

Abstract
Listeria monocytogenes is an environmental bacterium that becomes a pathogen following ingestion by a mammalian host. The transition from environmental organism to pathogen requires significant changes in gene expression, including the increased expression of gene products that contribute to bacterial growth within host cells. PrfA is a L. monocytogenes transcriptional regulator that becomes activated upon bacterial entry into mammalian cells and induces the expression of gene products required for virulence. How PrfA activation occurs is not known, however several mutations have been identified that increase PrfA activity in strains grown in vitro (prfA* mutations). Here we describe a novel prfA mutation that enhances extracellular PrfA-dependent gene expression but in contrast to prfA* mutants inhibits the cytosol-mediated induction of virulence genes. prfA Y154C strains entered cells and escaped from phagosomes with an efficiency similar to wild type bacteria, however the mutation prevented efficient L. monocytogenes actin polymerization and reduced spread of bacteria to adjacent cells. The prfA Y154C mutation severely attenuated bacterial virulence in mice but the mutant strains did generate target antigen specific CD8+ effector cells. Interestingly, the prfA Y154C mutant was significantly less cytotoxic for host cells than wild type L. monocytogenes. The prfA Y154C mutant strain may therefore represent a novel attenuated strain of L. monocytogenes for antigen delivery with reduced host cell toxicity.
Keywords: PrfA, ActA, virulence regulation, gene expression
1. Introduction
Listeria monocytogenes is a gram-positive bacterium that survives and flourishes in a myriad of environments that range from soil and decaying vegetation to the cytosol of infected mammalian cells. The bacterium mediates the transition from environmental organism to mammalian pathogen via the regulated expression of gene products that promote survival in specific environmental niches. In the environment of a human host L. monocytogenes is responsible for serious food-borne infections resulting in bacteremia, meningitis, and neonatal death [1–4]. Fundamental to the transition of L. monocytogenes from environmental organism to pathogen is the transcriptional up-regulation of a number of gene products that facilitate host cell invasion, bacterial entry into the cytosol, intracellular replication, and spread to adjacent cells [2, 5–8]. The central virulence regulatory protein PrfA appears to function as a key switch to help mediate the transition from bacterial survival in the outside environment to replication within the human host [9]. It is required for the expression of most known L. monocytogenes virulence determinants [9–13].
PrfA is a 27 kD protein that is a member of the Crp/Fnr family of transcriptional activators [14]. Members of this family require the binding of small molecule co-factors (such as cAMP or carbon monoxide) or some form of post-translational modification for full activity [15, 16]. In some cases, mutations have been identified in these regulatory proteins that enable activation in the absence of co-factor binding or modification. For example, several mutations have been identified within crp (known as crp* mutations) that provide for activation of Crp-dependent gene expression in the absence of co-factor cAMP binding [17, 18]. No co-factor or post-translation modification has as of yet been identified for PrfA, but mutationally activated forms of PrfA (PrfA* mutants) have been isolated that provide for increased levels of PrfA-dependent gene expression outside of mammalian cells [19–22]. Strains containing prfA* mutations are fully virulent in mouse models of infection [21, 23]. In contrast, L. monocytogenes strains lacking functional PrfA remain trapped within host cell vacuoles and are severely attenuated for virulence in mice [24]. This study describes the isolation of a unique prfA mutant that facilitates L. monocytogenes entry into the cytosol of infected host cells but inhibits bacterial cell-to-cell spread and attenuates bacterial virulence.
2. Results
2.1 Isolation and identification of a L. monocytogenes isolate containing the prfA Y154C mutation
As part of an approach to study the effects of PrfA activation on L. monocytogenes pathogenesis, a genetic screen was undertaken to identify L. monocytogenes prfA mutant strains with altered expression of the PrfA-dependent virulence gene actA. actA is normally expressed at low to undetectable levels during bacterial growth in broth culture but its expression is highly induced (> 200-fold) in the mammalian cell cytosol [30, 37, 42, 43]. A plasmid (pNF1019) [19] containing the prfA promoters and the prfA coding region was maintained for several generations within a hypermutator strain of Escherichia coli and then introduced into a L. monocytogenes ΔprfA strain containing an actA transcriptional fusion to the reporter gene gus, encoding β-glucuronidase (GUS) [44]. Bacterial colonies with altered actA expression were identified via their blue color (indicating GUS activity) on indicator media plates. Under these conditions bacteria containing wild type prfA form white colonies (M. Miner, unpublished results). Approximately 40,000 bacterial colonies were examined, and one mutant (NF-L1215) was selected for further characterization based on its intermediate blue color. Mutant NF-L1215 exhibited an approximately four-fold increase in levels of actA expression in broth culture as measured by GUS activity in comparison to isogenic strains containing wild type prfA (Fig. 1A and B), and sequencing of the NF-L1215 prfA gene revealed a coding change resulting in the substitution of a cysteine for tyrosine at amino acid position 154 (Y154C). To confirm that the prfA Y154C mutation was responsible for the mutant phenotype observed, the mutation was introduced into the chromosome of the L. monocytogenes in place of the wild type copy of prfA by allelic exchange (resulting in strain NF-L1213) and the patterns of actA expression in NF-L1213 were found to be identical to those of the originally isolated NF-L1215 plasmid-insertion mutant, thus confirming the role of prfA Y154C in stimulating a moderate enhancement of actA expression in vitro (Fig. 1A and B).
Fig. 1. prfA Y154C strains exhibit enhanced virulence gene expression in vitro.

(A) Bacterial actA expression levels as measured by monitoring GUS activity. Levels of actA expression are indicated by symbols denoting GUS activity as measured at the indicated time points. GUS activity was measured for bacterial cultures grown in BHI broth and the units of GUS were normalized for optical density as described by Shetron-Rama et al., [38]. Each time point was measured in triplicate, and the data shown is representative of three experiments ± SEM. ◆, parent strain NF-L1124;  , prfA* mutant strain NF-L943 (prfA G155S);
, prfA* mutant strain NF-L943 (prfA G155S);  , original plasmid integration prfA Y154C mutant (NF-L1215);
, original plasmid integration prfA Y154C mutant (NF-L1215);  , isogenic chromosomal prfA Y154C (NF-L1213). Large symbols indicate GUS activity, the smaller symbols reflect bacterial growth as measured by optical density. (B) Detailed view of NF-L1124, NF-L1213, and NF-L1215 from panel A. (C) Measurement of phospholipase activity. Bacteria were grown on media containing egg-yolk; the white precipitate surrounding bacterial growth is indicative of PC-PLC activity. Both NF- L1213 and NF-L1215 prfA Y154C strains exhibit enhanced PC-PLC activity in comparison to wild type (NF-L1124) or ΔprfA (NF-L1123) strains, but have lower levels than the prfA* mutant strain NF-L943 (prfA G155S). Data shown is representative of two experiments. (D) Secreted hemolytic activity as measured by erythrocyte (RBC) lysis. Serial dilutions of bacterial supernatants were incubated with RBCs and cell lysis was determined by measuring absorbance at 450 nm (OD450). Data shown is representative of three experiments. Symbols: ◆, parent strain NF-L1124;
, isogenic chromosomal prfA Y154C (NF-L1213). Large symbols indicate GUS activity, the smaller symbols reflect bacterial growth as measured by optical density. (B) Detailed view of NF-L1124, NF-L1213, and NF-L1215 from panel A. (C) Measurement of phospholipase activity. Bacteria were grown on media containing egg-yolk; the white precipitate surrounding bacterial growth is indicative of PC-PLC activity. Both NF- L1213 and NF-L1215 prfA Y154C strains exhibit enhanced PC-PLC activity in comparison to wild type (NF-L1124) or ΔprfA (NF-L1123) strains, but have lower levels than the prfA* mutant strain NF-L943 (prfA G155S). Data shown is representative of two experiments. (D) Secreted hemolytic activity as measured by erythrocyte (RBC) lysis. Serial dilutions of bacterial supernatants were incubated with RBCs and cell lysis was determined by measuring absorbance at 450 nm (OD450). Data shown is representative of three experiments. Symbols: ◆, parent strain NF-L1124;  , isogenic chromosomal prfA Y154C (NF-L1213); ● ΔprfA (NF-L1123);
, isogenic chromosomal prfA Y154C (NF-L1213); ● ΔprfA (NF-L1123);  , prfA G155S (NF- L943).
, prfA G155S (NF- L943).
2.2 The prfA Y154C mutation results in enhanced expression of additional PrfA-dependent gene products in vitro
To determine if the prfA Y154C mutation influenced the expression of genes in addition to actA, two additional PrfA-dependent gene products were examined. Levels of phosphatidylcholine phospholipase C (PC-PLC), encoded by plcB, were enhanced (Fig. 1C) as anticipated given that plcB is co-transcribed with actA. The expression of listeriolysin O (LLO, encoded by hly) was also enhanced approximately three-fold in comparison to wild type strains [NF-L1124 (wild type prfA), 17 units +/− units; NF-L1213 (prfA Y154C), 50 units +/− 5 units] (Fig. 1D).
The levels of PrfA-dependent gene expression observed in BHI broth culture for the prfA Y154C strain were found to be modestly higher than wild type levels of expression but lower than those observed for the prfA* mutant prfA G155S [21] (Fig. 1A). When the mutant strains were grown in BHI broth treated with charcoal (BHIC), a condition known to enhance PrfA-dependent gene expression in vitro [23, 45], strains containing prfA Y154C exhibited only a modest increase in actA expression (an approximate 7 to 9-fold increase compared to BHI levels as measured by GUS activity) in comparison to the large increase (up to 93-fold) observed for the prfA* strain prfA E77K and a 33-fold increase observed for wild type L. monocytogenes (Table 3). The maximum actA expression levels as measured by GUS activity for both prfA G155S and prfA E77K strains grown in BHIC were significantly higher than either wild type or prfA Y154C strains (7312 units ± 137units and 7031 units ± 84 units versus 848 units ± 52 units and 1175 units ± 61 units, respectively) (Table 3). The prfA Y154C mutation thus differs from the prfA* mutants prfA G155S and prfA E77K in that it failed to stimulate high level expression of actA in BHIC. The prfA Y154C mutant also exhibited a lower fold induction of actA than wild type in BHIC but was similar to wild type with regards to the maximum levels of actA expression achieved in this media.
Table 3.
Fold change of actA/gus expression in BHIC compared to BHI and maximum GUS units produced during exponential growth
| Time (hours)a | OD600b | NF-L1123 (ΔprfA) | NF-L1124 (prfA) | NF-L924 (prfA E77K) | NF-L943 (prfA G155S) | NF-L1213 (prfA Y154C) |
|---|---|---|---|---|---|---|
| 1 | ~0.03 | 0.5c (26 ± 9.2)d | 1.1 (107 ± 13) | 3.8 (2980 ± 98) | 2.5 (5205 ± 409) | 0.9 (659 ± 0) |
| 3 | ~0.2 | 0.5 (6.7 ± 0) | 11 (404 ± 8.7) | 36 (7031 ± 84) | 20 (7312 ± 137) | 5.1 (735 ± 3.1) |
| 5 | ~1.2 | 0.5 (3.8 ± 0.4) | 33 (848 ± 52) | 77 (3764 ± 25) | 10 (4377 ± 184) | 6.9 (1175 ± 61) |
| 7 | ~2.0 | 1 (2.3 ± 0) | 17 (87 ± 0.4) | 93 (2654 ± 232) | 31 (874 ± 19) | 8.8 (190 ± 0.7) |
Hours growth in broth culture
Approximate bacterial culture optical density at 600 nm
Fold increase in GUS activity observed for growth in BHIC compared to growth in BHI at the same time point.
(Maximum GUS units in BHIC + SD)
2.3 PrfA Y154C fails to induce L. monocytogenes virulence gene expression within the cytosol of mammalian cells
While bacterial growth in BHIC for wild type strains results in a significant induction of actA expression in comparison to strains grown in BHI, expression levels are still low in comparison to the levels of actA expression achieved by bacteria located in the cytosol of infected host cells [38]. To determine if strains containing the prfA Y154C mutation were capable of fully inducing actA expression in the cytosol, mutant bacteria were first examined for their ability to gain entry and replicate within the cytosol of infected host cells (Fig. 2). Following bacterial uptake by J774 murine macrophage-like tissue culture cells, the prfA Y154C mutant was indistinguishable from wild type L. monocytogenes for intracellular growth (Fig. 2A). Based on rate and amount of intracellular bacterial replication, the prfA Y154C strain appeared to enter host cells and escape from cell vacuoles with an efficiency similar to that of wild type L. monocytogenes (Fig. 2A) and this same pattern was observed for the infection of epithelial cells (PtK2 cells) (Fig. 2B). In contrast, L. monocytogenes strains lacking functional PrfA remain trapped within host cell phagosomes and are unable to replicate [24]. However, when the NF-L1213 prfA Y154C strain was used to infect monolayers of murine fibroblast L2 cells, defects in bacterial cell-to-cell spread were readily observed (Fig. 2C). It has previously been demonstrated that defects in bacterial cell-to-cell spread can be visualized by the failure of a mutant strain to form plaques in infected monolayers or by an overall reduction in plaque size [27]. Surprisingly, despite the enhanced expression of hly and actA in BHI broth culture, the prfA Y154C mutant failed to form any visible plaques in cell monolayers after three days incubation (Fig. 2C). Only after prolonged incubation (seven days) were tiny plaques detectable (M. Miner, unpublished data). In contrast, wild-type L. monocytogenes and the prfA* mutants prfA G155S and prfA E77K {[19, 21] and M. Miner, unpublished results} formed multiple plaques following a three day incubation in cell monolayers.
Fig. 2. prfA Y154C strains gain entry and replicate within the cytosol of host cells.

L. monocytogenes strains were grown to stationary phase and used to infect tissue culture cells grown on glass coverslips. At 30 minutes (J774 cells) or 60 minutes (PtK2 cells) post-infection, monolayers were washed, gentamicin was added, and at the indicated time points cell monolayers were lysed and the number of bacteria per coverslip was determined. The results are expressed as the mean number of bacterial CFU + the standard deviation for three coverslips per time point. The data for one of three experiments with similar results is shown. (A) Bacterial intracellular growth in J774 macrophage-like tissue culture cells. (B) Bacterial intracellular growth in PtK2 epithelial cells. ◆, parent strain NF-L1124;  , prfA Y154C (NF-L1213). (C) Intracellular growth and cell-to-cell spread of L. monocytogenes in murine L2 fibroblasts, as indicated by plaque formation. The prfA Y154C strain failed to form visible plaques after three days of incubation, whereas multiple plaques were visible in monolayers infected with wild type bacteria. Data shown is representative of three experiments.
, prfA Y154C (NF-L1213). (C) Intracellular growth and cell-to-cell spread of L. monocytogenes in murine L2 fibroblasts, as indicated by plaque formation. The prfA Y154C strain failed to form visible plaques after three days of incubation, whereas multiple plaques were visible in monolayers infected with wild type bacteria. Data shown is representative of three experiments.
Consistent with its inability to form visible plaques after three days in cell monolayers, the mutant exhibited a profound delay in bacterial actin accumulation and polymerization in infected cells (Fig. 3A–G). At three hours post infection, approximately 70% of wild type bacteria were observed surrounded by actin clouds while 13% had progressed to having associated actin tails (Fig. 3A). In contrast, the prfA Y154C mutant at the same time point failed to accumulate actin of any form (Fig. 3B), and a trend of delayed actin association was seen for each time point examined (Fig. 3G). At 7 hours post-infection, small numbers of bacteria were observed associated with short actin tails (Fig. 3F), indicating that the prfA Y154C strain did eventually express sufficient intracellular actA to promote low amounts of actin polymerization.
Fig. 3. The prfA Y154C mutation reduces intracellular actA expression and bacterial actin association.

(A–F) Visualization by fluorescence microscopy of polymerized actin in PtK2 epithelial cells infected with L. monocytogenes. PtK2 cells were infected with either NF-L1124 (wild type prfA) (A, C, E) or NF-L1213 (prfA Y154C) (B, D, F) bacteria at an MOI of ~5:1. At one hour post-infection the monolayers were washed and gentamicin was added to kill any extracellular bacteria. At 3 hr (A, B), 5 hr (C, D), and 7 hr (E, F) post-infection the monolayers were fixed, permeabilized, and stained with NBD-phallicidin for filamentous actin (green) and an anti-Listeria antibody and tetramethylrhodamine goat anti-rabbit conjugated secondary antibody to detect bacteria (red) as described in Materials and Methods. Results shown are representative of two independent experiments. (G) Quantitation of actin association for wild type NF-L1124 and prfA Y154C strain NF-L1213. Actin association in the form of clouds (polymerized actin surrounding bacteria) or tails was scored for approximately 50 bacteria in infected cells from at least 10 independent microscopic fields for each time-point listed. Numbers listed represent the mean ± standard error derived from two independent experiments. (H) Bacterial intracellular actA expression as measured by GUS activity. Tissue culture dishes containing monolayers of PtK2 cells were infected with the indicated L. monocytogenes strains for 8 h as described in Materials and Methods. GUS activity was determined following lysis of the monolayers. The number of CFU per dish was determined by lysing infected PtK2 cells grown on coverslips in duplicate dishes and plating a portion of the lysates. Units of GUS are as described by Shetron-Rama et al. [38]. Each assay was done in triplicate and the data represent the mean ± standard deviation for at least three individual experiments.
The ability of prfA Y154C mutant strains to gain access to and replicate within the host cytosol enabled direct comparison of the intracellular levels of actA expression between wild type and mutant strains. The ability of L. monocytogenes to direct intracellular actin-based bacterial motility is dependent upon the abundant expression of the actA gene product in the host cytosol [35] and actA expression has been shown to increase at least 200-fold following bacterial escape from the phagosome [30, 37, 42, 43]. Intracellular actA expression was therefore measured in wild type and prfA Y154C strains following the infection of PtK2 epithelial cells. Although the L. monocytogenes prfA Y154C mutant entered the cytosol and replicated with kinetics that were comparable to the wild type strain (Fig. 3A – F and M. Miner, unpublished results), the mutant exhibited only a modest increase in actA expression (an approximately 10-fold increase in actA expression in comparison to BHI) (Fig. 3H). In contrast, levels of cytosolic actA expression increased in wild type strains over 1000-fold in comparison to BHI grown cultures (Fig. 3H)
2.4 The prfAY154C mutant is less cytotoxic to infected host cells
Interestingly, the prfA Y154C mutant was also found to be significantly less cytotoxic to infected host cells than wild type bacteria based on the observed release of cellular lactate dehydrogenase (LDH) (P < 0.001) and on microscopic examination of infected cells (Fig. 4A–C). This reduction in cytotoxicity was not associated with the failure of the mutant to efficiently spread to adjacent cells as an actA deletion mutant completely defective for cell-to-cell spread exhibited levels of cytotoxicity essentially equivalent to those observed for wild type strains (P > 0.05) (Fig. 4A). Two PrfA-dependent gene products, LLO and PC-PLC, have been demonstrated to be major contributors to L. monocytogenes-mediated host cell cytotoxicity in multiple cell types, including PtK2 cells [30, 40, 46]. plcB (encoding PC- PLC) is co-transcribed with actA and intracellular transcripts directed by the actA promoter are reduced in the PrfA Y154C mutant strain (Fig. 3H). It was hypothesized that a block in PrfA Y154C activation would result not only in a decrease in PC-PLC activity but also in intracellular LLO production. Immunoprecipitation of radioactively labeled LLO from infected cells indicated that intracellular LLO production was indeed reduced by approximately 64% in PrfA Y154C mutants in comparison to wild type bacteria (Fig. 4D). These data are consistent with the failure of prfA Y154C to up- regulate PrfA-dependent target gene expression within the cytosol of infected host cells.
Fig. 4. The prfA Y154C mutation reduces bacterial cytotoxicity for host cells and intracellular LLO expression.

(A) Cytotoxicity was measured by lactate dehydrogenase (LDH) release from bacteria-infected PtK2 epithelial cells at 24 hours post-infection with the indicated strains. Maximum release (max) was determined from complete host cell lysis and set to 100%; all values are expressed as a percentage of max ± standard deviation. The data shown is representative of three independent experiments. (B–C) Cytotoxicity visualized by microscopy of PtK2 cells infected with the parent strain (NF- L1124) (B) or prfA Y154C (NF-L1213) (C) at 24 hours post-infection. PtK2 cells were infected with bacteria at an MOI of ~5:1. At one hour post-infection the monolayers were washed and gentamicin was added to kill any extracellular bacteria. At 24 hr post-infection the monolayers were fixed and stained with Diff Quik stain. Data shown is representative of three independent experiments. (D) Intracellular expression of LLO by L. monocytogenes in PtK2 cells. PtK2 cells were infected with L. monocytogenes: at 5 hr post-infection the infected cells were starved for methionine and cysteine and host protein synthesis was blocked as described in Materials and Methods. Infected cells were incubated with [35S]-methionine for 1 hr prior to host cell lysis and immunoprecipitation with monoclonal anti-LLO antibody B3–19 [41]. The amount of sample loaded per lane was normalized for the number of bacteria. An autoradiograph representative of two independent experiments is shown, which was exposed for 100 hours using the Storm Phosphorimager and analyzed with ImageQuant software. The arrow indicates the position of the LLO band, which is approximately 58 kD.
2.5 The L. monocytogenes prfA Y154C mutant is severely attenuated for virulence in mice but generates antigen specific CD8+ T cells
Given that the prfA Y154C mutation reduced the ability of L. monocytogenes to spread within infected cell monolayers, the capacity of the mutant strain to cause disease in infected animals was examined by determining the LD50 value following intravenous infection of BALB/c mice. Whereas the LD50 for wild type L. monocytogenes was approximately 2 × 104 CFU, the LD50 for the prfA Y154C mutant was approximately 3 × 106 CFU, which equates to a 150-fold reduction in virulence. This level of attenuation differs from that observed for strains completely lacking prfA as the absence of prfA or of actA expression results in severe attenuation [LD50 values of 3×109 and 2×107 for ΔprfA and ΔactA, respectively [26, 47]]. Consistent with low level PrfA activity (versus no PrfA activity) the prfA Y154C mutant was capable of bacterial replication in the spleens of infected mice at days one and two post-infection, with a decline in bacterial numbers occurring at day three (Fig. 5, left panel). In contrast, no bacterial replication was observed for the ΔactA mutant even when animals were inoculated with larger numbers of bacteria (Fig. 5, left panel). In order to assess whether diminished in vivo replication altered the subsequent stimulation of antigen specific CD8+ cells following injection, mice were immunized with either wildtype or the prfA Y154C strain, and 7 days later, the frequency of LLO91–99 or p60217–225 specific IFN-γ secreting cells determined by intracellular cytokine staining. Target antigen specific IFN-γ secreting CD8+ cells were found to be stimulated following immunization, and the levels of the responses were not statistically different from the levels detected following immunization with wild type (Fig. 5, right panel). However, the response stimulated by prfA Y154C strains is dependent upon the immunization dose, as indicated by negligible levels of peptide specific cells measured at the 520 CFU dose. Collectively these results show that the prfA Y154C mutation allows for in vivo bacterial replication and stimulation of target antigen specific CD8+ effector cells while reducing host cell cytotoxicity and bacterial virulence.
Fig. 5. The prfA Y154C mutant is capable of limited growth within the spleens of infected animals and stimulates target antigen specific CD8+ effector cells.
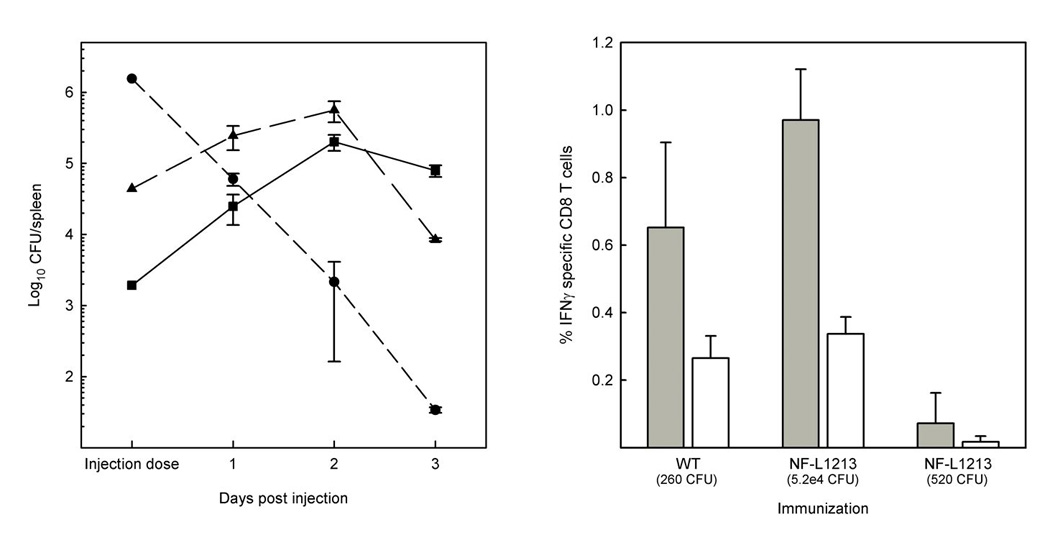
Left panel: BALB/c mice were injected intravenously with wild type (■, 1900 CFU), ΔactA (●, 1.55 × 106 CFU), or prfA Y154C (▲, 4.4 × 104 CFU). On the days indicated the spleens from 2–3 mice were homogenized individually and numbers of CFU/spleen determined by dilution plating. Right panel. BALB/c mice were immunized with either wildtype L. monocytogenes or prfA Y154C (NF-L1213) at the indicated doses. Seven days later, spleens were removed, a single cell suspensions prepared and the frequency of LLO91–99 (grey bars) or p60217–225 (white bars) specific IFN-γ producing CD8+ cells by determined by intracellular cytokine staining.
3. Discussion
This study describes the isolation of a novel prfA mutation that promotes L. monocytogenes entry into the cytosol but inhibits actin-based bacterial motility, thereby attenuating bacterial virulence. Although the PrfA Y154C mutation stimulates modestly enhanced levels of PrfA-dependent gene expression during bacterial growth in BHI, the mutation is distinct from all other prfA mutations in that it facilitates phagosome lysis but inhibits PrfA-dependent gene expression within the host cell cytosol. The reduced expression of bacterial virulence genes within the cytosol decreased bacterial toxicity to infected host cells. The prfA Y154C mutant therefore represents a novel attenuated form of L. monocytogenes that is capable of antigen delivery into the cytosol in the absence of LLO and PC-PLC-mediated cytotoxicity.
The prfA Y154C mutation is notably distinct from a prfA loss of function mutation as demonstrated by the ability of prfA Y154C strains to invade host cells and enter the cytosol at rates equivalent to wild type bacteria, a phenotype not shared by prfA loss of function strains [10, 47]. This indicates that the prfA Y154C mutation provides sufficient expression of PrfA-dependent gene products for host cell invasion and phagosomal escape. How might the prfA Y154C mutation modify PrfA function so as to impede cytosolic induction of PrfA-dependent genes? The PrfA Y154C mutation is located in a region of the protein associated with amino acid substitutions resulting in the mutational activation of PrfA (PrfA* mutants), including the PrfA G145S, PrfA G155S, and PrfA L140F mutations [19, 21, 22, 48, 49]. Studies by Eiting et al [48] have compared the structures of wild type PrfA with the PrfA G145S mutant protein and their data suggests that PrfA activation results in a conformational change that repositions the helix-turn-helix DNA binding region of PrfA such that it becomes more accessible for substrate. Eiting et al. [48] and additional investigators [22] have shown that PrfA G145S has a higher binding affinity for PrfA target promoters. It is thus possible, based on its location that the PrfA Y154C mutation interferes with or prevents the complete repositioning of the helix-turn-helix domain that occurs following co-factor binding. The potential failure of PrfA Y154C to undergo a conformational shift following bacterial entry into the cytosol could account for the low levels of actA expression observed for the mutant in infected cells. Alternatively, it is possible that the Y154C mutation may somehow decrease PrfA protein stability in cytosolic bacteria.
L. monocytogenes strains lacking functional ActA are also capable of reaching the host cell cytosol and are defective for cell-to-cell spread [50]. However, ΔactA strains differ from prfA Y154C in that these strains maintain wild type L. monocytogenes secretion levels of LLO and PC-PLC and show levels of cytotoxicity that are not significantly different from wild type strains (P > 0.05) (Fig. 4). In addition, ΔactA strains do not exhibit an increase in bacterial CFU in liver and spleen following infection of mice (Fig. 5A). The prfA Y154C strain may thus present a novel means of delivering antigen to the cytosol in that, in contrast to ΔactA (and presumably auxotrophic mutants as well), the mutant replicates but does not efficiently induce high levels of LLO or PlcB secretion thereby potentially extending the amount of time that antigen could be delivered in the absence of host cell lysis.
4. Materials and Methods
4.1 Bacterial strains
L. monocytogenes strains used in this study are listed in Table 1. All L. monocytogenes strains were derived from 10403S (serotype 1/2a) and unless specified were grown in brain-heart infusion (BHI) broth. Bacteria containing integrated copies of plasmid vector pPL2 [25]were grown in 7.5 ug/ml chloramphenicol for vector maintenance. α-select E. coli (Bioline) was used for cloning and maintenance of plasmid constructs and was grown in Luria Broth (LB) at 37° C. SM10 E. coli was used for pPL2 vector-derived plasmid conjugation into L. monocytogenes.
Table 1.
L. monocytogenes and E. coli strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| NF-L100 | 10403S WT | [39] |
| DP-L1492 | 10403S ΔactA | [26] |
| DP-L1261 | 10403S Δhly | [51] |
| NF-E470 | E. coli pKSV7-actA-gus-plcB | [30] |
| NF-L476 | NF-L100 actA-gus-plcB | [30] |
| NF-L890 | NF-L100 ΔprfA | [19] |
| NF-E908 | E. coli pBEST501 | [29] |
| NF-L924 | NF-L476 prfA E77K | [21] |
| NF-L943 | NF-L476 prfA G155S | [21] |
| NF-L1003 | NF-L890 actA-gus-plcB | [19] |
| NF-E1019 | E. coli pPL2-pplcA-pprfA-prfA | [19] |
| NF-L1123 | NF-L890 actA-gus-neo-plcB | This study |
| NF-L1124 | NF-L100 actA-gus-neo-plcB | This study |
| NF-L1215 | NF-L1003 pPL2-pplcA-pprfA-prfA Y154C | This study |
| NF-L1213 | NF-L1124 prfA Y154C | This study |
| NF-E1232 | E. coli pKSV7-prfA Y154C | This study |
| NF-E1118 | E. coli pKSV7-actA-gus-neo-plcB | This study |
| NF-E1146 | E. coli pKSV7-prfA | This study |
4.2 Tissue culture cells
The murine macrophage-like cell line J774 and L2 mouse fibroblasts were grown in DMEM plus 10% FBS plus mM glutamine at 37° C with 5% CO2 [26, 27]. The Potoroo rat kidney cell line PtK2 was grown in MEM plus 10% FBS plus 2 mM glutamine at 37° C with 5% CO2 [28].
4.3 Genetic screen for prfA mutations that lead to enhanced actA expression
Plasmid pNF1019 containing prfA under the control of the prfAP1, prfAP2, and plcA promoters in the integrative plasmid vector pPL2 (pNF1019) [19] was transformed into chemically competent XL1 Red E. coli hypermutator bacterial cells (Stratagene). Selected transformants were inoculated into LB at 1:1000 dilution and grown with shaking to stationary phase at 37°C with shaking. Cultures were repeatedly diluted and grown to stationary phase for a total of four cycles. The pNF1019 plasmid was then purified from XL1 Red and introduced via electroporation into conjugation competent SM10 cells. Transfer of pNF1019 from SM10 into L. monocytogenes ΔprfA was carried out as described previously [19]. Transconjugant prfA mutants exhibiting enhanced actA expression were identified as blue colonies on BHI plates containing 7.5 ug/ml chloramphenicol, 200 ug/ml streptomycin, and 50 ug/ml 5-bromo-4-chloro-3-indolyl- beta-D-glucuronic acid (X-gluc).
4.4 Construction of plasmids and L. monocytogenes recombinant strains
pNF1118, containing transcriptional fusions of gus and neo to L. monocytogenes actA in the temperature sensitive shuttle vector pKSV7 (pKSV7-actA-gus-neo-plcB), was generated by cloning the gene for neomycin resistance derived from pBEST501 [29] downstream of gus in plasmid pNF470 [30] using primers NEO-RBS and NEO-END (Table 2). Strains NF-L1123 (ΔprfA, actA-gus-neo-plcB) and NF-L1124 (actA-gus-neo-plcB) were constructed by introducing the actA-gus-neo-plcB fusion into the L. monocytogenes chromosome by allelic exchange using pNF1118 in NF-L890 (ΔprfA) and 10304S respectively. For the construction of an isogenic prfA Y154C strain, pNF1146 (pKSV7-prfA) was generated by PCR amplification of prfA coding sequences (from nucleotide +3 to +841 relative to the ATG start codon) from genomic DNA isolated from 10403S using primers prfA-pstI-Amplify-+3F and prfA-pstI-Amplify-+841R (Table 2). The PCR product was digested with PstI and ligated into PstI-digested pKSV7. The prfA Y154C mutations was introduced into pNF1146 using the Quik Change site directed mutagenesis (Stratagene) with plasmid pNF1146 (pKSV7-prfA) serving as a template for primers Y154C quik 5’ and Y154C quik 3’ (Table 2). The resulting plasmid, pNF1232, contained the prfA Y154C mutation and was used to replace the wild type chromosomal copy of prfA in NF-L1124 via allelic exchange as previously described [21].
Table 2.
Oligonucleotide primers used in this study
| Name | Sequence 5’-3’ | Characteristics |
|---|---|---|
| NEO-RBS | GCGCTGCAGAGGAGGAAAAATATGAATGGACCAATAATAATGACT | Neo 5’ RBS and PstI site |
| NEO-END | GCGCTGCAGCGTTCAAAATGGTATGCGTTTTGA | Neo 3’ PstI site |
| Y154C quik 5’ | TCCTGACCTATGTGTGTGGTAAAGAAACTCCTG | Y154C 5’ quik change |
| Y154C quik 3’ | CAGGAGTTTCTTTACCACACACATAGGTCAGGA | Y154C 3’ quik change |
| prfA-PstI-amplify-+3F | GCGCTGCAGGAACGCTCAAGCAGAAGAATTC | prfA 5’ PstI site |
| prfA-PstI-amplify-+841R | GCGCTGCAGGGAACAACTATCTGTTGCAGCTC | prfA 3’ PstI site |
4.5 Phospholipase activity
Phospholipase activity was measured as previously described [31]. Egg yolk overlay plates were made by mixing egg yolks 1:1 with PBS and added to BHI agar at 5% (vol/vol). Bacteria were streaked on egg yolk agar plates and incubated at 37° C overnight.
4.6 Hemolytic activity
Hemolytic activity was measured as previously described [32,33]. Briefly, bacteria were grown without shaking overnight at 30° C, the bacterial cells were removed using centrifugation, and two-fold serial dilutions of bacterial supernatants were incubated with PBS-washed sheep red blood cells for 30 minutes at 37° C. After incubation, cells were recovered by centrifugation to measure 50% lysis and supernatants were read in a spectrophotometer plate reader at OD450. Hemolytic units are reported in the text as the reciprocal of the supernatant dilution resulting in 50% lysis of red blood cells [34].
4.7 Measurement of in vitro GUS activity
GUS activity was measured as previously described [35]. In short, bacteria were grown overnight in BHI at 30° C without shaking, diluted 1:25 in fresh media and grown with shaking at 37° C. For media treated with activated charcoal, a heat-sterilized 5% (w/v) stock suspension of HCl washed activated charcoal powder (Sigma) in ultrapure water was added to BHI broth to 0.2% final volume before autoclaving, stirred for 1 h, then autoclaved and filtered through a 0.22 µm membrane. At the indicated time points, the OD600 was determined for each culture using a spectrophotometer (UV-1201 by Shimadzu). Bacterial pellets were harvested from 500ul of culture and resuspended in ABT buffer (0.1 M potassium phosphate, pH 7.0, 0.1 M NaCl, .1% Triton-X 100). GUS activity was measured as described by Youngman [36] with the substitution of 4-methylumbelliferyl-β-d-glucuronide (Sigma) in place of 4-methylumbelliferyl- β-d-galactoside [37].
4.8 Intracellular GUS assay
Measurement of intracellular GUS activity was carried out as described, modified for PtK2 cells [37]. Monolayers of PtK2 cells were grown to confluency on treated 60-mm petri dishes (Nunclon by Nunc). PtK2 cells were also grown on coverslips in duplicate 60-mm dishes. Monolayers were lysed at 8 hours post-infection in 200ul ABT buffer. 4-methylumbelliferyl- β -d-galactoside was replaced with 4-methylumbelliferyl-β-d-glucuronide (Sigma). At 2, 4, 6, and 8 hours post-infection, three coverslips were removed, cells were lysed, and bacteria plated for CFUs. GUS units were determined based on the number of bacterial CFU recovered as previously described [38].
4.9 Plaque formation
Plaque assays were performed with murine L2 fibroblasts as described, infecting 6-well plates of confluent cells with 20ul of 1:250 diluted overnight culture grown at 30° C in BHI without shaking [27].
4.10 Intracellular bacterial growth and fluorescence
Intracellular growth in J774 macrophage-like cells were performed as previously described [21, 39] with an MOI of ~1:5. Intracellular growth and fluorescence in PtK2 cells were performed as previously described [21] with an MOI of ~5:1. Actin association was quantified by fluorescence microscopy with a 100X objective by counting bacterial cells either enveloped in actin clouds or associated with actin tails. Ten separate fields were counted for a minimum of fifty bacteria within infected cells for each strain and the numbers were averaged for two independent experiments.
4.11 Cytotoxicity measurement
Cytotoxicity was measured by lactate dehydrogenase (LDH) release from infected PtK2 cells as described [40] using CytoTox 96 Non- Radioactive Cytotoxicity Assay (Promega) according to manufacturer’s instructions. PtK2 cells were infected with an MOI of 100:1 and LDH release was measured 24 hours post-infection. Percent cytotoxicity = 100 × [(experimental LDH release) - (spontaneous LDH release)]/[(maximum LDH release) − (spontaneous LDH release)].
4.12 Intracellular metabolic labeling of LLO
[35S]-methionine labeling of bacterial proteins was performed as previously described [37]. PtK2 cells were infected with an MOI of 100:1 in 35-mm dishes. At 1 hour post-infection, cells were washed 3 times with PBS and media replaced with gentamicin (10 ug/ml). At hours p.i. the media was replaced with methionine-free media (methionine, cysteine-free DMEM, 10% dialyzed FBS, gentamicin) that contained protein synthesis inhibitors cycloheximide (22.5 ug/ml) and anisomycin (30 ug/ml). Thirty minutes later, fresh methionine-free media was added containing 200 uCi of [35S]-methionine (EasyTag Express35S Protein Labeling Mix, NEN, PerkinElmer) for 1 hour. Monolayers were then washed 3 times with PBS and lysed with 500 microliters RIPA buffer (150 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS) containing protease inhibitors (Mini Complete, Roche, and 10 mM EDTA) and incubated rotating at 4° for 30 minutes. Lysates were then passed through 25 5/8 gauge needle and then centrifuged for 30 minutes at 4° to pellet bacteria. LLO was then immunoprecipitated using monoclonal antibody B3–19 [41] as described previously [26]. Immunoprecipitated samples were subjected to SDS-PAGE, transferred to nitrocellulose and processed for autoradiography using Storm phosphorimager and ImageQuant software. In a parallel experiment, PtK2 monolayers on coverslips were infected, the monolayers were lysed at 5 hours post-infection, and bacterial CFUs were enumerated on solid media to enable normalization of bacterial numbers.
4.13 Mouse infections
Fifty percent lethal doses (LD50) were determined in BALB/c mice by intravenous injection as described previously according to IACUC approved procedures [39]. To evaluate in vivo growth, naïve BALB/c mice were intravenously infected with L. monocytogenes wild type 10403S, prfA Y154C, or ΔactA strains. At various time-points post-infection, spleens from three mice were homogenized and serial dilutions of the homogenate were plated onto BHI agar to enumerate the bacterial load.
4.14 Intracellular cytokine staining and flow cytometry
BALB/c mice were immunized with wildtype of NF-L1213, and 7 days later, spleens were removed and single cell suspensions prepared by passing the tissue over a cell strainer. Intracellular cytokine staining was performed by culturing splenocytes ex vivo for 5 h with 10−6 M peptide in the presence of Brefeldin A (BioLegend). Surface staining for CD3 and CD8 and intracellular staining for IFNγ was performed with the Cytofix/Cytoperm kit according to the manufacturer’s directions (BD Biosciences). Cells were stained with FITC, PE, Pacific Blue and/or APC-conjugated antibodies specific for CD8α (clone 53–6.7, eBioscience), CD3 (clone 145-2C11, eBioscience), and IFNγ(clone XMG1.2, eBioscience). Data acquisition was performed on a BD LSR II flow cytometer, and was analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Acknowledgements
We thank Dr. Patrick Piggot for the pBEST501 plasmid, Dr. Hao Shen and Dr. Jeff Miller for the L. monocytogenes ΔprfA strain, Dr. Amy Decatur for her kind gift of the B3–19 antibody, and members of the Freitag lab for helpful discussions. This work was supported by Public Health Service grants AI41816 (N.E.F), by AI056446 (H.G.A.B.), by a VA Merit Award (H.G.A.B.), by a NIAID Bacterial Pathogenesis training grant fellowship AI55396 (M.D.M), a National Science Foundation Graduate Research Fellowship (NSF-GRF) (G.C.P.), and by the M. J. Murdock Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gellin BG, Broome CV. Listeriosis. Jama. 1989;261:1313–1320. [PubMed] [Google Scholar]
- 2.Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramaswamy V, Cresence VM, Rejitha JS, Lekshmi MU, Dharsana KS, Prasad SP, Vijila HM. Listeria - review of epidemiology and pathogenesis. J Microbiol Immunol Infect. 2007;40:4–13. [PubMed] [Google Scholar]
- 4.Posfay-Barbe KM, Wald ER. Listeriosis. Pediatr Rev. 2004;25:151–159. doi: 10.1542/pir.25-5-151. [DOI] [PubMed] [Google Scholar]
- 5.Kreft J, Vazquez-Boland JA. Regulation of virulence genes in Listeria. Int J Med Microbiol. 2001;291:145–157. doi: 10.1078/1438-4221-00111. [DOI] [PubMed] [Google Scholar]
- 6.Sheehan B, Kocks C, Dramsi S, Gouin E, Klarsfeld AD, Mengaud J, Cossart P. Molecular and genetic determinants of the Listeria monocytogenes infectious proces. Curr Top Microbiol Immunol. 1994;192:187–216. doi: 10.1007/978-3-642-78624-2_9. [DOI] [PubMed] [Google Scholar]
- 7.Dussurget O, Pizarro-Cerda J, Cossart P. Molecular determinants of Listeria monocytogenes virulence. Annu Rev Microbiol. 2004;58:587–610. doi: 10.1146/annurev.micro.57.030502.090934. [DOI] [PubMed] [Google Scholar]
- 8.Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- 9.Gray MJ, Freitag NE, Boor KJ. How the bacterial pathogen Listeria monocytogenes mediates the switch from environmental Dr. Jekyll to pathogenic Mr. Hyde. Infect Immun. 2006;74:2505–2512. doi: 10.1128/IAI.74.5.2505-2512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leimeister-Wachter M, Haffner C, Domann E, Goebel W, Chakraborty T. Identification of a gene that positively regulates expression of listeriolysin, the major virulence factor of Listeria monocytogenes. Proc Natl Acad Sci U S A. 1990;87:8336–8340. doi: 10.1073/pnas.87.21.8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leimeister-Wachter M, Goebel W, Chakraborty T. Mutations affecting hemolysin production in Listeria monocytogenes located outside the listeriolysin gene. FEMS Microbiol Lett. 1989;53:23–29. doi: 10.1016/0378-1097(89)90360-1. [DOI] [PubMed] [Google Scholar]
- 12.Chakraborty T, Leimeister-Wachter M, Domann E, Hartl M, Goebel W, Nichterlein T, Notermans S. Coordinate regulation of virulence genes in Listeria monocytogenes requires the product of the prfA gene. J Bacteriol. 1992;174:568–574. doi: 10.1128/jb.174.2.568-574.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miner MD, Port GC, Freitag NE. Regulation of Listeria monocytogenes virulence genes. In: Shen H, Goldfine H, editors. Listeria monocytogenes : pathogenesis and host response. Springer-Verlag: 2007. pp. 139–158. [Google Scholar]
- 14.Lampidis R, Gross R, Sokolovic Z, Goebel W, Kreft J. The virulence regulator protein of Listeria ivanovii is highly homologous to PrfA from Listeria monocytogenes and both belong to the Crp-Fnr family of transcription regulators. Mol Microbiol. 1994;13:141–151. doi: 10.1111/j.1365-2958.1994.tb00409.x. [DOI] [PubMed] [Google Scholar]
- 15.Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 16.Korner H, Sofia HJ, Zumft WG. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev. 2003;27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- 17.Garges S, Adhya S. Sites of allosteric shift in the structure of the cyclic AMP receptor protein. Cell. 1985;41:745–751. doi: 10.1016/s0092-8674(85)80055-6. [DOI] [PubMed] [Google Scholar]
- 18.Harman JG, Peterkofsky A, McKenney K. Arginine substituted for leucine at position 195 produces a cyclic AMP-independent form of the Escherichia coli cyclic AMP receptor protein. J Biol Chem. 1988;263:8072–8077. [PubMed] [Google Scholar]
- 19.Wong KK, Freitag NE. A novel mutation within the central Listeria monocytogenes regulator PrfA that results in constitutive expression of virulence gene products. J Bacteriol. 2004;186:6265–6276. doi: 10.1128/JB.186.18.6265-6276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ripio MT, Dominguez-Bernal G, Lara M, Suarez M, Vazquez-Boland JA. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol. 1997;179:1533–1540. doi: 10.1128/jb.179.5.1533-1540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shetron-Rama LM, Mueller K, Bravo JM, Bouwer HG, Way SS, Freitag NE. Isolation of Listeria monocytogenes mutants with high-level in vitro expression of host cytosol-induced gene products. Mol Microbiol. 2003;48:1537–1551. doi: 10.1046/j.1365-2958.2003.03534.x. [DOI] [PubMed] [Google Scholar]
- 22.Vega Y, Rauch M, Banfield MJ, Ermolaeva S, Scortti M, Goebel W, Vazquez-Boland JA. New Listeria monocytogenes prfA* mutants, transcriptional properties of PrfA* proteins and structure-function of the virulence regulator PrfA. Mol Microbiol. 2004;52:1553–1565. doi: 10.1111/j.1365-2958.2004.04052.x. [DOI] [PubMed] [Google Scholar]
- 23.Ripio MT, Dominguez-Bernal G, Suarez M, Brehm K, Berche P, Vazquez-Boland JA. Transcriptional activation of virulence genes in wild-type strains of Listeria monocytogenes in response to a change in the extracellular medium composition. Res Microbiol. 1996;147:371–384. doi: 10.1016/0923-2508(96)84712-7. [DOI] [PubMed] [Google Scholar]
- 24.Freitag NE, Rong L, Portnoy DA. Regulation of the prfA transcriptional activator of Listeria monocytogenes: multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect. Immun. 1993;61:2537–2544. doi: 10.1128/iai.61.6.2537-2544.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage Integration Vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci U S A. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun AN, Camilli A, Portnoy DA. Isolation of Listeria monocytogenes small- plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun. 1990;58:3770–3778. doi: 10.1128/iai.58.11.3770-3778.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA. The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- 29.Chary VK, Amaya EI, Piggot PJ. Neomycin- and spectinomycin-resistance replacement vectors for Bacillus subtilis. FEMS Microbiol Lett. 1997;153:135–139. doi: 10.1111/j.1574-6968.1997.tb10474.x. [DOI] [PubMed] [Google Scholar]
- 30.Shetron-Rama LM, Marquis H, Bouwer HG, Freitag NE. Intracellular induction of Listeria monocytogenes actA expression. Infect Immun. 2002;70:1087–1096. doi: 10.1128/IAI.70.3.1087-1096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mueller KJ, Freitag NE. Pleiotropic enhancement of bacterial pathogenesis resulting from the constitutive activation of the Listeria monocytogenes regulatory factor PrfA. Infect Immun. 2005;73:1917–1926. doi: 10.1128/IAI.73.4.1917-1926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kingdon GC, Sword CP. Biochemical and Immunological Effects of Listeria monocytogenes Hemolysin. Infect Immun. 1970;1:363–372. doi: 10.1128/iai.1.4.363-372.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Camilli A, Paynton CR, Portnoy DA. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc Natl Acad Sci U S A. 1989;86:5522–5526. doi: 10.1073/pnas.86.14.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camilli A, Paynton CR, Portnoy DA. Intracellular methicillin selection of Listeria monocytogenes mutants unable to replicate in a macrophage cell line. Proc. Natl. Acad. Sci. USA. 1989;86:5522–5526. doi: 10.1073/pnas.86.14.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wong KK, Bouwer HG, Freitag NE. Evidence implicating the 5' untranslated region of Listeria monocytogenes actA in the regulation of bacterial actin-based motility. Cell Microbiol. 2004;6:155–166. doi: 10.1046/j.1462-5822.2003.00348.x. [DOI] [PubMed] [Google Scholar]
- 36.Youngman P. Plasmid Vectors for Recovering and Exploiting Tn917 Transpositions in Bacillus and Other Gram-Positive Bacteria. In: Hardy K, editor. Plasmids: a Practical Approach. Oxford: IRL Press; 1987. pp. 79–103. 79–103. [Google Scholar]
- 37.Moors MA, Levitt B, Youngman P, Portnoy DA. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect Immun. 1999;67:131–139. doi: 10.1128/iai.67.1.131-139.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shetron-Rama LM, Marquis H, Bouwer HGA, Freitag NE. Intracellular induction of Listeria monocytogenes actA expression. Infect Immun. 2002;70:1087–1096. doi: 10.1128/IAI.70.3.1087-1096.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Portnoy DA, Jacks SP, Hinrichs DJ. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med. 1988;167:1459–1771. doi: 10.1084/jem.167.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Decatur AL, Portnoy DA. A PEST-like sequence in listeriolysin O essential for Listeria monocytogenes pathogenicity. Science. 2000;290:992–995. doi: 10.1126/science.290.5493.992. [DOI] [PubMed] [Google Scholar]
- 41.Nato F, Reich K, Lhopital S, Rouyre S, Geoffroy C, Mazie JC, Cossart P. Production and characterization of neutralizing and nonneutralizing monoclonal antibodies against listeriolysin O. Infect Immun. 1991;59:4641–4646. doi: 10.1128/iai.59.12.4641-4646.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bubert A, Sokolovic Z, Chun SK, Papatheodorou L, Simm A, Goebel W. Differential expression of Listeria monocytogenes virulence genes in mammalian host cells. Mol Gen Genet. 1999;261:323–336. doi: 10.1007/pl00008633. [DOI] [PubMed] [Google Scholar]
- 43.Freitag NE, Jacobs KE. Examination of Listeria monocytogenes intracellular gene expression by using the green fluorescent protein of Aequorea victoria. Infect Immun. 1999;67:1844–1852. doi: 10.1128/iai.67.4.1844-1852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karow ML, Piggot PJ. Construction of gusA transcriptional fusion vectors for Bacillus subtilis and their utilization for studies of spore formation. Gene. 1995;163:69–74. doi: 10.1016/0378-1119(95)00402-r. [DOI] [PubMed] [Google Scholar]
- 45.Ermolaeva S, Novella S, Vega Y, Ripio MT, Scortti M, Vazquez-Boland JA. Negative control of Listeria monocytogenes virulence genes by a diffusible autorepressor. Mol Microbiol. 2004;52:601–611. doi: 10.1111/j.1365-2958.2004.04003.x. [DOI] [PubMed] [Google Scholar]
- 46.Glomski IJ, Gedde M, Tsang AW, Swanson JA, Portnoy DA. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol. 2002;156:1029–1038. doi: 10.1083/jcb.200201081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freitag NE, Rong L, Portnoy DA. Regulation of the prfA transcriptional activator of Listeria monocytogenes: multiple promoter elements contribute to intracellular growth and cell-to-cell spread. Infect Immun. 1993;61:2537–2544. doi: 10.1128/iai.61.6.2537-2544.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eiting M, Hageluken G, Schubert WD, Heinz DW. The mutation G145S in PrfA, a key virulence regulator of Listeria monocytogenes, increases DNA- binding affinity by stabilizing the HTH motif. Mol Microbiol. 2005;56:433–446. doi: 10.1111/j.1365-2958.2005.04561.x. [DOI] [PubMed] [Google Scholar]
- 49.Ripio M-T, Dominguez-Bernal G, Lara M, Suarez M, Vazquez-Boland JA. A Gly145Ser substitution in the transcriptional activator PrfA causes constitutive overexpression of virulence factors in Listeria monocytogenes. J Bacteriol. 1997;179:1533–1540. doi: 10.1128/jb.179.5.1533-1540.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci USA. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones S, Portnoy DA. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun. 1994;62:5608–5613. doi: 10.1128/iai.62.12.5608-5613.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]