaki A, Kawano K, Kitano S. Inhibition of indoleamine 2,3-dioxygenase suppresses NK cell activity and accelerates tumor growth. J Exp Ther Oncol. 2003;3:336–345. doi: 10.1111/j.1533-869x.2003.01108.x. [DOI] [PubMed] [Google Scholar]
- 27.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 28.Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med. 2004;10:15–18. doi: 10.1016/j.molmed.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 30.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson EJ, Shanmugam K, Hattrup CL, Kotlarczyk KL, Gutierrez A, Bradley JM, Mukherjee P, Gendler SJ. Tyrosines in the MUC1 cytoplasmic tail modulate transcription via the extracellular signal-regulated kinase 1/2 and nuclear factor-kappaB pathways. Mol Cancer Res. 2006;4:489–497. doi: 10.1158/1541-7786.MCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 32.Basu GD, Liang WS, Stephan DA, Wegener LT, Conley CR, Pockaj BA, Mukherjee P. A novel role for cyclooxygenase-2 in regulating vascular channel formation by human breast cancer cells. Breast Cancer Res. 2006;8:R69. doi: 10.1186/bcr1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hattrup CL, Gendler SJ. MUC1 alters oncogenic events and transcription in human breast cancer cells. Breast Cancer Res. 2006;8:R37. doi: 10.1186/bcr1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukherjee P, Tinder TL, Basu GD, Gendler SJ. MUC1 (CD227) interacts with lck tyrosine kinase in Jurkat lymphoma cells and normal T cells. J Leukoc Biol. 2005;77:90–99. doi: 10.1189/jlb.0604333. [DOI] [PubMed] [Google Scholar]
- 35.Gion M, Mione R, Leon AE, Dittadi R. Comparison of the diagnostic accuracy of CA27.29 and CA15.3 in primary breast cancer. Clin Chem. 1999;45:630–637. [PubMed] [Google Scholar]
- 36.Mukherjee P, Ginardi AR, Madsen CS, Sterner CJ, Adriance MC, Tevethia MJ, Gendler SJ. Mice with spontaneous pancreatic cancer naturally develop MUC1-specific CTLs that eradicate tumors when adoptively transferred. J Immunol. 2000;165:3451–3460. doi: 10.4049/jimmunol.165.6.3451. [DOI] [PubMed] [Google Scholar]
- 37.Torres MI, Lopez-Casado MA, Lorite P, Rios A. Tryptophan metabolism and indoleamine 2,3-dioxygenase expression in coeliac disease. Clin Exp Immunol. 2007;148:419–424. doi: 10.1111/j.1365-2249.2007.03365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukherjee P, Madsen CS, Ginardi AR, Tinder TL, Jacobs F, Parker J, Agrawal B, Longenecker BM, Gendler SJ. Mucin 1-specific immunotherapy in a mouse model of spontaneous breast cancer. J Immunother. 2003;26:47–62. doi: 10.1097/00002371-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, Yeo CJ, Hruban RH. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902–912. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 40.Agrawal B, Krantz MJ, Reddish MJ, Longenecker BM. Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nature Medicine. 1998;4:43–49. doi: 10.1038/nm0198-043. [DOI] [PubMed] [Google Scholar]
- 41.Chan AK, Lockhart DC, von Bernstorff W, Spanjaard RA, Joo HG, Eberlein TJ, Goedegebuure PS. Soluble MUC1 secreted by human epithelial cancer cells mediates immune suppression by blocking T-cell activation. Int J Cancer. 1999;82:721–726. doi: 10.1002/(sici)1097-0215(19990827)82:5<721::aid-ijc16>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 42.Hinoda Y, Ikematsu Y, Horinochi M, Sato S, Yamamoto K, Nakano T, Fukui M, Suehiro Y, Hamanaka Y, Nishikawa Y, Kida H, Waki S, Oka M, Imai K, Yonezawa S. Increased expression of MUC1 in advanced pancreatic cancer. J Gastroenterol. 2003;38:1162–1166. doi: 10.1007/s00535-003-1224-6. [DOI] [PubMed] [Google Scholar]
- 43.Nassar H, Pansare V, Zhang H, Che M, Sakr W, Ali-Fehmi R, Grignon D, Sarkar F, Cheng J, Adsay V. Pathogenesis of invasive micropapillary carcinoma: role of MUC1 glycoprotein. Mod Pathol. 2004;17:1045–1050. doi: 10.1038/modpathol.3800166. [DOI] [PubMed] [Google Scholar]
- 44.Giorgadze TA, Peterman H, Baloch ZW, Furth EE, Pasha T, Shiina N, Zhang PJ, Gupta PK. Diagnostic utility of mucin profile in fine-needle aspiration specimens of the pancreas: an immunohistochemical study with surgical pathology correlation. Cancer. 2006;108:186–197. doi: 10.1002/cncr.21913. [DOI] [PubMed] [Google Scholar]
- 45.Moniaux N, Andrianifahanana M, Brand RE, Batra SK. Multiple roles of mucins in pancreatic cancer, a lethal and challenging malignancy. Br J Cancer. 2004;91:1633–1638. doi: 10.1038/sj.bjc.6602163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gold DV, Modrak DE, Ying Z, Cardillo TM, Sharkey RM, Goldenberg DM. New MUC1 serum immunoassay differentiates pancreatic cancer from pancreatitis. J Clin Oncol. 2006;24:252–258. doi: 10.1200/JCO.2005.02.8282. [DOI] [PubMed] [Google Scholar]
- 47.Mukherjee P, Ginardi AR, Tinder TL, Sterner CJ, Gendler SJ. MUC1-specific CTLs eradicate tumors when adoptively transferred in vivo. Clin Can Res. 2001;7:848s–855s. [PubMed] [Google Scholar]
- 48.Jerome KR, Barnd DL, Bendt KM, Boyer CM, Taylor-Papadimitriou J, McKenzie IF, Bast RC, Jr, Finn OJ. Cytotoxic T-lymphocytes derived from patients with breast adenocarcinoma recognize an epitope present on the protein core of a mucin molecule preferentially expressed by malignant cells. Cancer Res. 1991;51:2908–2916. [PubMed] [Google Scholar]
- 49.Schmitz-Winnenthal FH, Volk C, Z'Graggen K, Galindo L, Nummer D, Ziouta Y, Bucur M, Weitz J, Schirrmacher V, Buchler MW, Beckhove P. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65:10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 50.Hamanaka Y, Suehiro Y, Fukui M, Shikichi K, Imai K, Hinoda Y. Circulating anti-MUC1 IgG antibodies as a favorable prognostic factor for pancreatic cancer. Int J Cancer. 2003;103:97–100. doi: 10.1002/ijc.10801. [DOI] [PubMed] [Google Scholar]
- 51.Inaba T, Sano H, Kawahito Y, Hla T, Akita K, Toda M, Yamashina I, Inoue M, Nakada H. Induction of cyclooxygenase-2 in monocyte/macrophage by mucins secreted from colon cancer cells. Proc Natl Acad Sci U S A. 2003;100:2736–2741. doi: 10.1073/pnas.0435410100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohno S, Ohno Y, Nakada H, Suzuki N, Soma G, Inoue M. Expression of Tn and sialyl-Tn antigens in endometrial cancer: its relationship with tumor-produced cyclooxygenase-2, tumor-infiltrated lymphocytes and patient prognosis. Anticancer Res. 2006;26:4047–4053. [PubMed] [Google Scholar]
- 53.Pinho S, Marcos NT, Ferreira B, Carvalho AS, Oliveira MJ, Santos-Silva F, Harduin-Lepers A, Reis CA. Biological significance of cancer-associated sialyl-Tn antigen: modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett. 2007;249:157–170. doi: 10.1016/j.canlet.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 54.Sewell R, Backstrom M, Dalziel M, Gschmeissner S, Karlsson H, Noll T, Gatgens J, Clausen H, Hansson GC, Burchell J, Taylor-Papadimitriou J. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J Biol Chem. 2006;281:3586–3594. doi: 10.1074/jbc.M511826200. [DOI] [PubMed] [Google Scholar]
- 55.Carlos CA, Dong HF, Howard OM, Oppenheim JJ, Hanisch FG, Finn OJ. Human tumor antigen MUC1 is chemotactic for immature dendritic cells and elicits maturation but does not promote Th1 type immunity. J Immunol. 2005;175:1628–1635. doi: 10.4049/jimmunol.175.3.1628. [DOI] [PubMed] [Google Scholar]
- 56.Rughetti A, Pellicciotta I, Biffoni M, Backstrom M, Link T, Bennet EP, Clausen H, Noll T, Hansson GC, Burchell JM, Frati L, Taylor-Papadimitriou J, Nuti M. Recombinant tumor-associated MUC1 glycoprotein impairs the differentiation and function of dendritic cells. J Immunol. 2005;174:7764–7772. doi: 10.4049/jimmunol.174.12.7764. [DOI] [PubMed] [Google Scholar]
- 57.Hiltbold EM, Vlad AM, Ciborowski P, Watkins SC, Finn OJ. The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol. 2000;165:3730–3741. doi: 10.4049/jimmunol.165.7.3730. [DOI] [PubMed] [Google Scholar]
- 58.Monti P, Leone BE, Zerbi A, Balzano G, Cainarca S, Sordi V, Pontillo M, Mercalli A, Di Carlo V, Allavena P, Piemonti L. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10highIL-12low regulatory dendritic cell. J Immunol. 2004;172:7341–7349. doi: 10.4049/jimmunol.172.12.7341. [DOI] [PubMed] [Google Scholar]
- 59.Botti C, Seregni E, Ferrari L, Martinetti A, Bombardieri E. Immunosuppressive factors: role in cancer development and progression. Int J Biol Markers. 1998;13:51–69. doi: 10.1177/172460089801300201. [DOI] [PubMed] [Google Scholar]