

SUMMARY
Mechanisms that prevent inappropriate or excessive interleukin-17-producing T helper (Th17) cell responses after microbial infection may be necessary to avoid autoimmunity. Here, we define a pathway initiated by engagement of Type-I IFN receptor (IFNAR) expressed by dendritic cells (DC) that culminated in suppression of Th17 cell differentiation. IFNAR-dependent inhibition of an intracellular translational isoform of Osteopontin, termed Opn-i, de-repressed interleukin-27 (IL-27) secretion and prevented efficient Th17 responses. Moreover, Opn-i expression in DC and microglia regulated the type and intensity of Experimental Autoimmune Encephalomyelitis (EAE). Mice containing DC deficient in Opn-i produced excessive amounts of IL-27 and developed a delayed disease characterized by an enhanced Th1 response compared with the dominant Th17 response of Opn sufficient mice. Definition of the IFNAR Opn-i axis that controls Th17 development provides insight into regulation of Th sublineage development and the molecular basis of Type I interferon therapy for MS and other autoimmune diseases.
INTRODUCTION
Efficient protection against microbial infection depends on an interaction between the innate and adaptive immune systems. Recognition of microbial pathogens by pattern recognition receptors expressed by dendritic cells (DC) activates these cells to efficiently present pathogen-derived antigens to T cells, which initiate adaptive immune responses. Whereas the task of CD8+ T cells is limited primarily to lysis of cells that display peptides derived from intracellular pathogens, the more complex activities of CD4+ T cells are divided among several functionally distinct sublineages of CD4+ T cells. T helper 1 (Th1) and T helper 2 (Th2) cells, discovered some twenty years ago, have evolved to enhance clearance of intracellular pathogens, including viruses and parasites, respectively. A recently-defined third sublineage of Th cells termed Th17 cells may not be equipped to protect against infection by extracellular bacterial and fungal pathogens including Mycobacteria, Borrelia burgdorferi, and Candida albicans (Burchill et al., 2003; Khader et al., 2007; Umemura et al., 2007). The nature of the Th17 response reflects, in part, the ability of its signature cytokine – interleukin-17 (IL-17) to attract extensive inflammatory cellular infiltrates through interactions with chemokine receptors. Excessive Th17 responses after infection with, e.g. B. burgdorferi, can result in widespread inflammatory tissue destruction and chronic autoimmune disease.
These considerations suggest that mechanisms that prevent inappropriate or excessive Th17 responses after microbial infection may be essential to avoid host immunopathology and associated autoimmunity. For example, DC may not produce sufficient quantities of cytokines that promote Th17 development unless particular infectious stimuli are present. A more reliable mechanism for controlling Th17 responses depends on active suppression of Th17 differentiation by antigen-presenting DC that may selectively allow anti-viral Th1 responses.
The ability of antigen-presenting DC to differentially regulate generation of Th subsets is not well understood. Although recent reports have indicated that dectin-1-Syk-CARD9 signaling, triggered by detection of fungal β-glucans, in DC promotes Th17 responses (Leibundgut-Landmann et al., 2007), whether activated DC to inhibit Th17 responses has not been established. We reasoned that because a molecular signature of viral infection is early expression of Type I interferons (IFN-I), engagement of the IFN-I receptor (IFNAR) on antigen-presenting DC might constitutively pre-empt the ability of these DC to promote Th17 responses in the course of viral infections.
Here, we show that robust Th17 responses depended on the ability of a newly-defined translational intracellular isoform of Osteopontin (Shinohara et al., 2008) – Opn-i – to inhibit DC-dependent secretion of the potent inhibitory cytokine IL-27. IFNAR-dependent inhibition of Opn-i expression resulted in de-repression of IL-27 secretion and inhibition of the Th17 response in vitro and in vivo. Mice that contain Opn-i-deficient DC developed increased serum IL-27 amounts and a dominant pathogenic Th1 response in the spinal cord compared with the central nervous system (CNS) infiltrates that developed in Opn wt control mice. Definition of an IFNAR Opn-i axis that regulates the Th17 response provides insight into the therapeutic impact of IFN-I treatment of Multiple Sclerosis and other Th17 diseases (Paty and Li, 1993; Yu et al., 1996) as well as a rationale for new approaches to identify IFNAR ligands with increased therapeutic efficacy in MS and related diseases.
RESULTS
IFNAR engagement inhibits Opn-i-dependent generation of Th17 cells
Although some forms of microbial infection can efficiently stimulate robust Th17 responses, the Th17 response following conventional immunization is normally meager compared to the Th1 response (Park et al., 2005; Gocke et al., 2007). We investigated whether Th1 dominance is imprinted by DC at the outset of a polyclonal T cell response. In the absence of T cell polarizing reagents, cultures containing resting DC or DC that had been pre-activated with LPS contained >10-fold greater numbers of IFNγ-producing CD4+ T cells compared with IL-17-producing cells (Fig 1A, upper panel). Culture conditions that contained rIL-23, IL-4 and IFNγ antibodies only modestly increased the Th17 response. However, the Th17/Th1 ratio increased substantially in cultures containing T cells and DC deficient in IFNAR (Fig. 1A, lower panel).
Figure 1. IFNAR signaling in DC suppresses Th17 generation.

(A) Polyclonal T cell activation. (upper panel) After CD4+ T cells (2×105 T cells/well) incubated with soluble CD3ε Ab (1 μg/ml) and splenic DC. Intracellular cytokines were stained with PE-conjugated IL-17 Ab or biotin-conjugated IFNγ Ab followed by CyChrome-conjugated streptavidin. The proportions of IL-17+CD4+ and IFNγ+ CD4+ T cells are shown. DC were either unactivated or pre-activated for 24h with LPS (1 μg/ml); (lower panel) cell cultures were polarized to Th17 with rIL-23 and IFNγ + IL-4 Abs; data are representative of 3 experiments. (B) Antigen-specific T cell activation of MOG-specific CD4+ T cells expressing the 2D2 TCR transgene. 2D2 T cells were activated by BM-derived DC (Ifnar1+/+ vs. Ifnar1−/−) with 1 μg/ml of MOG peptide. The proportions of IL-17+CD4+ and IFNγ+CD4+ T cells are shown. Culture conditions were Th17-polarized with rIL-23, and IFNγ + IL-4 Ab. DC were pre-treated with the indicated reagents for 4 hrs. Data are representative of 4 experiments. (C, D) CD4+ T cells expressing the OT-2 TCR transgene were activated by BM-DC (Ifnar1+/+ vs. Ifnar1−/−) and 1 μg/ml OT-2 peptide antigen. Cultures were skewed to Th17 with rIL-6, rTGFβ, rIL-23, and IFNγ + IL-4 Abs (C), or not (D) with the indicated amounts of rIFNα added to DC/T cell co-culture. Data are representative of 5 identical experiments. Culture supernatants were harvested 24 h after T cell re-stimulation with soluble CD3 Ab and the amounts of IL-17 were analyzed by ELISA in triplicate wells shown as mean ± SEM.
IFNAR expression by DC suppresses the Th17 response
Expression of the IL-6 and TGFβ cytokines is necessary for generation of Th17 cells from naïve T cell precursors (Veldhoen et al., 2006; Bettelli et al., 2006), whereas IL-23 contributes to expansion of this RORγt-dependent Th subset (Park et al., 2005; Harrington et al., 2005; Ivanov et al., 2006) from activated or memory T cells (reviewed in (Kastelein et al., 2007). To test the possibility that the above result reflected, in part, an increase in memory Th17 cells in Ifnar1−/− mice, we asked whether IFNAR-deficient antigen-presenting DC alone could drive T cells from IFNAR wt donors to differentiate into Th17 cells. To this end, we compared the ability of Ifnar1−/− DC or Ifnar1+/+ DC to present MOG peptide to (Ifnar1+/+) CD4+ T cells expressing the MOG-specific T cell receptor (2D2) transgene (Fig. 1B). Inclusion of Ifnar1−/− DC altered the Th1 and Th17 balance in favor of the latter (Fig. 1B). Indeed, the Th17:Th1 ratios were increased approximately 3–5-fold in cultures containing Ifnar1−/− DC, independent of its state of activation. Enhancement of Th17 responses could not be attributed to IFNAR-dependent changes in inhibitory Th1 and Th2 cytokines (IFNγ, IL-4) or to IL-23, since these cultures included IL-4 and IFNγ antibodies and exogenous IL-23 (Fig. 1B). IFNAR expression by DC was also sufficient to fully inhibit the MOG-specific Th17 response in the absence of Th17 polarizing conditions (Figure S1 in Supplemental Data). We also examined Th17 cell expansion in cultures containing Ifnar1+/+ DC and Ifnar1−/− T cells. Under both Th17 neutral and Th17 polarizing culture conditions, with or without activation of DC by rIFNα, CpG and LPS, cultures containing IFNAR-deficient T cells did not display substantially enhanced Th17 responses (Fig. S2A and S2B in Supplemental Data).
We then tested the impact of Ifnar1−/− DC on activation of CD4+ T cells expressing an OVA-specific TCR transgene, OT-2, in cultures containing IL-6, TGFβ, IL-23 and neutralizing IFNγ and IL-4 antibodies, i.e. under conditions engineered to fully optimize Th17 development from both naïve and memory T cell pools. Presentation of the OVA peptide by Ifnar1−/− DC provoked substantially enhanced Th17 responses compared to cultures containing Ifnar1+/+ DC (Fig. 1C), indicating that supplementation of cultures with exogenous IL-6 and IL-23 or inhibition of IFNγ and IL-4 does not enhance responses of cultures containing Ifnar1+/+ DC to the degree seen in cultures containing Ifnar1−/− DC, i.e. increased Th17 responses promoted by Ifnar1−/− DC do not reflect enhanced production of IL-6 or IL-23 or inhibition of IFNγ and IL-4.
Since DC used in the experiment were not activated, autocrine amounts of IFN-I, which are constitutively expressed by unstimulated DC (Taniguchi and Takaoka, 2001), may be sufficient to inhibit Th17 generation. Indeed, in the absence of exogenous rIFNα, cultures containing Ifnar1−/− DC generated enhanced Th17 responses even under non-polarized conditions (Fig. 1D). We further examined the impact of increasing concentrations of IFNα on the Th17 response of CD4+ cells expressing the OT-2 TCR transgene. Increasing concentrations of IFNα resulted in progressively reduced Th17 responses in cultures containing Ifnar1+/+ DC, whereas addition of this cytokine did not suppress the Th17 response in cultures containing Ifnar1−/− DC (Fig. 1D). These data suggest that constitutive autocrine IFNAR signaling in DC is sufficient to suppress Th17 responses. In sum, IFNAR on DC negatively regulates Th17 responses upon antigen presentation to CD4+ T cells.
IFNAR expression by DC alters the balance between the Th17 and Th1 response
We then asked whether the impact of Ifnar1−/− DC on the IL-17 response reflected a generalized increase in potency of APC activity. IFNAR-deficiency did not alter expression of DC cell surface molecules involved in T cell activation including MHC class I, class II, CD80, and CD40 (splenic DC, Fig. S3A and BM-derived DC, Fig. S3B in Supplemental Data) and did not affect development of plasmacytoid DC (pDC) or conventional DC (cDC) (data not shown). T cell proliferation to antigen-pulsed Ifnar1−/− and Ifnar1 wt DC was not distinguishable under non-polarized or two different Th17-polarizing conditions (rIL-23 and IL-4 + IFNγ Ab and rIL-6 + rTGFβ) (Fig. S3C). Although strong Th17 responses were observed after stimulation of OT-2 T cells by Ifnar1−/− DC in cultures containing IL-6 and TGFβ (Fig. 2A), T cell stimulation under Th1-polarizing conditions (IL-12 and IL-4 Ab) by Ifnar1−/− DC produced substantially weaker Th1 responses compared to cultures containing Ifnar1+/+ DC (Fig. 2B). These findings indicate that enhanced induction of Th17 generation by Ifnar1−/− DC does not reflect a generalized increase in antigen-presenting activity. Instead, Ifnar1−/− DC acquire the ability to skew T cells towards the Th17 pathway and away from Th1 development.
Figure 2. IFNAR expression by DC alters the balance between the Th17 and Th1 response.
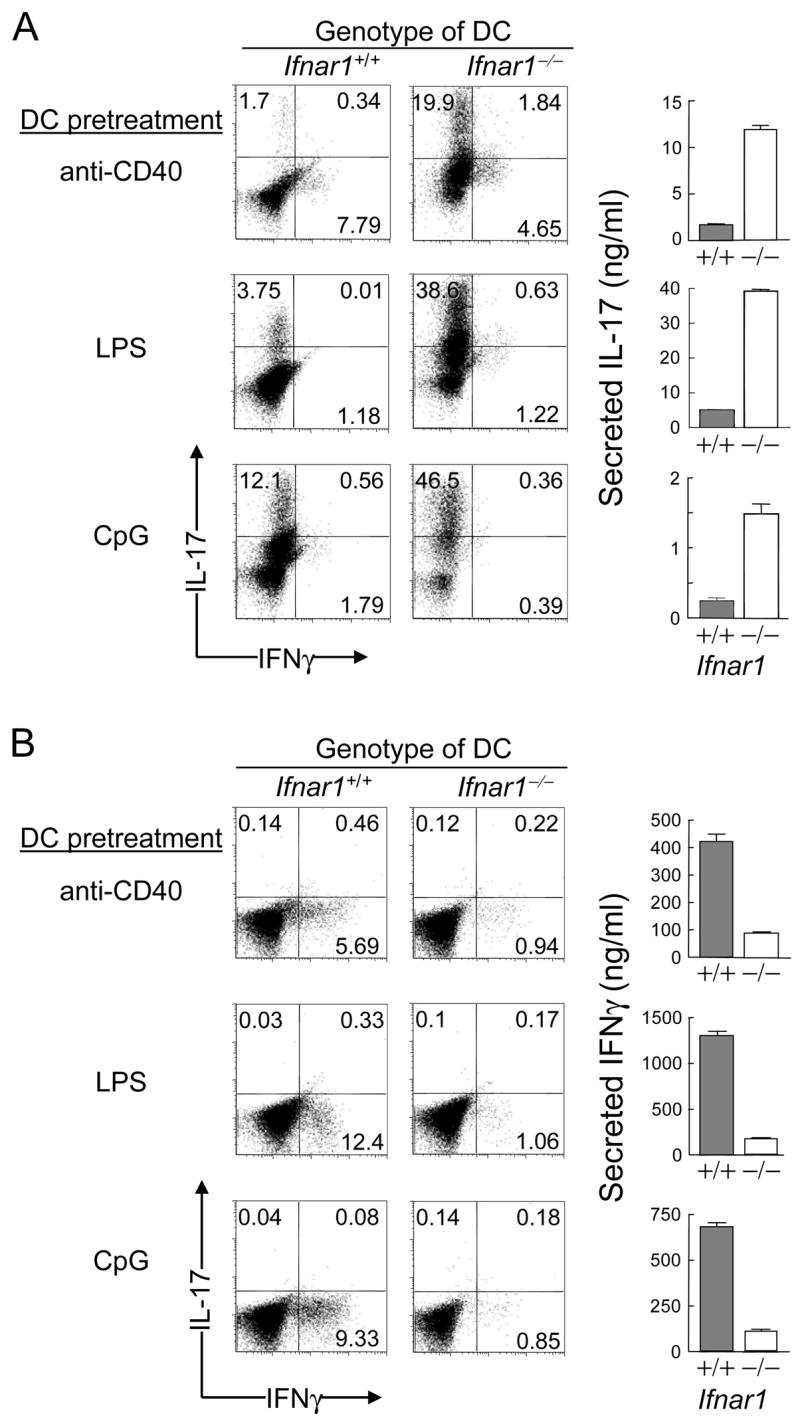
2D2 T cells were incubated with BM-derived DC and 1 μg/ml of OT-2 peptide in cultures skewed to Th17 [rIL-6 and rTGFβ (A)] or Th1 [rIL-12 and IL-4 Ab (B)]. Supernatants harvested 24h after re-stimulation with soluble CD3 Ab were analyzed by ELISA in triplicate wells shown as mean ± SEM; representative of 5 and 3 experiments for (A) and (B), respectively.
IFNAR-dependent regulation of Opn-i expression in DC and the Th17 response
Opn-deficient (Spp1−/−) mice develop milder EAE compared to Opn-sufficient mice and this has been attributed to expression of Opn-s by activated T cells (Chabas et al., 2001; Jansson et al., 2002; Shinohara et al., 2005; Shinohara et al., 2006; Hur et al., 2007). Since we have defined a translational isoform of Opn that is expressed in the cytosol of DC subsets, termed Opn-i (see above), we asked whether Opn-i expression by DC contributes to the development of Th17 cells. IFNAR expression reduced the expression of Opn-i in DC to 20–25%, as judged by immunoblotting (Fig. 3A). Constitutive expression of IFNAR in the absence of added ligand (recombinant) IFNα inhibits Opn-i expression, whereas addition of limiting concentrations of IFN [10 U/ml of IFNα or IFNβ] results in a further increase (up to 2-fold) in suppression). Reduced Opn-i expression in Ifnar1+/+ DC was associated with reduced promotion of the Th17 response; expression of Opn by cDC, rather than pDC, was critical to Th17 generation (Fig. 3B–D). We also noted that Opn-deficient T cells produced less IL-17 than Opn wt T cells, in cultures containing Opn wt DC, opening the possibility that secreted Opn (Opn-s) produced by activated T cells may augment the Th17 response, possibly via inhibition of T cell apoptosis, as delineated by Hur et al. (2007) (Fig. 3B). Opn-deficient T cells were routinely used in these co-cultures to study the isolated Opn-dependent contribution of DC to the response (Fig. 3C, 3D). Generation of Foxp3+ regulatory T cells (Treg) was not affected by the amount of Opn expression in DC in the absence of TGFβ (Figs. 3C, S4A), suggesting that Opn-i expression in DC regulates Th17 generation without affecting Treg production under these conditions. In addition, Opn expression in DC did not alter the ability of T cells to proliferate under IL-23-mediated Th17 polarizing conditions (Fig. S4B). These findings indicate that IFNAR-dependent regulation of the Th17 response reflects inhibition of Opn-i expression in DC.
Figure 3. IFNAR signaling suppresses Opn-i expression and Th17 cell generation.
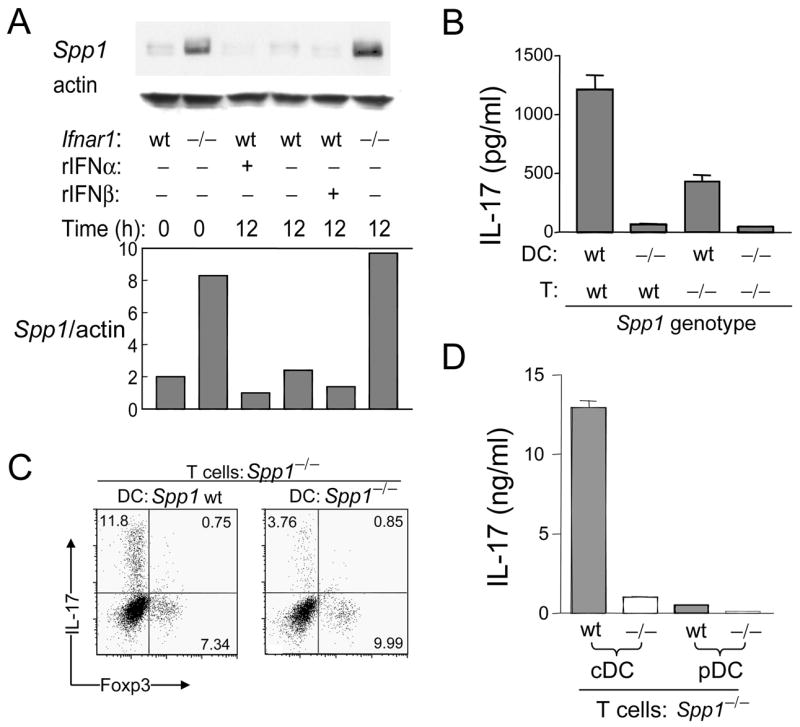
(A) Opn in lysates of BM-derived DC was analyzed by immunoblot and quantified by densitometry after incubation with or without rIFNα or rIFNβ (10 U/ml), as indicated. The bar graph summarizes the amounts of Opn normalized to actin; data are representative of 3 identical experiments. (B–D) CD4+ 2D2 Tg cells were stimulated by DC with 1 μg/ml MOG and rIL-23, and IFNγ + IL-4 Abs. (B) Spleen-derived DC and 2D2 T cells harvested on day 6 after 24h re-stimulation with soluble CD3 Ab (1 μg/ml) before the amount of IL-17 in supernatants was evaluated by ELISA in triplicate wells shown as mean ± SEM and representative of 7 identical experiments. (C) Opn-deficient (Spp1−/−) CD4+ 2D2 Tg T cells incubated with BM-derived DC and 1 μg/ml MOG peptide, before gating on CD4+ cells; representative of 7 identical experiments. (D) BM-DC sorted by FACS into pDC and cDC. The amount of IL-17 in T cell re-stimulated culture supernatants determined by ELISA in triplicate wells is shown as mean ± SEM and represent 4 identical experiments.
If suppression of Th17 development by IFNAR signaling is mediated by inhibition of Opn-i expression, reconstitution of Opn-i expression in Opn-deficient DC should restore Th17 generation. We transduced Opn-deficient DC with a lentiviral Opn-i vector, lenti-ΔOpn, which restores expression of intracellular but not secreted Opn (Shinohara et al., 2006). Incubation of T cells with lenti-ΔOpn-transduced DC or lenti-GFP-transduced DC resulted in a 7-fold increase in the Th17 response and a 10-fold increase in IL-17 concentrations in culture supernatants. However, IL-10 was not detectable in either culture supernatant and <1% of CD4+ cells expressed intracellular IL-10 (Fig. 4A and Fig. S5 in Supplemental data). These data suggested that IFNAR-dependent down-regulation of Th17 development reflects inhibition of Opn-i expression in DC.
Figure 4. Reconstitution of Opn-i expression in Opn-deficient DC rescues Th17 cell generation in vitro and in vivo.
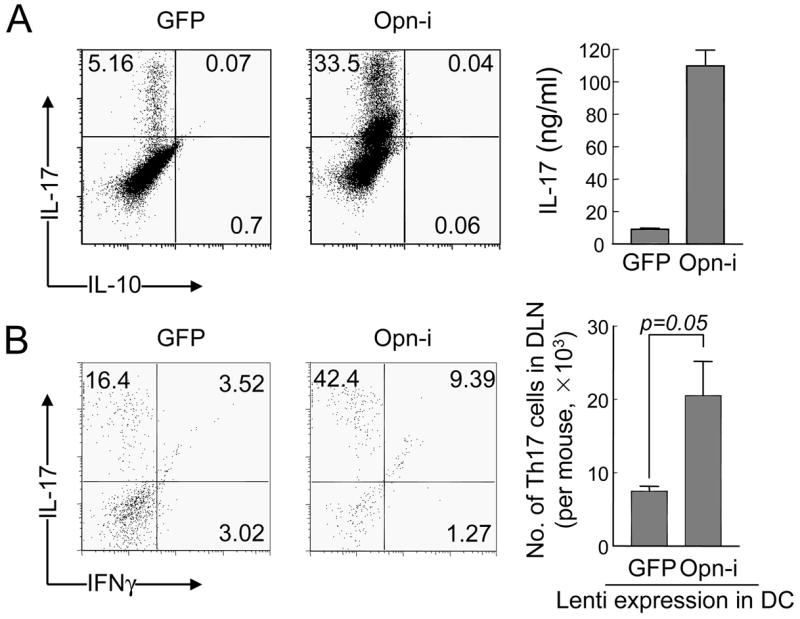
(A) BM-derived Opn-deficient DC transduced with lentivirus encoding either Opn-i or GFP (negative control) were incubated with Opn-deficient CD4+ 2D2+ cells with 1 μg/ml MOG peptide under Th17-polarizing conditions (rIL-23, and IFNγ + IL-4 Abs). Cells were stained with IL-17 Ab on day 6 and supernatants harvested 24h after re-stimulation with CD3 Ab and analyzed by ELISA; representative of 2 experiments. (B) Rag2−/− hosts reconstituted i.v. with naïve (CD44−CD62L+) CD4+ 2D2 Tg T cells (1.5×105/mouse) immunized by s.c. injection of MOG-pulsed BM (2×106/mouse). DLN cells harvested 7 days later were treated immediately with PMA and ionomycin ×4h for FACS analysis. Dot plot shows IL-17 and IFNγ immunofluorescence of DLN cells gated on CD4+. Numbers of DLN Th17 cells per mouse (n=3) calculated from total numbers of DLN cells and proportions of CD4+IL-17+ double positive cells. Error bars represent mean ± S.D.
In vivo Th17 polarization is enhanced by Opn-i expression in antigen-presenting DC
To test this pathway in vivo, we evaluated the ability of Opn-deficient and Opn-i-reconstituted DC to induce Th17 responses to the MOG peptide. Opn-deficient DC transduced with lenti-Opn-i were pulsed with MOG peptide before injection into Rag2−/− mice along with naïve 2D2 T cells (sorted as CD4+CD62LhiCD44lo). One week later, the Th17 response was defined immediately after harvesting cells from draining LN after a 4h pulse with PMA and ionomycin before FACS analysis. In the absence of pertussis toxin, no sign of EAE onset was observed in these mice. Immunization of mice with Opn-i-transduced DC provoked a 2~3-fold increase in the Th17 response compared with GFP-transduced Opn−/− DC (Fig. 4B).
Inhibition of Th17 differentiation by an IFNAR Opn-i pathway is mediated by IL-27
IFNAR-dependent regulation of the Th17 response did not reflect the impact of this receptor on cytokines that might require the Th17 response, including IL-6, TGFβ, IL-23, and IFNγ (Figs. 1A–C; 2A,B; 3B–D; 4A). Because expression and secretion of IL-27 by DC can exert potent suppressive effects on Th17 generation (Batten et al., 2006; Stumhofer et al., 2006), we asked whether IFNAR-mediated inhibitory pathway depicted in Fig. S6A resulted in increased IL-27 expression. Activation of Ifnar1+/+ but not Ifnar1−/− DC by LPS resulted in progressively increasing concentrations of IL-27 (Fig. 5A). Addition of limiting concentrations of IFNα to Opn-deficient DC did not further enhance IL-27 responses. In contrast, addition of the same concentrations of IFNα to Opn wt DC resulted in a dose-dependent induction in IL-27 secretion (Fig. 5B, right panel), suggesting that Opn-i inhibition is a key element in enhanced IL-27 expression under these conditions. Although co-cultures contain too few DC (2.5×105/ml) to produce amounts of IL-27 detectable by ELISA, examination of the IL-27 response of (~10×) higher concentrations of Opn-deficient Ifnar1−/− or wt DC also showed that Opn-deficient DC produced substantially more IL-27 than Opn wt, whereas Ifnar1−/− DC produced substantially less IL-27 than Ifnar1 wt DC (Fig. 5C). Induction of IL-27p28 mRNA expression was also confirmed by quantitative PCR in IFNAR-deficient and Opn-sufficient DC (Fig. 5D).
Figure 5. Suppression of IL-27 expression in DC contributes to Opn-i-mediated Th17 differentiation.

(A) BM-derived DC (Ifnar1+/+ [●] vs. Ifnar1−/− [○]) were incubated (2×106 DC/ml) with the indicated concentration of LPS before 30h supernatants were evaluated for IL-27p28 amounts by ELISA in triplicate well; representative of 3 experiments. (B) BM-derived DC (Opn wt [●] vs. Opn−/− [○]) incubated with rIFNα (5×106 DC/ml). 42h later IL-27-p28 amounts were determined and represent 2 experiments. (C) BM-derived DC incubated ×48h before IL-27-p28 amounts were determined by ELISA in triplicate wells. (left panel) Comparison of IL-27 secretion by Ifnar1+/+ and Ifnar1−/− DC (107 DC/ml) in 44h culture supernatants. (middle panel) Comparison of IL-27 secretion by Opn wt and Opn-deficient (Spp1−/−) DC (2×106 DC/ml). (right panel) Comparison of IL-27 secretion by Opn-deficient DC transduced with lenti-GFP (control) or lenti-ΔOpn-i (2×106 DC/ml) in 24h culture supernatants; data representative of 2 experiments. (D) Comparison of the amounts of IL-27p28 mRNA in resting BM-derived DC from Ifnar1+/+ or Ifnar1−/− donors. Real-time PCR of total cDNA and results obtained from cycling threshold (−ΔΔCt) values normalized to β-actin. (E–F) Two different Th17 skewing conditions were tested: (upper panel) rIL-23 and IFNγ + IL-4 Abs; (lower panel) rIL-6 and rTGFβ. (E) 2D2 T cells were incubated with Opn-deficient BM-DC, 1 μg/ml MOG peptide and 20 μg/ml IL-27 Ab or isotype IgG. 24h after T cell re-stimulation with soluble CD3 Ab, the amount of IL-17 was measured (mean ± SEM) and is representative of 4 experiments. (F) DC and OT-2 T cell co-cultures under the same skewing conditions used in (E) were treated with rIFNα with or without IL-27 Ab (20 μg/ml) and stimulated with OVA peptide ×5d, followed by 24h T cell re-stimulation with soluble CD3 Ab. The amount of IL-17 analyzed by ELISA in triplicate wells is shown (mean ± SEM); data are representative of 2 experiments.
To directly test the hypothesis that the IFNAR Opn-i axis resulting in enhanced expression of IL-27 was responsible for suppression of Th17 responses, we measured the effects of neutralizing IL-27 antibody in these cultures. Addition of IL-27 neutralizing antibody to cultures containing Opn-deficient unstimulated DC, under two different Th17-polarizing culture conditions successfully restored the Th17 response (Fig. 5E), i.e. reversed suppression of the Th17 response in cultures containing Opn-deficient DC. In contrast, treatment of cultures containing Opn wt DC with IL-27 neutralizing antibody had no effect on the Th17 response in these cultures (Fig. S6B). Moreover, addition of IL-27 antibody to cultures containing IFNα also restored the Th17 response (Fig. 5F). These findings indicated that IFNAR-dependent suppression of Th17 responses is mediated by enhanced secretion of the inhibitory IL-27 cytokine.
Delayed EAE onset in Opn-deficient hosts associated with increased IL-27 expression and altered ratio of Th1 and Th17 autoreactive cells
These analyses suggested that promotion of Th17 responses by DC requires expression of Opn-i in these APC. Early experiments of Langrish et al. showed that IL-17 is essential to the development of severe EAE (Langrish et al., 2005). We therefore investigated whether presentation of autoantigen by Opn-deficient APC might impair development of EAE. Opn-deficient Rag2−/− hosts reconstituted with naïve (CD4+CD62LhiCD44lo) MOG-specific 2D2 T cells displayed a substantially delayed onset of EAE compared to Opn wt Rag2−/− hosts (Fig. 6A) characterized by delayed lymphocyte and macrophage infiltration into spinal cord (Fig. 6B). We also analyzed mice on days 12 (Opn wt) and 17 (Opn-deficient) in a separate experiment from Fig. 6A when Opn wt and Opn-deficient hosts displayed a clinical score of 3–3.5 for analysis of serum IL-27. Serum IL-27 concentrations of Opn wt hosts were 5–10 pg at day 5 and 17, whereas serum IL-27 amounts in Opn-deficient hosts were 40 pg/ml at day 5 and 80 pg/ml at day 17 (Fig. 6C). Splenic DC from Opn-deficient Rag2−/− hosts also expressed higher amounts of IL-27p28 than DC from Opn wt Rag2−/− hosts (Fig. S7) and brains of Opn-deficient hosts expressed 10-fold higher IL-27p28 mRNA amounts than Opn wt hosts (data not shown).
Figure 6. Opn-deficiency in non-T cells delays EAE onset.

(A) EAE development in Rag2−/− hosts. Naive CD4+ 2D2 Tg Opn-deficient T cells (1.4×105 cells/mouse) were adoptively transferred into Opn wt or Opn-deficient B6.Rag2−/− hosts. Disease development and incidence are shown for each group. (Opn wt, ●, n=9; Opn−/−, ○, n=8; error bars denote mean + SEM. Data are representative of 3 experiments. (B–E) Analysis of mice at different time-points in separate experiments. (B) CD4+ 2D2 T cells from B6 2D2 Tg mice were adoptively transferred to either Opn wt or Opn-deficient B6 Rag2−/− mice, as described in (A). Twenty days later, the total number of spinal cord-infiltrated cells, spinal cord-infiltrated macrophages (%) (F4/80+CD45hi), and total CD4+ T cells were enumerated. Increased lymphocyte infiltration was observed in Opn wt hosts (●) compared with Opn-deficient (○) hosts. Negative control (✳) mice are Rag2−/− mice without T cell transfer. Average disease scores of the Opn wt and Opn-deficient (Spp1−/−) hosts used for these experiments (at day 20) were 3.8 and 1.2 for Opn wt and Opn-deficient hosts, respectively. Horizontal lines denote mean values. (C) Serum IL-27 analysis. Rag2−/− hosts were analyzed when EAE scores were either 0 (day 5) or 3~3.5 (day 12 and day 17 for Opn wt and Opn-deficient hosts, respectively). Error bars represent mean ± S.D. from values of 3 mice (score 0) or 4 mice (score 3~3.5) each of Opn wt and Opn-deficient Rag2−/− hosts. (D) Th17 development. Cells from DLN were harvested from mice on day 5 (score =0), treated immediately with PMA and ionomycin and incubated with IL-17 or IFNγ Abs and subject to FACS analysis; dot plots were gated on CD4+ cells. (E) Th subsets in CNS. Cells from spinal cord or brain were harvested from score 3~3.5 mice on day 12 and 17 from Opn wt and Opn-deficient Rag2−/− hosts, respectively, treated immediately with PMA and ionomycin and incubated with IL-17 or IFNγ Ab. Panels represent results of 3 mice (score 0) or 4 mice (score 3~3.5) each from Opn wt and Opn-deficient Rag2−/− hosts.
Increased amount of serum IL-27 in Opn-deficient hosts beginning at day 5 (Fig. 6C) was associated with substantially reduced Th17 responses in draining LN – 2.54% (Opn-deficient) vs. 17.6% (Opn wt) Th17 cells (Fig. 6D). Analysis of the spinal cord and brain later in the course of EAE in Opn-deficient hosts revealed a 2-fold increase in the proportions of Th1 cells (~20% to 48%) and a doubling of CD4+ T cells (approximately 9×105 to 2.1×105), representing a 4-fold enrichment of the CNS Th1 response in Opn-deficient hosts (Fig. 6E). This is consistent with findings that IL-27 can enhance Th1 responses in other settings (Pflanz et al., 2002; Cao et al., 2008) and suggests that suppression of the Th17 response in Opn-deficient Rag2−/− hosts is apparent before onset of disease.
Opn expression in microglia is essential for Th17-mediated progression of EAE
Since restimulation of T cells by APC in CNS is necessary for full development of EAE (Tompkins et al., 2002; Kawakami et al., 2004), we asked whether microglia also required expression of Opn-i to induce Th17 responses to MOG peptide (Fig. 7A). Lentiviral-mediated reconstitution of Opn-deficient microglia with Opn-i resulted in substantially increased production of IL-17 and decreased production of IL-27p28 (Fig. 7B).
Figure 7. Microglia require Opn-i expression to induce Th17 cells.
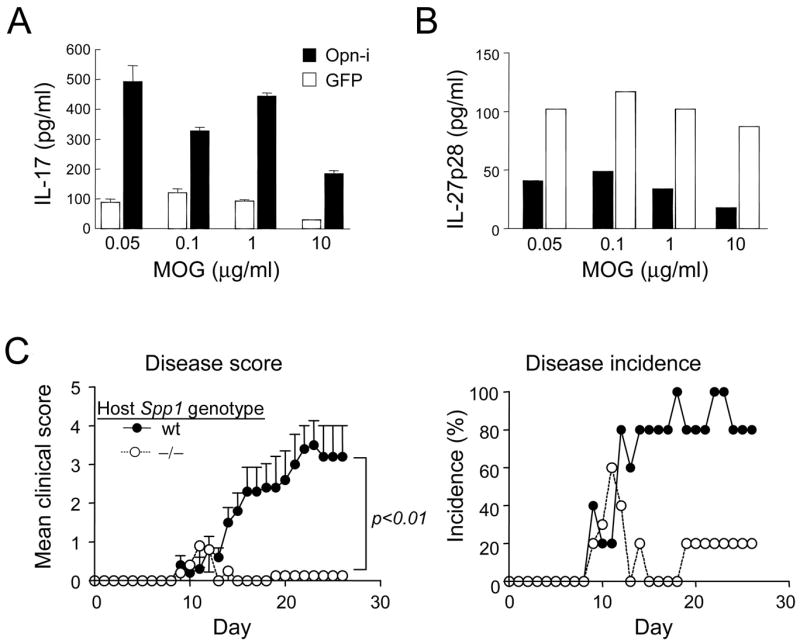
Reconstitution of Opn-deficient microglia with Opn-i rescues IL-17 production by CD4+ 2D2 T cells. (A,B) Opn-deficient microglia were infected with lenti-Opn-i or lenti-GFP and incubated 2D2+ CD4− T cells and MOG peptide. (A) IL-17 concentrations in culture supernatants 24h after T cell restimulation on day 4 were determined by ELISA in triplicate wells. (B) Concentrations of IL-27p28 in day 4 cultures before T cell restimulation. The amounts of IL-27p28 were determined by ELISA in triplicate wells; empty and filled bars denote supernatants from Opn-i-transfected and GFP-transfected DC, respectively, and data are representative of 2 experiments. (C) EAE development is inhibited in irradiated Opn-deficient hosts. Total BM cells (1.7×107 cells/mouse) from 2D2+ Tg Opn-deficient mice were i.v. transferred to irradiated (800 rads) hosts. Ten weeks later, EAE was induced. Disease development (mean clinical score) and incidence (%) is shown for each group (n=5). Error bars in disease score denote mean ± SEM and T-test values on the last day of observation are shown. Mice displayed >95% engraftment of donor bone marrow according to immunofluorescence with Ly5.2 Ab (donor BM cells were obtained from B6.Ly5.2+ Opn-deficient mice expressing the 2D2 TCR transgene) 10 weeks after reconstitution and before disease induction, as judged by immunofluorescence of peripheral blood cells.
To test the contribution of Opn expression by CNS-resident microglia to EAE, we induced EAE in irradiation BM chimeras, in which host (radiation-resistant) microglia were Opn wt or Opn-deficient whereas peripheral hematopoietic APC were derived from donor (Opn+/+) BM cells (Greter et al., 2005; Heppner et al., 2005). Opn-deficiency in the CNS compartment dramatically reduced the intensity of EAE in radiation chimeras in which the hematopoietic compartment of Opn-deficient or Opn wt hosts was reconstituted with 1.7×107 Opn-deficient BM cells (Fig. 7C), suggesting that expression of Opn in microglia is necessary for efficient development of EAE. These findings may also be relevant to MS pathogenesis, since overexpression of Opn in MS plaques is a notable feature of active disease (Steinman and Zamvil, 2003).
DISCUSSION
It has taken two to three decades for Th17 cells to be identified as a new lineage of effector CD4+ T cells following the original description of Th1 and Th2 cells. This delayed recognition reflects, in part, the relative paucity of Th17 cells after conventional immunization with antigen. In the absence of a cytokine milieu required to induce and expand Th17 development, stimulation of T cells from antigen-immunized mice often generates less than 1% Th17 cells, compared with much higher proportions of Th1 cells (Park et al., 2005; Gocke et al., 2007). Because generation of murine Th17 cells from naïve cells requires IL-6 and TGFβ (Veldhoen et al., 2006; Bettelli et al., 2006), and IL-23 for Th17 expansion (Park et al., 2005; Harrington et al., 2005), a relative insufficiency of these cytokines may dampen the Th17 response. However, a more efficient mechanism responsible for pre-emption of Th17 responses probably depends on the IFNAR-dependent inhibitory pathway described in this report.
The negative impact of IFNAR engagement on Th17 responses was mediated through inhibition of Opn-i expression, which in turn enhanced IL-27-dependent inhibition of Th17 development. We have not ruled out the possibility that IFNAR signaling in T cells may also inhibit expansion or survival of Th17 cells. Although extremely low amounts of IFN-I were sufficient to effectively initiate this IFNAR-dependent pathway in DC and attenuate Th17 development, increasing amounts of IFN-I, e.g. after certain viral infections, may be necessary to fully inhibit Th17 responses. According to this view, IFN-I constitutively produced at low amounts in the absence of viral infection may chronically dampen Th17 responses, whereas increased IFNAR signaling secondary to rapid increases in IFN-I amounts after viral infections may be necessary for increased IL-27-dependent inhibition of Th17 responses. In contrast, certain bacterial or fungal infections that disrupt this inhibitory pathway may provoke robust Th17 responses and increase the risk of autoimmune disease. It may be relevant that Th17 generation is also suppressed by expression of the IFNγ receptor on APC (Batten et al., 2006), and the potential similarities between IFNα and IFNγ-dependent suppression of the Th17 response via interactions with DC deserve further investigation.
IL-27, a potent inhibitor of Th17 cell development, can also induce IL-10-producing T cells (Awasthi et al., 2007; Fitzgerald et al., 2007). Our DC-T cell co-cultures do not produce sufficiently high IL-27 concentrations to enhance IL-10-producing cells, suggesting that inhibition of Th17 responses that we observe does not reflect enhanced production of IL-10. In addition to being a potent inhibitor of Th17 cell development (Stumhofer et al., 2006), Huber et al. have shown that IL-27 also inhibits TGFβ-mediated expansion of Treg (Huber et al., 2008). Low concentrations of IL-27 sufficient to suppress Th17 responses in cultures containing Opn-deficient DC did not substantially alter TGFβ-dependent generation of Foxp3+ Treg cells in this study, emphasizing the striking susceptibility of the Th17 response to low concentrations of IL-27 and also indicating that Treg induction is not required to inhibit the Th17 response in our experiments.
Previous studies have indicated that Opn-i expression in pDC promotes expression of IFNα and contributes to early protective responses against HSV-1 infection and tumor growth (Shinohara et al., 2006). Here we show that Opn-i expression by conventional DC (cDC) (and microglia) contributes to Th17 commitment in vitro and in vivo in the setting of EAE. Since Opn expression is induced after TLR engagement of pDC but not cDC (Shinohara et al., 2006), Th17 generation may be negatively regulated by pDC-dependent production of IFNα through engagement of IFNAR expressed by cDC. Additional experiments are necessary to evaluate the contribution of an inhibitory interaction between DC subsets: IFN-I-producing pDC (which do not themselves promote detectable Th17 responses) and IFNAR-producing cDC in regulating the Th17 response.
Opn (Spp1) gene expression in T cells is essential for efficient Th1 development (Ashkar et al., 2000; O’Regan et al., 2000; Chabas et al., 2001; Shinohara et al., 2005; Sato et al., 2005; Renkl et al., 2005), whereas expression in cDC promotes Th17 differentiation. The contribution of Opn to the development of these Th lineages reflects cell-type specific expression of the two Opn isoforms. Secretion of Opn (Opn-s) by T cells leads to an interaction with its receptors on macrophages that upregulates IL-12 production and enhances Th1 development. In addition, Opn-deficient T cells produced less IL-17 than Opn wt T cells in cultures containing Opn wt DC, suggesting that secreted Opn (Opn-s) may also augment the Th17 response under some circumstances, possibly through a direct interaction that inhibits apoptosis (Hur et al., 2007). In addition to inhibition of IL-27, Opn-i may also promote Th17 responses through enhancement of DC-T cell interactions. Opn-i-dependent polarization of the actin cytoskeleton may enhance formation of immunological synapses necessary for efficient T cell activation and helper lineage commitment (Al Alwan et al., 2003; Shapiro et al., 2003; Benvenuti et al., 2004a; Benvenuti et al., 2004b; Maldonado et al., 2004; West et al., 2004).
We tested two different models of EAE: (1) naïve 2D2 CD4+ T cell transferred into Rag2−/− hosts and (2) irradiation BM chimera reconstituted with 2D2 BM cells (in both models, transferred cells were Opn-deficient to rule out excess Opn-s). EAE development was robust in the former system, characterized by efficient T cell activation and infiltration into the CNS (data not shown), allowing us to examine full-blown EAE in Opn-deficient mice. The latter model, which allowed analysis of the role of Opn in APC, revealed that Opn-deficient APC promoted a dominant Th1-type of EAE. Enhanced Th1 responses in the CNS of Opn-deficient Rag2−/− hosts were associated with increased IL-27 secretion by DC and/or microglia, that may have resulted in enhanced IFNγ responses (Pflanz et al., 2002; Cao et al., 2008). Analyses of Th17 and Th1 components of CNS cellular infiltrates at different stages of EAE induced by different autoantigens may define clinically distinct subsets of this murine model of MS, and provide insight into clinical subtypes of MS.
Definition of the IFNAR signaling pathway in DC leading to suppression of Th17 responses described in this report provides insight into the essential elements that govern the Th17 response. In addition, these findings are relevant to the use of IFNβ as the leading treatment for patients suffering from MS (Paty and Li, 1993; Hafler et al., 2005). The ability of IFNβ, an innate immune cytokine with anti-viral properties, to inhibit autoimmune inflammation and reduce the incidence of disease relapse has been poorly understood. Suppression of the development of pro-inflammatory Th17 cells provides a mechanistic explanation for the therapeutic effects of Type I interferon therapy in MS. These findings also suggest that analysis of the effects of IFNAR engagement by antibodies or mutant IFNs that differential engage the Opn IL-27 pathway described here may yield new and effective therapies for MS.
EXPERIMENTAL PROCEDURES
Animals and reagents
Opn-deficient (Spp1−/−) mice (Rittling et al., 1998) were backcrossed to C57BL/6 (B6) for 15 generations; B6 2D2 MOG-specific T cell receptor transgenic mice (Bettelli et al., 2003) a gift from V.K. Kuchroo (Brigham and Women’s Hospital, Boston, MA). B6 OT-2 TCR transgenic mice and B6 Ly5.2 mice (Jackson Labs, Bar Harbor, ME) and 129/SvEv IFNAR1-deficient mice (B&K Universal, Hull, England) were used. Antibodies to IL-17 (TC11-18H10) and IFNγ (XMG1.2) for flow cytometry from BD Pharmingen; Foxp3 (FJK-16s) and IL-10 (JES5-16E3) Abs from eBioscience; IL-27p28 Ab (BAF1834), recombinant proteins for mouse IL-23, IL-6, Flt-3L and human TGFβ1 from R&D systems; recombinant mouse IFNα from HyCult. Lentiviral constructs of Opn without the signal peptide (ΔOpn) as described (Shinohara et al., 2006). MOG35–55 peptide (MEVGWYRSPFSRVVHo9LYRNGK) and OVA329–339 peptide (ISQAVHAAHAEINEAGR) from New England Peptides. All work involving vertebrate animals was reviewed and approved by the DFCI IACUC and conducted in accordance with AAALAC guidelines.
EAE induction
As described (Shinohara et al., 2005) with MOG peptide in CFA s.c. day 0, and pertussis toxin i.v. day 0 and 2. Mice were assessed daily for clinical signs of disease in a blinded fashion and graded as described previously. Mean clinical scores and mean maximal scores were recorded daily and calculated by adding scores of individual mice and dividing with number of mice in each group, including mice with no sign of disease.
Preparation of CD4+ T cells, DC, microglia and spinal cord lymphocytes
Performed as described previously (Shinohara et al., 2006). Naïve T cells were FACS-sorted as CD4+CD62LhiCD44lo and splenic DC were derived after collagenase treatment and positive selection with CD11c MACS beads (Miltenyi). BM-derived DC were prepared according to an in vitro culture method (Gilliet and Liu, 2002) with modifications (Shinohara et al., 2006), whereas further purification of pDC and cDC is described in Shinohara et al. (2006). Primary microglia were prepared as previously described (Dalpke et al., 2002). Briefly, after treatment of spinal cords with collagenase for 50 m at 37°C, single cells were separated by Percoll spin, resulting in >98% Mac-1+F4/80+ cells.
APC-T cell co-culture
1×105 DC/well and 2×105 CD4+ T cells/well were incubated in 200 μl/well of complete RPMI media with antigen or soluble CD3ε Ab (BD Pharmingen). For microglia–T cell co-cultures, 2.5×105/well each of microglia and 2D2 T cells were incubated in 500 μl/well with RPMI media. Th17 polarization entailed rIL-23 (100 ng/ml) and IFNγ + IL-4 Ab (10 μg/ml each) or rIL-6 (20 ng/ml) and rTGFβ (3 ng/ml) at culture initiation. Culture supernatants in Fig. 1C included rIL-23 (10 ng/ml), rIL-6 (20 ng/ml), rTGFβ (1 ng/ml), and IFNγ + IL-4 antibodies (10 μg/ml each). In some experiments, IL-27 was neutralized with IL-27 Ab (20 μg/ml). For Th1 polarization, rIL-12 (5 ng/ml) and IL-4 Ab (10 μg/ml) were added during set up of co-cultures.
Irradiation BM chimeric mice
B6 Opn wt and -deficient mice were irradiated (800 rads) 24h before i.v.-transfer of total BM cells from Opn-deficient 2D2 mice (1.7×107 cells/mouse). Ten weeks later, EAE was induced as indicated above.
In vivo development of Th17 cells
B6 Rag2−/− mice were transferred i.v. with Opn-deficient naïve 2D2 CD4+ T cells (1.5×105/mouse; CD4+CD62LhiCD44lo). BM-derived DC (infected by lentivirus, below) were pulsed with 20 μg/ml of MOG ×3h at 37°C and then extensively washed. Mice were immunized with 2×106/mouse of MOG-pulsed DC by s.c. injection. Draining LN cells were excised 7d later and treated with PMA and ionomycin (×4h) and brefeldin A (in the last 2h) before FACS analysis.
Lentiviral Opn-i transfection
Opn-i and GFP constructs were lentivirally-transfected into DC or microglia as described (Shinohara et al., 2006)
ELISA and intracellular cytokine flow cytometry
Following antigen-specific stimulation with DC for 5–6 days, T cell were restimulated ×24h with soluble CD3ε Ab (145-2C11, BD Pharmingen) before culture supernatants were analyzed for IL-17 and IL-27p28 concentrations by ELISA (R&D Systems). Intracellular cytokine and Foxp3 staining was performed on cells treated with ionomycin (500 ng/ml) and PMA (50 ng/ml) ×4h and brefeldin A (10 μg/ml) for the last 2h. Cells were stained for cell surface markers, fixed/permeabilized with Fix/Perm solution (eBioscience), followed by FcBlock (2.4G2, BD Pharmingen) and intracellular staining. Gating of intracellular cytokine staining was determined by immunofluorescence with an isotype-matched control Ig (Ab control) and, in some experiments, immunofluorescence of an (IL-17−) CD8+ T cell line with IL-17 Ab (cell control) used as well.
Immunoblotting of DC lysates
BM-derived DC were prepared as described above. On day 7 of culture, DC were harvested and cell lysates were either immediately prepared (“0 h” samples) or prepared 12 h after replating cells in RPMI complete medium with or without recombinant IFNα or IFNβ (“12 h” samples). Ten μg/lane of DC lysate was applied to an SDS-PAGE gel and proteins were blotted onto a PDVF membrane before immunodetection was performed with Opn Ab (O-17, IBL, America).
RNA cDNA preparation and real-time PCR
After washing DC with PBS, total RNA was extracted from cells using an RNeasy kit (Qiagen). cDNA synthesis was initiated by priming total RNA with oligo (dT) before RNA was reverse-transcribed with MMLV RT (Ambion). The resultant cDNA was used for real-time PCR analyses using an ABI 7700 (Applied Biosystems). QuantiTect SYBR Green PCR (Qiagen) was used to detect IL-27p28, and β-actin was used as an internal control. (IL-27p28 forward: 5′-CTC TGC TTC CTC GCT ACC AC-3′, reverse; 5′-GGG GCA GCT TCT TTT CTT CT -3′; β-actin primers forward: 5′-TGT TAC CAA CTG GGA CGA CA-3′, reverse; 5′-CTG GGT CAT CTT TTC ACG GT-3′; primer sequences for detecting subsets of IFNα were obtained from Clontech). Error bars indicate the maximum and minimum values calculated from SD and -ΔΔCt values from triplicate PCR reactions, according to Applied Biosystems protocols.
Supplementary Material
Acknowledgments
The authors wish to thank S.J. Turley and K.W. Wucherpfennig for critical reading of the manuscript, D. Laznik for technical assistance, and A. Angel for assistance with the manuscript and figures. This work was supported in part by research grants from the National Institutes of Health (AI 12184, AI 48125) to HC; an NRSA Fellowship (T32 CA70083) to MLS.
References
- Al Alwan MM, Liwski RS, Haeryfar SM, Baldridge WH, Hoskin DW, Rowden G, West KA. Cutting edge: dendritic cell actin cytoskeletal polarization during immunological synapse formation is highly antigen-dependent. J Immunol. 2003;171:4479–4483. doi: 10.4049/jimmunol.171.9.4479. [DOI] [PubMed] [Google Scholar]
- Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MG, Cantor H. Eta-1 (osteopontin): an early component of Type 1(cell-mediated) immunity. Science. 2000;287:860–864. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–36. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, Tybulewicz VL, Amigorena S. Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science. 2004a;305:1150–1153. doi: 10.1126/science.1099159. [DOI] [PubMed] [Google Scholar]
- Benvenuti F, Lagaudriere-Gesbert C, Grandjean I, Jancic C, Hivroz C, Trautmann A, Lantz O, Amigorena S. Dendritic cell maturation controls adhesion, synapse formation, and the duration of the interactions with naive T lymphocytes. J Immunol. 2004b;172:292–301. doi: 10.4049/jimmunol.172.1.292. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchill MA, Nardelli DT, England DM, DeCoster DJ, Christopherson JA, Callister SM, Schell RF. Inhibition of interleukin-17 prevents the development of arthritis in vaccinated mice challenged with Borrelia burgdorferi. Infect Immun. 2003;71:3437–3442. doi: 10.1128/IAI.71.6.3437-3442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Doodes PD, Glant TT, Finnegan A. IL-27 induces a Th1 immune response and susceptibility to experimental arthritis. J Immunol. 2008;180:922–930. doi: 10.4049/jimmunol.180.2.922. [DOI] [PubMed] [Google Scholar]
- Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT, Sobel RA, Lock C, Karpuj M, Pedotti R, Heller R, Oksenberg JR, Steinman L. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–1735. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- Dalpke AH, Schafer MK, Frey M, Zimmermann S, Tebbe J, Weihe E, Heeg K. Immunostimulatory CpG-DNA activates murine microglia. J Immunol. 2002;168:4854–4863. doi: 10.4049/jimmunol.168.10.4854. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DC, Zhang GX, El Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CJ, Gran B, Ciric B, Rostami A. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol. 2007;8:1372–1379. doi: 10.1038/ni1540. [DOI] [PubMed] [Google Scholar]
- Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med. 2002;195:695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, Lovett-Racke AE. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–1348. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–34. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Slavik JM, Anderson DE, O’Connor KC, De Jager P, Baecher-Allan C. Multiple sclerosis. Immunol Rev. 2005;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med. 2005;11:146–152. doi: 10.1038/nm1177. [DOI] [PubMed] [Google Scholar]
- Huber M, Steinwald V, Guralnik A, Brustle A, Kleemann P, Rosenplanter C, Decker T, Lohoff M. IL-27 inhibits the development of regulatory T cells via STAT3. Int Immunol. 2008;20:223–234. doi: 10.1093/intimm/dxm139. [DOI] [PubMed] [Google Scholar]
- Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol. 2007;8:74–83. doi: 10.1038/ni1415. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Jansson M, Panoutsakopoulou V, Baker J, Klein L, Cantor H. Attenuated experimental autoimmune encephalomyelitis in Eta-1/osteopontin-deficient mice. J Immunol. 2002;168:2096–2099. doi: 10.4049/jimmunol.168.5.2096. [DOI] [PubMed] [Google Scholar]
- Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–242. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- Kawakami N, Lassmann S, Li Z, Odoardi F, Ritter T, Ziemssen T, Klinkert WE, Ellwart JW, Bradl M, Krivacic K, Lassmann H, Ransohoff RM, Volk HD, Wekerle H, Linington C, Flugel A. The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J Exp Med. 2004;199:185–197. doi: 10.1084/jem.20031064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibundgut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E, Tybulewicz V, Brown GD, Ruland J, Reis e Sousa Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630–638. doi: 10.1038/ni1460. [DOI] [PubMed] [Google Scholar]
- Maldonado RA, Irvine DJ, Schreiber R, Glimcher LH. A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature. 2004;431:527–532. doi: 10.1038/nature02916. [DOI] [PubMed] [Google Scholar]
- O’Regan AW, Nau GJ, Chupp GL, Berman JS. Osteopontin (Eta-1) in cell-mediated immunity: teaching an old dog new tricks. Immunol Today. 2000;21:475–478. doi: 10.1016/s0167-5699(00)01715-1. [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paty DW, Li DK. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial UBC MS/MRI Study Group and the IFNB Multiple Sclerosis Study Group. Neurol. 1993;43:662–667. doi: 10.1212/wnl.43.4.662. [DOI] [PubMed] [Google Scholar]
- Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, Blumenschein WM, Mattson JD, Wagner JL, To W, Zurawski S, McClanahan TK, Gorman DM, Bazan JF, de Waal MR, Rennick D, Kastelein RA. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity. 2002;16:779–790. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- Renkl AC, Wussler J, Ahrens T, Thoma K, Kon S, Uede T, Martin SF, Simon JC, Weiss JM. Osteopontin functionally activates dendritic cells and induces their differentiation toward a Th1-polarizing phenotype. Blood. 2005;106:946–955. doi: 10.1182/blood-2004-08-3228. [DOI] [PubMed] [Google Scholar]
- Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone & Min Res. 1998;13(7):1101–1111. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- Sato T, Nakai T, Tamura N, Okamoto S, Matsuoka K, Sakuraba A, Fukushima T, Uede T, Hibi T. Osteopontin/Eta-1 upregulated in Crohn’s disease regulates the Th1 immune response. Gut. 2005;54:1254–1262. doi: 10.1136/gut.2004.048298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro S, Galboiz Y, Lahat N, Kinarty A, Miller A. The ‘immunological-synapse’ at its APC side in relapsing and secondary-progressive multiple sclerosis: modulation by interferon-beta. J Neuroimmunol. 2003;144:116–24. doi: 10.1016/j.jneuroim.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Shinohara ML, Jansson M, Hwang ES, Werneck MBF, Glimcher LH, Cantor H. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci U S A. 2005;102:17101–17106. doi: 10.1073/pnas.0508666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara ML, Kim HJ, Kim JH, Garcia VA, Cantor H. Alternative translation of Osteopontin generates intracellular and secreted isoforms that mediate distinct biological activities in dendritic cells. Proc Natl Acad Sci U S A. 2008;105:7235–7239. doi: 10.1073/pnas.0802301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara ML, Lu L, Bu J, Werneck MBF, Kobayashi K, Glimcher LH, Cantor H. Osteopontin expression is essential for IFN-α production by plasmacytoid dendritic cells. Nat Immunol. 2006;7:498–506. doi: 10.1038/ni1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L, Zamvil S. Transcriptional analysis of targets in multiple sclerosis. Nat Rev Immunol. 2003;3:483–492. doi: 10.1038/nri1108. [DOI] [PubMed] [Google Scholar]
- Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O’Shea J J, Hennighausen L, Ernst M, Hunter CA. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–45. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- Tompkins SM, Padilla J, Dal Canto MC, Ting JP, Van Kaer L, Miller SD. De novo central nervous system processing of myelin antigen is required for the initiation of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:4173–4183. doi: 10.4049/jimmunol.168.8.4173. [DOI] [PubMed] [Google Scholar]
- Umemura M, Yahagi A, Hamada S, Begum MD, Watanabe H, Kawakami K, Suda T, Sudo K, Nakae S, Iwakura Y, Matsuzaki G. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J Immunol. 2007;178:3786–3796. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- Yu M, Nishiyama A, Trapp BD, Tuohy VK. Interferon-beta inhibits progression of relapsing-remitting experimental autoimmune encephalomyelitis. J Neuroimmunol. 1996;64:91–100. doi: 10.1016/0165-5728(95)00160-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.