

Abstract
The periodontal pathogen Porphyromonas gingivalis is implicated in certain systemic diseases including atherosclerosis and aspiration pneumonia. This organism induces innate responses predominantly through TLR2, which also mediates its ability to induce experimental periodontitis and accelerate atherosclerosis. Using a validated mouse model of intratracheal challenge, we investigated the role of TLR2 in the control of P. gingivalis acute pulmonary infection. TLR2-deficient mice elicited reduced proinflammatory or antimicrobial responses (KC, MIP-1α, TNF-α, IL-6, IL-12p70, and NO) in the lung and exhibited impaired clearance of P. gingivalis compared to normal controls. However, the influx of polymorphonuclear leukocytes (PMN) into the lung and the numbers of resident alveolar macrophages (AM) were comparable between the two groups. TLR2 signaling was important for in vitro killing of P. gingivalis by PMN or AM and, moreover, the AM bactericidal activity required NO production. Strikingly, AM were more potent than peritoneal or splenic macrophages in P. gingivalis killing, attributed to diminished AM expression of complement receptor-3 (CR3) which is exploited by P. gingivalis to promote its survival. The selective expression of CR3 by tissue macrophages and the requirement of TLR2 inside-out signaling for CR3 exploitation by P. gingivalis (Wang et al, 2007, J Immunol 179:2349) suggest that the role of TLR2 in host protection may be contextual. Thus, although TLR2 may mediate destructive effects, as seen in models of experimental periodontitis and atherosclerosis, we have now shown that the same receptor confers protection against P. gingivalis in acute lung infection.
Keywords: monocytes/macrophages, neutrophils, phagocytosis, lung, bacterial (infections), rodent (model)
Introduction
Porphyromonas gingivalis is a gram-negative oral anaerobic bacterium that is strongly associated with human periodontitis (1, 2) and implicated in several systemic diseases including aspiration pneumonia (3–5), which is responsible for significant morbidity and mortality in the elderly (6). In this regard, it is thought that the dental plaque biofilm serves as a persistent reservoir for respiratory infections and that oral pathogens can be aspirated into the lung to cause aspiration pneumonia (7–9). Indeed, certain oral pathogens and especially P. gingivalis are common isolates from aspiration pneumonia and lung abscesses (5, 10, 11). Recently, periodontitis was epidemiologically implicated as a mortality risk factor for aspiration pneumonia in the elderly (12).
To facilitate the study of host pulmonary-bacterial interactions and the pathogenesis of anaerobic pulmonary infections, a well-defined mouse model was established by one of the coauthors (13). In this model, intratracheal challenge with Bacteroides (Porphyromonas) gingivalis causes inflammation in the lung leading to severe bronchopneumonia and lung abscess (13). The inflammation is characterized by a marked recruitment of polymorphonuclear leukocytes (PMN)3 and significant bacterial killing (13). Subsequent mouse studies by independent groups characterized the inflammatory pulmonary response against P. gingivalis, which includes induction of TNF-α, IL-1β, IL-6, and KC, a functional homologue of human IL-8 (14, 15). However, the host receptor(s) involved in inflammation and control of P. gingivalis lung infection have not been addressed.
In the absence of lung infection or inflammation, PMN are essentially absent and alveolar macrophages (AM) constitute more than 98% of the total leukocyte population in the alveolar space. AM form the first line of pulmonary defense at the air-tissue interface and their role lies in phagocytosis of airborne particles or invading microorganisms, killing, and coordination of the innate immune response (16). For instance, when the invading pathogen represents an overwhelming load or is too virulent to be contained by the AM alone, AM-released chemokines and other inflammatory mediators recruit and activate large numbers of PMN from the pulmonary vasculature into the alveolar space (17). Studies in humans and experimental animals have underscored the importance of certain cytokines/chemokines in pulmonary host defense, including TNF-α, IL-1, IL-6, IL-8, IL-10, IL-12, MIP-1α, and IL-17 (16–20). Also important for host defense of the lung are microbicidal products such as phagocyte-derived reactive oxygen and nitrogen intermediates (21). Recruited PMN and resident AM utilize these mechanisms to mediate early bacterial clearance (19, 21).
These antimicrobial activities and the pulmonary innate response in general are mediated, in large part, by TLRs (21, 22). TLRs are expressed on a number of immune effector cells, including AM and PMN, and serve as sensors of infection for triggering cytokine and antimicrobial responses (16, 21). Studies with different lung pathogens suggest that the role of TLRs in the host response to lung infection ranges from resistance to susceptibility (22–26). In general, TLRs can mediate critical antimicrobial responses, although they may be exploited by certain pathogens for induction of IL-10-mediated immunosuppression (27, 28).
The objective of this study was to determine whether TLR2 plays a role in the control of P. gingivalis acute infection in the lung. TLR2 was selected for investigation owing to its importance in P. gingivalis-induced innate immune responses. Specifically, we have shown that TLR2-deficient cells exhibit diminished induction of NF-κB activation and cytokine production in response to P. gingivalis or purified components thereof (29, 30). The TLR2 dependence of the innate response to P. gingivalis was confirmed in vivo by an independent group (31). Indeed, subcutaneous infection with P. gingivalis elicited high levels of cytokines in wild-type or TLR4-deficient mice, although in TLR2-deficient mice cytokine responses were hardly detectable or delayed (31). In the same study, TLR2 was associated with exacerbation of P. gingivalis-induced periodontal bone loss (31). This finding is supported by another group, which attributed periodontal bone loss to TLR2-induced TNF-α-dependent osteoclastogenesis (32, 33). TLR2 has moreover been implicated in the acceleration of atherosclerotic heart disease by P. gingivalis (34).
In this study, we show for the first time a protective role for TLR2 in acute lung infection with P. gingivalis. Indeed, TLR2-deficient mice elicited reduced proinflammatory or antimicrobial responses and impaired clearance of P. gingivalis compared to wild-type controls. At an in vitro mechanistic level, both PMN and AM exhibited TLR2-dependent killing of P. gingivalis, and the bactericidal activity of the latter required production of reactive nitrogen species. Interestingly, AM were more potent in P. gingivalis killing than peritoneal or splenic macrophages. This is probably because most tissue macrophages, but not AM, express CR3 which is exploited by P. gingivalis to promote its survival in host tissues and cause disease (35–37). The selective expression of CR3 by tissue macrophages (37, 38) and the requirement of TLR2 inside-out signaling for CR3 exploitation by P. gingivalis (36, 39, 40) suggest that TLR2 deficiency may not always predispose to increased host susceptibility to P. gingivalis. Indeed, our results in conjunction with those of others (31, 32, 34) suggest that TLR2 may play disparate roles in P. gingivalis infections, exacerbating certain chronic conditions (e.g., periodontitis, atherosclerosis) while providing protection against acute pulmonary infection.
Materials and Methods
Mice, bacteria, and other reagents
Female TLR2−/−, CD11b−/−, or wild-type control mice (C57BL/6) were obtained from the Jackson Laboratory (Bar Harbor, ME). The mice were housed in specific pathogen-free rooms in the Animal Care Facilities of Louisiana State University or the University of Louisville and were used when they were 10–12 week-old, in compliance with established federal guidelines and institutional policies. P. gingivalis ATCC 33277 was grown anaerobically at 37°C in brain heart infusion broth supplemented with hemin (5 μg/ml) and menadione (l μg/ml). Diphenylene iodonium (DPI), N(G)-nitro-L-arginine methyl ester (L-NAME) and its inactive enantiomer N(G)-nitro-D-arginine methyl ester (D-NAME) were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant mouse TNF-a was from R&D Systems (Minneapolis, MN).
Lung infection model
Mice were intratracheally infected with P. gingivalis essentially as previously described (13, 41). Briefly, following mouse anesthesia with intraperitoneal ketamine/xylazine injection, the trachea was aseptically exposed and a P. gingivalis inoculum (108 CFU in 40 μl PBS) was administered via a 28-gauge needle. Unchallenged naïve mice were administered PBS only. Groups of animals were sacrificed immediately after infection (time 0) or at later time points, 5h or 24h postinfection. To determine lung bacterial clearance, the lungs were aseptically removed, homogenized in a tissue homogenizer, and serial 10-fold dilutions were plated onto hemin/menadione-supplemented blood agar plates and cultured anaerobically for enumerating recovered CFU. To determine induction of cytokine production and recruitment of inflammatory cells, each pair of lungs (from naïve or infected mice not used for assessing lung bacterial clearance) was surgically removed in toto and lavaged with 1.0-ml aliquots of PBS/0.5 mM EDTA up to 10 ml. The first ml of recovered bronchoalveolar lavage fluid (BALF) was centrifuged at 500 g and the supernatant was stored at −80°C for subsequent cytokine analysis. The cell pellet was combined with additional lavage fluid obtained from the same pair of lungs and analyzed for total cell count and differential cell counts performed in cytospins stained with Wright-Giemsa (Diff-Quick, Baxter McGaw Park, IL).
Cytokine and antimicrobial molecule induction analysis
Cytokine levels in the collected BALF samples were assessed by means of a Bio-Plex cytokine bead array using a multiplex for eight cytokines: IL-1β, IL-6, IL-10, IL-12p70, IL-17, TNF-α, KC, and MIP-1α (Bio-Rad Laboratories, Hercules, CA). Induction of cytokine production in cell culture supernatants was determined using ELISA kits (eBioscience, San Diego, CA). Myeloperoxidase (MPO) levels in BALF were determined using an ELISA kit (Hycult Biotechnology, Uden, The Netherlands). NO production was assessed by measuring the amount of nitrite (NO2−, a stable oxidative metabolite of NO) in lung tissue homogenates using an assay kit based on the Griess reaction (Cayman, Ann Arbor, MI).
Cell isolation and killing assays
AM were obtained for cell culture from the BALF of naive mice. The BALF cell population contained more than 98% macrophages as determined by morphology upon cytologic examination. The cells were cultured at 37°C and 5% CO2 atmosphere, in complete RPMI (RPMI 1640 containing 10% heat-inactivated FBS, 2 mM L-glutamine, 10 mM HEPES, 100 units/ml penicillin G, 100 ng/ml streptomycin; InVitrogen/Gibco, Carlsbad, CA). Mouse macrophages were also isolated from the peritoneal cavity via peritoneal lavage with ice-cold HBSS or from splenic cell suspensions by positive selection using anti-CD11b magnetic bead cell sorting (Miltenyi Biotec, Auburn, CA) (42). Cells were washed and incubated in complete RPMI. After 2h, aspiration and three successive washes with HBSS were performed to remove any residual non-adherent cells, resulting in a population containing more than 95% macrophages. To determine intracellular killing of P. gingivalis, an antibiotic protection-based intracellular survival assay was used, as we previously described (36). Briefly, following incubation of P. gingivalis with macrophages (at a multiplicity of infection [MOI] of 10:1) for various time points, extracellular bacteria were eliminated by washing and antibiotic treatment. Viable internalized bacteria were released by macrophage lysis and serial dilutions of the lysates were plated onto blood agar plates for anaerobic culture and CFU enumeration.
PMN were purified from mouse blood through three-layer Percoll gradient centrifugation as previously described (43). This method yielded about 200,000 cells per mouse containing more than 95% PMN. Killing of P. gingivalis by mouse PMN was determined according to the method of Mydel et al (44) with minor modifications. PMN-P. gingivalis suspensions (MOI = 1:1) were incubated at 37°C and 5% CO2 for 2h without washing to remove extracellular bacteria, and enumeration of viable CFU on anaerobically cultured blood agar plates after PMN lysis was used to determine the killing of both extracellular and intracellular bacteria. The killing index was calculated according to the following formula: [(CFU in the absence of PMN – CFU in the presence of PMN)/CFU in the absence of PMN] × 100. The selected 2-h time point was based on a time course study showing significant mouse PMN killing of P. gingivalis in the interval between 105 and 135 min (44).
Oxidative burst assay
The PMN oxidative burst (H2O2 production) was monitored as we described previously (45) following the Bass method (46). The assay is based on the ability of 2′,7′-dichlorofluorescin diacetate (DCFH-DA) to diffuse into the cells where it is hydrolyzed to 2′,7′-dichlorofluorescin (DCFH) and is thereby trapped within the cells. During the oxidative burst, non-fluorescent intracellular DCFH is oxidized to highly fluorescent dichlorofluorescein (DCF) and the generated fluorescent signal has been quantitatively correlated with the PMN oxidative burst (46). PMN in glucose-containing HBSS (InVitrogen) were preincubated at 37°C for 15 min with 100 nM DCFH-DA (InVitrogen/Molecular Probes) in microtiter culture plates (2 × 105 cells/well). The samples were then incubated with P. gingivalis (MOI = 1:1) at 37°C for various time points. The fluorescent signal resulting from the oxidation of DCFH into DCF was measured as relative fluorescence units on a microplate fluorescence reader (Bio-Tek, Winooski, VT) with excitation/emission wavelength settings of 485/530 nm.
Flow cytometric uptake assay
Mouse macrophages were incubated at 37ºC with FITC-labeled P. gingivalis at a MOI of 10:1 (36). Phagocytosis was stopped at various time points (5–60 min) by cooling the incubation tubes on ice. After cell washing to remove nonadherent bacteria, extracellular fluorescence (representing attached but not internalized bacteria) was quenched with 0.2% trypan blue. The cells were washed again, fixed with 1% paraforlmadehyde, and analyzed by flow cytometry (% positive cells for FITC-P. gingivalis) using the FACSCalibur and the CellQuest software (Becton-Dickinson).
Statistical analysis
Data were evaluated by ANOVA using the InStat v3.06 program (GraphPad Software, San Diego, CA). Where appropriate (comparison of two groups only), two-tailed t tests were conducted. Statistical differences were considered significant at the level of p < 0.05. The experiments were performed at least twice for verification.
Results
Impaired clearance of P. gingivalis acute lung infection and reduced cytokine responses in TLR2-deficient mice
To determine the role of TLR2 in the acute response to P. gingivalis, a 5-h time point was carefully selected as being the most appropriate for the purpose. Indeed, a previous time-course study, which established the P. gingivalis-induced aspiration pneumonia model, has demonstrated pronounced recruitment of PMNs and significant killing of P. gingivalis in wild-type mice at 5-h postinfection (13). A comparable time point (6 h) was shown to be ideal for simultaneously assessing cytokine induction and inflammatory cell influx (47). Moreover, we included a 24-h time point, which represents the peak of the PMN influx upon intratracheal P. gingivalis challenge (13). Preliminary experiments using wild-type mice confirmed that intratracheal infection with 108 P. gingivalis bacteria causes a marked increase in PMN recruitment at 5h postinfection. Although PMN were essentially undetectable in the BALF of naïve mice, their numbers increased to 4.5 (±1.1) ×106 in the BALF of infected mice (n =5), accounting for more than 80% of the total cell count. On the other hand, the number of AM remained virtually unaltered 5h post-infection compared to the levels seen in naive mice (0.9 [±0.2] ×106). Comparison of wild-type and TLR2-deficient (TLR2−/−) mice at 5h or 24h postinfection showed that TLR2 did not play a significant role in PMN recruitment since both groups showed comparable total cell count and differential cell counts (Table I). Consistent with this finding, no statistically significant differences were observed between wild-type and TLR2−/− mice in MPO levels (reflecting PMN activity) in the BALF (Table I). Mock inoculation (intratracheal injection of plain PBS) resulted in relatively low levels of PMN recruitment to the lungs after 5h or 24h (Table I). Indeed, PMN comprised only about 19–27 % of the total cell count in the BALF of mock-inoculated wild-type or TLR2−/− mice compared to 79–83 % in P. gingivalis-infected wild-type or TLR2−/− mice (Table I). As the BALF total cell count was four to six times higher in infected mice (Table I), the estimated PMN numbers in mock-inoculated mice were consistently below 8% of the numbers corresponding to infected mice. Consistent with this, the MPO levels in the BALF of mock-inoculated mice were less than 9% of the levels seen in infected mice (Table I).
Table I.
Inflammatory cell recruitment in wild-type and TLR2−/− mice intratracheally infected with P. gingivalisa.
| Parameters | 5 h | 24 h | ||||||
|---|---|---|---|---|---|---|---|---|
| Naïve | Infected | Naïve | Infected | |||||
| WT | TLR2−/− | WT | TLR2−/− | WT | TLR2−/− | WT | TLR2−/− | |
| TCC (×106) | 1.18 ± 0.44 | 1.07 ± 0.28 | 5.16 ± 1.51 | 4.02 ± 1.34 | 1.86 ± 0.47 | 1.48 ± 0.50 | 11.8 ± 2.46 | 9.82 ± 2.17 |
| % AM | 78.5 ± 3.24 | 80.9 ± 5.72 | 16.8 ± 6.42 | 20.6 ± 5.27 | 74.5 ± 4.26 | 73.4 ± 8.54 | 20.4 ± 4.87 | 18.7 ± 4.14 |
| % PMN | 21.5 ± 3.24 | 19.1 ± 5.72 | 83.2 ± 6.42 | 79.4 ± 5.27 | 25.5 ± 4.26 | 26.6 ± 8.54 | 79.6 ± 4.87 | 81.3 ± 4.14 |
| MPO (ng/ml) | 329 ± 76 | 266 ± 98 | 4775 ± 1519 | 3156 ± 1040 | 536 ± 223 | 472 ± 182 | 8938 ± 2527 | 7712 ± 2721 |
Wild-type (WT) or TLR2−/− mice (n = 5 for each group) were challenged intratracheally with P. gingivalis (108 CFU) or PBS only (mock-inoculated naïve mice) and sacrificed 5h or 24h postinfection. BALF was used for total cell counts (TCC) and differential cell counts, and for determination of MPO levels by ELISA.
Although TLR2−/− mice exhibited quite normal recruitment of PMNs, they were defective in controlling P. gingivalis. Indeed, although wild-type mice cleared about 80% of the bacteria by 5h postinfection, there was no sign of net killing activity in TLR2−/− mice (Fig. 1A). In this regard, no statistically significant differences were observed between P. gingivalis CFU recovered from the lungs of TLR2−/− mice and CFU counts from mice sacrificed immediately upon infection, representing the initial observed inoculum (Fig. 1A). In contrast, P. gingivalis CFU counts recovered from the lungs of wild-type mice were significantly lower compared to the initial inoculum or the CFU levels of TLR2−/− mice (p < 0.05; Fig. 1A). At 24-h postinfection, the viable P. gingivalis counts recovered from the lungs of wild-type mice were about 300 less than seen in TLR2−/− mice and 500 times less than the initial observed inoculum (p < 0.05; Fig. 1B). In contrast, an apparent modest reduction in viable CFU counts in TLR2−/−mice, relative to the initial observed inoculum, did not reach statistical significance (Fig. 1B). In conclusion, no significant net growth or killing of P. gingivalis was observed in the lungs of TLR2−/− mice, whereas wild-type mice readily clear the organism, suggesting that TLR2 mediates effective control of P. gingivalis lung infection.
Figure 1. Impaired clearance of P. gingivalis from the lungs of infected TLR2-deficient mice.
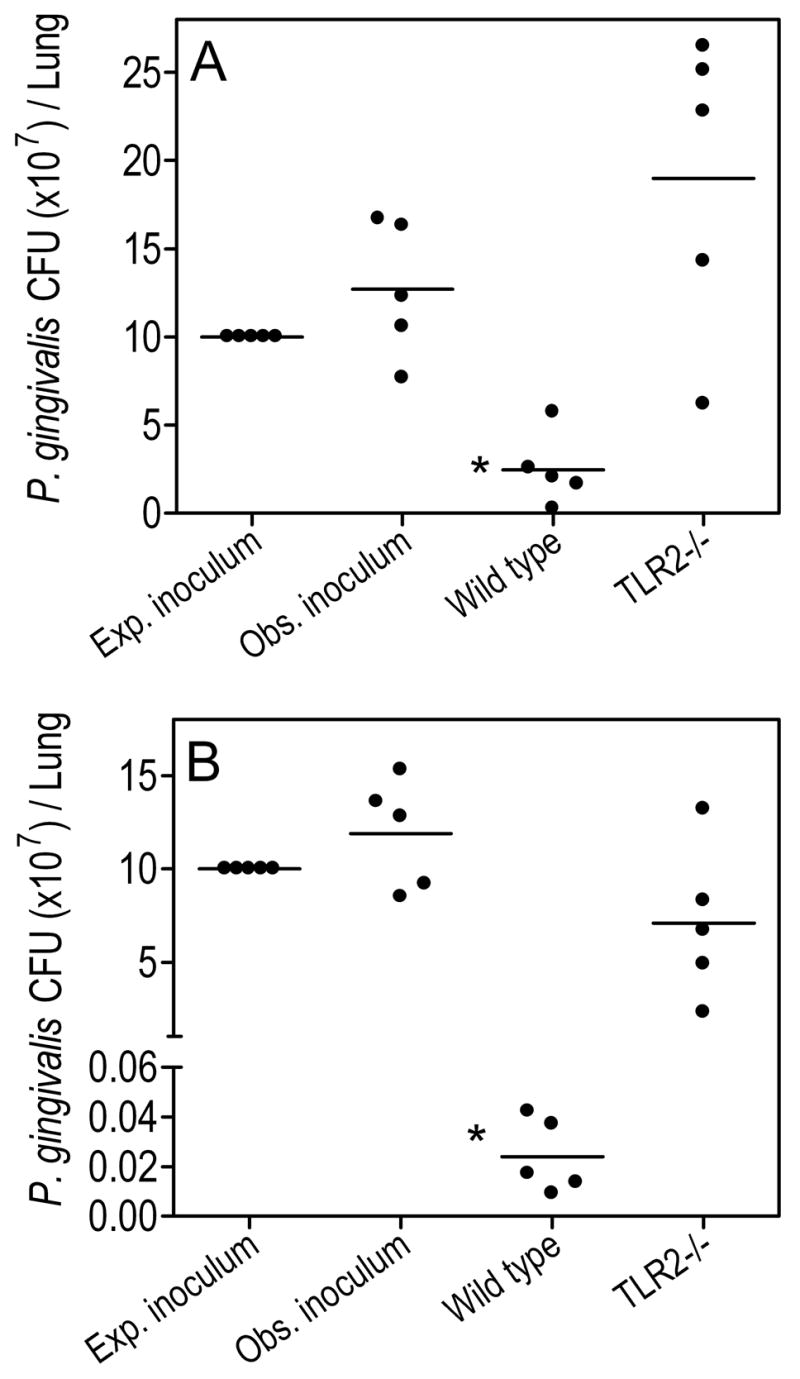
Wild-type or TLR2−/− mice were infected intratracheally with P. gingivalis (108 CFU; expected inoculum). The actual infecting dose (observed inoculum) was determined in mice sacrificed immediately after intratracheal administration of the organism. The wild-type and TLR2−/− experimental mice were sacrificed 5h (A) or 24h (B) postinfection and their lungs were aseptically removed. Serial 10-fold dilutions of lung homogenates were plated for anaerobic growth and enumeration of recovered CFU. Data are shown for each mouse and horizontal lines indicate the mean value. The asterisk indicates significantly (p < 0.05) lower CFU values compared to those seen in TLR2−/− mice and the observed inoculum. No statistically significant differences were found between TLR2−/− CFU and observed inoculum.
Cytokine responses in the BALF were generally significantly higher in wild-type mice compared to TLR2−/− mice at 5h postinfection. Significant differences (p < 0.05) were seen for KC, MIP-1α, TNF-α, IL-6, and IL-12p70, but not for IL-1β, IL-10, or IL-17, the latter being undetectable (Fig. 2A). At 24-h postinfection, TLR2−/− mice still exhibited significantly (p < 0.05) lower levels of KC, MIP-1α, and IL-12p70 compared to wild-type controls, but not of TNF-α or IL-6 (Fig. 2B). The relatively increased bacterial challenge in TLR2−/− mice (compared to wild-type controls which effectively eliminated most of the bacteria; Fig. 1B), and the possibility for TLR2-independent induction of certain cytokines, may have somewhat blunted differences in cytokine production between the two groups at 24h. Wild-type or TLR2−/− naïve mice had no or negligible cytokine responses (Fig. 2). With regard to induction of antimicrobial molecules, wild-type mice elicited significantly higher levels of NO at 5h or 24h postinfection (p < 0.05 vs. TLR2−/− mice), whereas NO production was undetectable in wild-type or TLR2−/−naïve mice (Fig. 3).
Figure 2. Cytokine levels in the BALF of wild-type and TLR2-deficient mice in response to P. gingivalis.

Wild-type or TLR2−/− mice were infected intratracheally with P. gingivalis (108 CFU) and sacrificed 5h (A) or 24h (B) postinfection. Cytokine responses in BALF samples were determined by means of a Bio-Plex cytokine bead array. Baseline levels were determined using uninfected (naïve) wild-type or TLR2−/− mice, intratracheally administered PBS only. Data are means ± SD (n = 5). Asterisks denote statistically significant (p < 0.05) reduction of cytokine responses in TLR2−/− mice compared to infected wild-type controls.
Figure 3. Induction of NO in the lungs of P. gingivalis-infected wild-type or TLR2−/− mice.
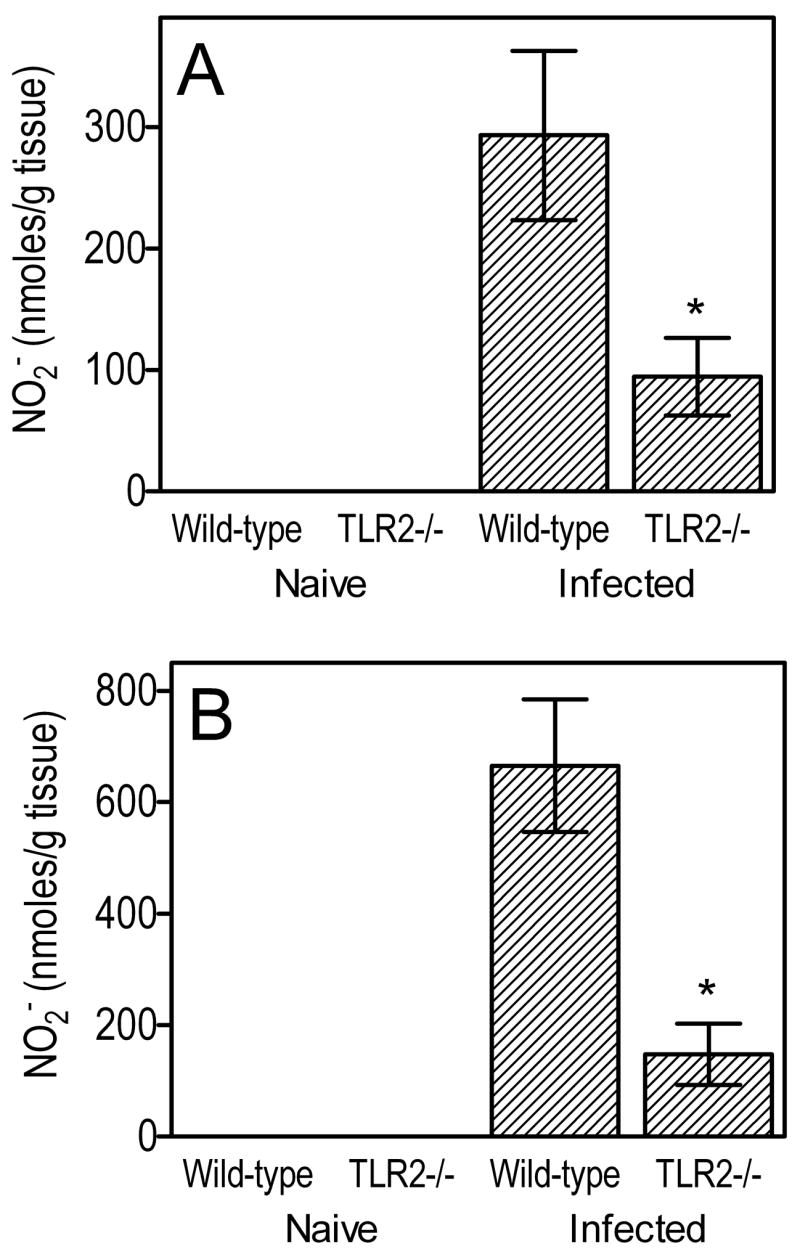
Wild-type or TLR2−/− mice were infected intratracheally with P. gingivalis (108 CFU) and sacrificed 5h (A) or 24h (B) postinfection. Wild-type or TLR2−/− mice that were intratracheally given PBS only are denoted as “naïve”. Levels of NO2− (a stable oxidative metabolite reflecting production of NO) in lung homogenate supernatants were measured by the Griess assay. Data are means ± SD (n = 5) and the asterisk indicates a statistically significant (p < 0.05) difference between wild-type and TLR2−/− mice.
In summary, TLR2 contributes to induction of innate immune responses in the lung and, though not essential for inflammatory cell recruitment, is required for efficient clearance of P. gingivalis infection.
TLR2-deficient PMN display reduced P. gingivalis killing
The impaired clearance of P. gingivalis in the lungs of TLR2−/− mice could be attributed to reduced bactericidal activities of their alveolar phagocytes (PMN and AM). We have thus used freshly explanted wild-type and TLR2−/− phagocytes to address this hypothesis. We first evaluated the ability of TLR2−/− PMN to kill P. gingivalis and found that their bactericidal capacity was reduced by 54% in comparison to wild-type controls (p < 0.05; Fig. 4A). Therefore, TLR2 signaling in PMN contributes significantly to P. gingivalis killing. An additional factor that could have affected the in vivo ability of TLR2−/− PMN to clear P. gingivalis may be related to reduced priming due to decreased cytokine responses in the lungs of TLR2−/− mice. Indeed, certain cytokines and especially TNF-α (which was reduced by 62% in TLR2−/− mice; Fig. 2) can effectively prime the bactericidal capacity of PMN, even though they do not directly activate killing mechanisms (48). Priming of wild-type neutrophils with recombinant mouse TNF-α (10 ng/ml) resulted in modest enhancement of their killing activity (not shown); however, priming of TLR2−/− PMN with TNF-α or even with activated culture supernatants from wild-type PMN did not significantly enhance their killing capacity against P. gingivalis, which ranged within 37–47% of the killing capacity of wild-type PMN (Fig. 4B).
Figure 4. Role of TLR2 in PMN killing of P. gingivalis.

(A) Comparative killing by wild-type (WT) or TLR2−/− PMN (2 × 105 cells) of P. gingivalis (MOI = 1:1) after 2-h incubation at 37°C. The killing index was calculated as described in Materials and Methods. (B) TLR2−/− PMN were primed, or not, for 30 min with recombinant mouse TNF-α (10 ng/ml) or with culture supernatants from activated wild-type or TLR2−/− PMN, prior to incubation with P. gingivalis for performing a killing assay as above. The killing activities of the variously primed TLR2−/− PMN were normalized to that of unprimed wild-type PMN. (C) Comparative induction of oxidative burst in WT or TLR2−/− PMN (2 × 105 cells) by P. gingivalis (MOI = 1:1) over time at 37°C. The oxidative burst was monitored using a quantitative DCFH assay, as explained in Material and Methods. (D) PMN oxidative burst and killing of P. gingivalis was determined as above in the presence or absence of 10 μM DPI. Data are means ± SD (A and D, n = 5; B and C, n = 3). Asterisks show statistically significant (p < 0.05) differences compared to wild-type controls (A, C) or to medium-only controls (B, D).
The oxidative burst is an important mechanism whereby PMN eliminate pathogens (49). However, TLR2−/− PMN did not display significantly reduced capacity for oxidative burst in response to P. gingivalis, when compared to their normal counterparts (Fig. 4C). Thus, the reduced bactericidal activity of TLR2−/− PMN may not be attributed to defective induction of the oxidative burst. Moreover, the killing of P. gingivalis by PMN was not significantly suppressed by the NADPH oxidase inhibitor DPI at 10 μM (Fig. 4D), a concentration demonstrated to abrogate the oxidative burst (Fig. 4D). Therefore, induction of reactive oxygen species may not be an effective mechanism to eliminate P. gingivalis. Finally, we could not detect production of NO in P. gingivalis-stimulated PMN (not shown), ruling out a possible role for inducible nitric oxide synthase (iNOS) in PMN killing of P. gingivalis.
In summary, the Fig. 4 data show that TLR2 signaling in PMN is required for maximal killing activity against P. gingivalis. Moreover, the differential killing capacity of wild-type and TLR2−/− PMN appears to involve NADPH oxidase- and iNOS-independent mechanisms.
TLR2 mediates killing of P. gingivalis in AM; comparison with other macrophage types
We have previously shown that TLR2 deficiency does not impair the ability of peritoneal macrophages for intracellular killing of P. gingivalis (36). In striking contrast, TLR2−/− AM exhibited defective intracellular killing of this pathogen compared to normal controls (p < 0.05; Fig. 5A). Specifically, using an antibiotic protection-based intracellular survival assay, we found that the recovery of viable P. gingivalis CFU from TLR2−/Δ AM was significantly higher by about 0.5 or 1.3 log10 units after 5h- or 24h-incubation, respectively, compared to CFU recovery from wild-type controls (p < 0.05; Fig. 5A). These data in conjunction with the Fig. 4A results indicate that TLR2 contributes to the killing of P. gingivalis and provide a mechanistic basis for the observed impaired clearance of this pathogen from the lungs of TLR2−/− mice (Fig. 1).
Figure 5. Intracellular killing of P. gingivalis by AM.
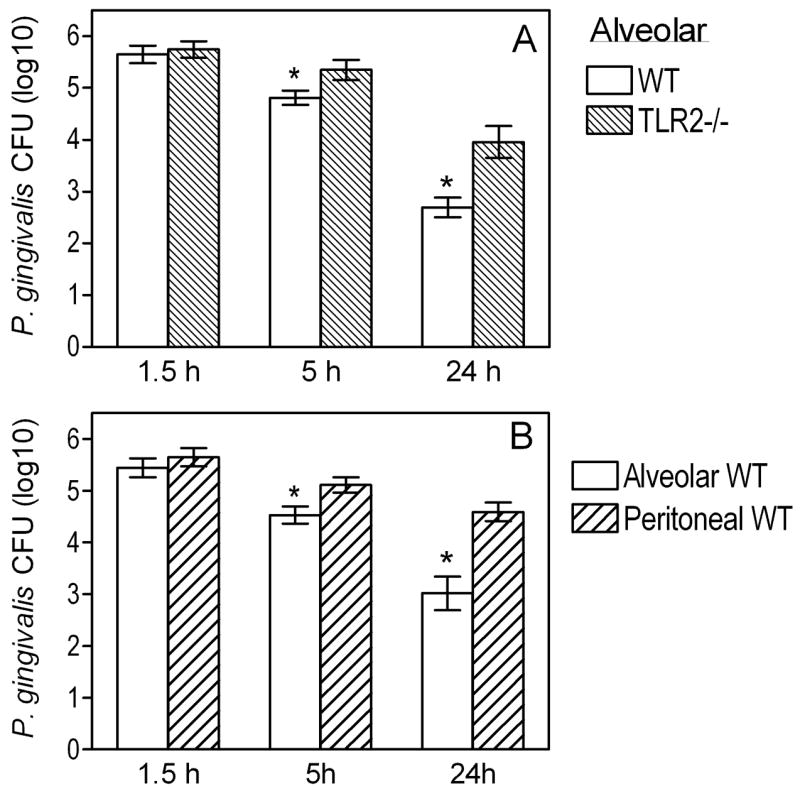
(A) Wild-type and TLR2−/− AM were incubated with P. gingivalis (MOI=10:1) for the indicated times. The persistence of viable internalized bacteria was determined using an antibiotic protection-based survival assay. (B) Similar procedures were followed to measure the intracellular killing of P. gingivalis by alveolar and peritoneal macrophages. Data are means ± SD (n = 5). Asterisks show significantly (p < 0.05) lower CFU levels (i.e., increased killing) in AM compared to their TLR2−/− counterparts (A) or to peritoneal macrophages (B).
However, it was puzzling that TLR2 appeared to play disparate roles in alveolar and peritoneal macrophages regarding the control of P. gingivalis intracellular infection. Side-by-side comparison conclusively showed that AM are more potent than peritoneal macrophages in P. gingivalis intracellular killing (Fig. 5B). Indeed, the recovery of viable P. gingivalis CFU from AM was significantly lower by 0.6 or 1.6 log10 units after 5h- or 24h-incubation, respectively, compared to CFU recovery from peritoneal macrophages (p < 0.05; Fig. 5B). Moreover, comparison of wild-type and TLR2−/− peritoneal macrophages confirmed our previous findings that TLR2 actually promotes rather than controls the intracellular persistence of P. gingivalis (36)(not shown here). This is attributed to TLR2 inside-out signaling which activates CR3-mediated uptake of P. gingivalis resulting in increased intracellular survival of this pathogen (36). By contrast, this CR3-dependent immune evasion mechanism of P. gingivalis is unlikely to operate in AM which express little or no CR3 (37, 38). Indeed, we found that CR3 does not play a significant role in the uptake of P. gingivalis by AM. Specifically, there were no significant differences between wild-type and CR3-deficient AM in P. gingivalis uptake, as opposed to significant (p < 0.05) differences between wild-type and CR3-deficient peritoneal macrophages (Fig. 6A). Moreover, although CR3 is associated with increased intracellular survival of P. gingivalis in peritoneal macrophages (36) (confirmed in Fig. 6B), as well as in splenic macrophages which also express CR3 (Fig. 6B), this was not the case with AM. Indeed, no significant differences were observed between wild-type and CR3-deficient AM in terms of their ability to control the intracellular fate of P. gingivalis (Fig. 6B). Thus, the TLR2/CR3 inside-out pathway is not exploited by P. gingivalis in AM, and this may explain at least partly why AM TLR2 inhibits, rather than promotes, the intracellular survival of P. gingivalis.
Figure 6. Role of CR3 in the uptake and intracellular survival of P. gingivalis: alveolar vs. peritoneal macrophages.
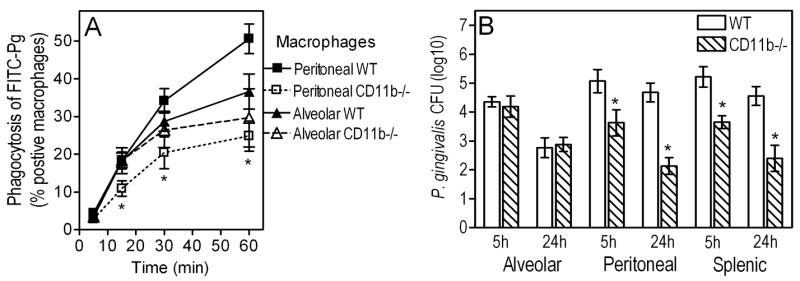
(A) Alveolar or peritoneal macrophages from wild-type (WT) or CR3-deficient (CD11b−/−) mice were incubated with P. gingivalis (MOI = 10:1) for the indicated times at 37°C. Uptake was assessed using FITC-labeled bacteria and flow cytometry after quenching extracellular fluorescence, and was expressed as percentage of FITC-positive macrophages. (B) The same macrophage groups, as well as wild-type and CD11b−/− splenic macrophages, were compared for their intracellular killing abilities against P. gingivalis (used at a MOI of 10:1) by means of an antibiotic protection-based survival assay. Results are presented as means ± SD (n = 3). In panel A, the asterisks show significantly (p < 0.05) reduced uptake by CD11b−/− AM compared to WT controls. In panel B, the asterisks show significantly (p < 0.05) reduced survival of P. gingivalis in the indicated CD11b−/− macrophages compared to WT controls.
TLR2-mediated killing of P. gingivalis in AM involves reactive nitrogen intermediates
NO is a key antimicrobial molecule produced by activated macrophages for pathogen killing (49). To determine whether AM utilize this mechanism for intracellular killing of P. gingivalis, we pretreated them with L-NAME, a specific inhibitor of NO synthesis, or with the inactive enantiomer D-NAME (50). The macrophages were then infected with P. gingivalis (MOI = 10:1) and its intracellular fate was monitored using the antibiotic-based intracellular survival assay. The pathogen was recovered at significantly higher CFU numbers from L-NAME-treated AM than from untreated cells (p < 0.05; Fig. 7A). In contrast, D-NAME had no effect in this regard (Fig. 7A). The increased recovery of P. gingivalis CFU from L-NAME-treated cells could not be attributed to increased uptake (relative to medium-only or D-NAME treated cells) since L-NAME had no effect on the ability of the AM to take up P. gingivalis (not shown). We also confirmed that L-NAME, but not D-NAME, dramatically diminished production of NO (measured as NO2−, its stable oxidative metabolite) by AM in response to P. gingivalis (p < 0.05; Fig. 8A). However, L-NAME did not affect P. gingivalis-induced cytokine production (Fig. 8A) thus ruling out potential toxic effects on AM.
Figure 7. Role of NO in P. gingivalis intracellular killing by AM.

AM from (A) wild-type or (B) TLR2−/− mice were pretreated with L-NAME (inhibitor of NO production) or the inactive enantiomer D-NAME (both at 1 mM) and then incubated with P. gingivalis (MOI=10:1) for 5 h. The persistence of viable internalized bacteria was determined using an antibiotic protection-based survival assay. Data are means ± SD (n = 5) and the asterisk indicates significant (p < 0.05) differences between L-NAME-pretreated and control macrophages that were pretreated with medium-only or D-NAME.
Figure 8. Regulation of P. gingivalis-induced NO and cytokine responses in AM.

(A) Wild-type AM were pretreated with L-NAME (inhibitor of NO production) or the inactive enantiomer D-NAME (both at 1 mM) and then stimulated for 5 h with P. gingivalis (MOI = 10:1). (B) Wild-type or TLR2−/− AM were stimulated for 5 h with P. gingivalis (MOI = 10:1). In both experiments, culture supernatants were assayed for production of NO2− (stable oxidative metabolite of NO), TNF-α, and MIP-1α. Data are means ± SD (n = 3). Asterisks denote statistically significant (p < 0.05) inhibition of NO or cytokine responses due to L-NAME (A) or TLR2 deficiency (B).
In contrast to the findings from wild-type AM, the intracellular survival of P. gingivalis in TLR2−/− AM was not significantly affected by L-NAME (Fig. 7B). Therefore, NO does not significantly contribute to the intracellular control of P. gingivalis in the absence of TLR2 signaling. In fact, P. gingivalis-induced NO levels were significantly diminished in TLR2−/− AM compared to wild-type controls (p < 0.05; Fig. 8B). Therefore, NO production in this in vitro system is heavily depended on TLR2, as seen in vivo (Fig. 3). The observed inhibition of NO production was about 75%, comparable to the inhibition of cytokine production (TNF-α and MIP-1α) in TLR2−/− AM (Fig. 8B).
In conclusion, the data from Figs. 7 and 8 suggest that induction of NO is at least one mechanism whereby AM control P. gingivalis lung infection. Moreover, this antimicrobial mechanism accounts, at least partly, for the differential capacities of wild-type and TLR2−/− AM to eliminate P. gingivalis.
Discussion
Our findings of impaired P. gingivalis clearance from the lungs of TLR2−/− mice after 5h or 24h postinfection support a protective role for TLR2 in the control of acute pulmonary infection with this pathogen. Given the mechanism of oral pathogen-induced aspiration pneumonia and the fact that 1 mm3 of dental plaque biofilm contains >108 viable bacteria (7–9), it is likely that the selected P. gingivalis dose (108 CFU) may represent a realistic aspirated inoculum in patients with severe periodontitis. TLR2 was also shown to confer protection against pulmonary infection with Francisella tularensis (24), Chlamydia pneumoniae (26), or Aspergillus fumigatus (51), although its role is relatively minor in infections with Cryptococcus neoformans (52) or Legionella pneumophila (53). Interestingly TLR2 signaling may, in contrast, exacerbate certain pulmonary infections. For example, TLR2−/− mice are more resistant to Acinetobacter baumannii-induced acute pneumonia than normal controls, although TLR4−/− mice are quite susceptible (23). In the same study, TLR2−/− mice exhibited enhanced innate responses in the lung and it was thus concluded that TLR2 mediated anti-inflammatory signaling (23). In a similar context, the virulence of Paracoccidioides brasiliensis in pulmonary infection of susceptible B10.A mice was attributed to TLR2-dependent induction of IL-10 (25). We did not observe significant differences in IL-10 production in the BALF of P. gingivalis-infected normal or TLR2−/− mice, both of which had very low levels of this immunosuppressive cytokine. In contrast, TLR2 signaling was associated with increased levels of chemokines, certain proinflammatory cytokines, and antimicrobial molecules (NO) in our P. gingivalis lung infection model.
The defective killing of P. gingivalis in the lungs of TLR2−/− mice could not be attributed to impaired recruitment of inflammatory cells. Indeed, despite reduced production of certain chemokines (KC, MIP-1α) in the BALF of P. gingivalis-infected TLR2−/− mice, and although TLR2 deficiency has occasionally been associated with decreased PMN influx (26, 54), we did not observe significant differences between TLR2−/− and wild-type control mice regarding PMN recruitment to the lungs after 5h or 24h postinfection. It is possible that TLR2-independent mechanisms have compensated for this function, such as stimulation by bacterially-derived formyl peptides of the formyl-peptide receptor, which plays a major role in PMN recruitment to infected alveoli (20).
In principle, the defective killing of P. gingivalis in the lungs of TLR2−/− mice could result, at least partly, from decreased priming of their alveolar phagocytes (PMN and AM) owing to reduced proinflammatory cytokine responses. Alternatively or in addition to the above, the lack of TLR2 signaling in the TLR2−/− alveolar phagocytes may have directly affected their ability to kill P. gingivalis. Our in vitro mechanistic data are consistent with the latter interpretation. TLR2 was also shown to mediate AM killing of Mycobacterium tuberculosis (55) and PMN killing of Streptococcus pneumoniae (56). In stark contrast, TLR2 promotes the survival of Staphylococcus aureus in peritoneal macrophages (57). It is unclear whether the outcome depends upon the phagocytic cell type or the pathogen involved (or both), but it should be noted that the antimicrobial role of TLR2 may be contextual (discussed in greater detail later in this section) and thus may not be predicted a priori in the absence of experimental evidence.
Our findings that the oxidative burst is not important for P. gingivalis killing are consistent with a recent study showing no difference in killing activity against this pathogen between wild-type and NADPH oxidase-deficient (Cybb−/−) PMN (44). It is thus possible that PMN use non-oxidative mechanisms to eliminate P. gingivalis, such as granule proteases or antimicrobial peptides which can be delivered to the phagosome or released to the extracellular space (58, 59). In contrast, NO may be an effective antimicrobial mechanism for the control of P. gingivalis since inhibition of its production in normal AM resulted in impaired intracellular killing of this pathogen. Therefore, the diminished capacity TLR2−/− AM to induce NO production may, at least partly, explain why TLR2−/− AM failed to efficiently control P. gingivalis. An additional mechanism may involve impaired phagosome maturation in the absence of TLR2 signaling (60), although a competing view supports that phagolysosomal maturation is a TLR-independent process (61). Our data are consistent with earlier findings that iNOS-deficient mice exhibit reduced killing of P. gingivalis in a subcutaneous chamber model (62). Our results are also in line with a recent study which demonstrated NO-dependent killing of P. gingivalis by mouse peritoneal macrophages (44).
Although we have previously demonstrated intracellular killing of P. gingivalis by peritoneal macrophages (36), we have now found that AM are even more potent than their peritoneal or splenic counterparts in this activity. This finding is at first sight surprising given that AM mediate critical immunosuppressive functions for maintaining respiratory tract homeostasis (16). This difference in P. gingivalis killing by distinct macrophage types may be attributed to selective expression of CR3 by tissue macrophages. Specifically, the fact that resident AM express little or no CR3 (37, 38) renders them resistant to a CR3-dependent immune evasion strategy of P. gingivalis (35, 36). It should be noted that the inhibitory effect of CR3 deficiency on the intracellular survival of P. gingivalis is quite dramatic (viability is reduced by up to 2.5 to 3.0 log10 units) which cannot be attributed to a ≈ 50% reduction in bacterial uptake ((36) and Fig. 6). Rather, the enhanced intracellular survival of P. gingivalis in CR3-expressing macrophages could be attributed to the notion that CR3 phagocytosis does not promote phagolysosomal fusion (63). Wild-type AM behave closer to CR3-deficient rather than wild-type peritoneal or splenic macrophages in terms of their ability to clear P. gingivalis. It should be pointed out that AM may not be inherently stronger than peritoneal macrophages in general microbicidal function, since alveolar and peritoneal macrophages display similar killing activities against Pneumocystis carinii (64).
In vitro, TLR2 inside-out signaling activates CR3-mediated uptake of P. gingivalis (39) leading to inhibition of IL-12p70 induction and enhanced intracellular persistence of the pathogen (36). In vivo, CR3 blockade leads to increased clearance of P. gingivalis from murine periodontal tissues and reduced periodontal disease activity (35). These observations, taken together with the selective expression of CR3 by tissue macrophages (37, 38), can at least partly explain why TLR2 deficiency may not necessarily predispose to increased susceptibility to P. gingivalis. For example, TLR2 deficiency has been associated with increased in vivo clearance of P. gingivalis and resistance to P. gingivalis-induced periodontitis in the mouse model (31). On the basis of available evidence [(31, 32, 34) and this study], we suggest that TLR2 may play diverse and contextual roles in P. gingivalis infections, exacerbating certain chronic conditions (e.g., periodontitis, atherosclerosis) while providing protection against acute pulmonary infection. Whether this depends on the nature of the infection (chronic vs. acute) or the selective expression of CR3, or even a combination of these and other factors, remains to be established.
P. gingivalis is a common isolate from aspiration pneumonia (5, 10, 11), which is usually seen in the elderly or the immunocompromised host (8, 65) and is epidemiologically associated with periodontal disease (12). Although TLR2 polymorphisms influence susceptibility to certain infectious or inflammatory diseases (66), there is currently no evidence linking hyporesponsive TLR2 polymorphisms to aspiration pneumonia. However, our findings from the TLR2-deficient mouse model suggest that compromised TLR2 signaling may increase a host’s susceptibility to lung infection with P. gingivalis. Among the immunocompromised population at high risk for aspiration pneumonia are alcohol-abusing patients (67). In addition to increased risk of oropharyngeal aspiration, these patients have impaired acute inflammatory response and ability to control infection (67), at least partly owing to detrimental alcohol effects on TLR-mediated innate immune response (68, 69). Therapeutic immunomodulation to stimulate protective TLR immunity in the immunocompromised patient may thus serve as an effective adjunct therapy. This warrants further investigations to clarify the role of TLR signaling in bacterial infections of the lung.
Acknowledgments
We thank Amy Weinberg and Pukar Ratti for excellent technical assistance.
Footnotes
This work was supported by U.S. PHS Grants DE015254 and DE018292 (to G.H.) and AA09803 (to S.N. and G.J.B.) from the National Institutes of Health.
Abbreviations used in this paper: PMN, polymorphonuclear leukocyte; AM, alveolar macrophage; DPI, diphenylene iodonium; L-NAME, N(G)-nitro-L-arginine methyl ester; D-NAME, N(G)-nitro-D-arginine methyl ester; BALF, bronchoalveolar lavage fluid; MPO, myeloperoxidase; MOI, multiplicity of infection; DCFH-DA, 2′,7′-dichlorofluorescin diacetate; NO2−, nitrite; iNOS, inducible nitric oxide synthase; CR3, complement receptor-3.
References
- 1.Zambon JJ, Grossi S, Dunford R, Harazsthy VI, Preus H, Genco RJ. Epidemiology of subgingival bacterial pathogens in periodontal diseases. In: Genco RJ, Hamada S, Lehrer JR, McGhee JR, Mergenhangen S, editors. Molecular Pathogenesis of Periodontal Disease. Am. Soc. Microbiol; Washington, D.C: 1994. pp. 3–12. [Google Scholar]
- 2.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 3.Okuda K, Kimizuka R, Abe S, Kato T, Ishihara K. Involvement of periodontopathic anaerobes in aspiration pneumonia. J Periodontol. 2005;76:2154–2160. doi: 10.1902/jop.2005.76.11-S.2154. [DOI] [PubMed] [Google Scholar]
- 4.Scannapieco FA. Role of oral bacteria in respiratory infection. J Periodontol. 1999;70:793–802. doi: 10.1902/jop.1999.70.7.793. [DOI] [PubMed] [Google Scholar]
- 5.Terpenning M. Editorial Commentary: Prevention of Aspiration Pneumonia in Nursing Home Patients. Clin Infect Dis. 2005;40:7–8. doi: 10.1086/426030. [DOI] [PubMed] [Google Scholar]
- 6.Janssens JP. Pneumonia in the elderly (geriatric) population. Curr Opin Pulm Med. 2005;11:226–230. doi: 10.1097/01.mcp.0000158254.90483.1f. [DOI] [PubMed] [Google Scholar]
- 7.Terpenning M, Bretz W, Lopatin D, Langmore S, Dominguez B, Loesche W. Bacterial colonization of saliva and plaque in the elderly. Clin Infect Dis. 1993;16(Suppl 4):S314–316. doi: 10.1093/clinids/16.supplement_4.s314. [DOI] [PubMed] [Google Scholar]
- 8.Paju S, Scannapieco FA. Oral biofilms, periodontitis, and pulmonary infections. Oral Dis. 2007;13:508–512. doi: 10.1111/j.1601-0825.2007.1410a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gendron R, Grenier D, Maheu-Robert L. The oral cavity as a reservoir of bacterial pathogens for focal infections. Microbes Infect. 2000;2:897–906. doi: 10.1016/s1286-4579(00)00391-9. [DOI] [PubMed] [Google Scholar]
- 10.Bartlett JG, Gorbach SL, Finegold SM. The bacteriology of aspiration pneumonia. Am J Med. 1974;56:202–207. doi: 10.1016/0002-9343(74)90598-1. [DOI] [PubMed] [Google Scholar]
- 11.Finegold SM, Strong CA, McTeague M, Marina M. The importance of black-pigmented gram-negative anaerobes in human infections. FEMS Immunol Med Microbiol. 1993;6:77–82. doi: 10.1111/j.1574-695X.1993.tb00306.x. [DOI] [PubMed] [Google Scholar]
- 12.Awano S, Ansai T, Takata Y, Soh I, Akifusa S, Hamasaki T, Yoshida A, Sonoki K, Fujisawa K, Takehara T. Oral health and mortality risk from pneumonia in the elderly. J Dent Res. 2008;87:334–339. doi: 10.1177/154405910808700418. [DOI] [PubMed] [Google Scholar]
- 13.Nelson S, Laughon BE, Summer WR, Eckhaus MA, Bartlett JG, Jakab GJ. Characterization of the pulmonary inflammatory response to an anaerobic bacterial challenge. Am Rev Respir Dis. 1986;133:212–217. doi: 10.1164/arrd.1986.133.2.212. [DOI] [PubMed] [Google Scholar]
- 14.Kimizuka R, Kato T, Ishihara K, Okuda K. Mixed infections with Porphyromonas gingivalis and Treponema denticola cause excessive inflammatory responses in a mouse pneumonia model compared with monoinfections. Microbes Infect. 2003;5:1357–1362. doi: 10.1016/j.micinf.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Petelin M, Naruishi K, Shiomi N, Mineshiba J, Arai H, Nishimura F, Takashiba S, Murayama Y. Systemic up-regulation of sTNFR2 and IL-6 in Porphyromonas gingivalis pneumonia in mice. Exp Mol Pathol. 2004;76:76–81. doi: 10.1016/j.yexmp.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. 2008;8:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- 17.Zhang P, Summer WR, Bagby GJ, Nelson S. Innate immunity and pulmonary host defense. Immunol Rev. 2000;173:39–51. doi: 10.1034/j.1600-065x.2000.917306.x. [DOI] [PubMed] [Google Scholar]
- 18.van der Poll T, Keogh CV, Guirao X, Buurman WA, Kopf M, Lowry SF. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis. 1997;176:439–444. doi: 10.1086/514062. [DOI] [PubMed] [Google Scholar]
- 19.Strieter RM, Belperio JA, Keane MP. Host innate defenses in the lung: the role of cytokines. Curr Opin Infect Dis. 2003;16:193–198. doi: 10.1097/00001432-200306000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Fillion I, Ouellet N, Simard M, Bergeron Y, Sato S, Bergeron MG. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J Immunol. 2001;166:7353–7361. doi: 10.4049/jimmunol.166.12.7353. [DOI] [PubMed] [Google Scholar]
- 21.Basu S, Fenton MJ. Toll-like receptors: function and roles in lung disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L887–892. doi: 10.1152/ajplung.00323.2003. [DOI] [PubMed] [Google Scholar]
- 22.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knapp S, Wieland CW, Florquin S, Pantophlet R, Dijkshoorn L, Tshimbalanga N, Akira S, van der Poll T. Differential roles of CD14 and toll-like receptors 4 and 2 in murine Acinetobacter pneumonia. Am J Respir Crit Care Med. 2006;173:122–129. doi: 10.1164/rccm.200505-730OC. [DOI] [PubMed] [Google Scholar]
- 24.Malik M, Bakshi CS, Sahay B, Shah A, Lotz SA, Sellati TJ. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect Immun. 2006;74:3657–3662. doi: 10.1128/IAI.02030-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreira KS, Bastos KR, Russo M, Almeida SR. Interaction between Paracoccidioides brasiliensis and pulmonary dendritic cells induces interleukin-10 production and toll-like receptor-2 expression: possible mechanisms of susceptibility. J Infect Dis. 2007;196:1108–1115. doi: 10.1086/521369. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez N, Wantia N, Fend F, Durr S, Wagner H, Miethke T. Differential involvement of TLR2 and TLR4 in host survival during pulmonary infection with Chlamydia pneumoniae. Eur J Immunol. 2006;36:1145–1155. doi: 10.1002/eji.200535152. [DOI] [PubMed] [Google Scholar]
- 27.Sing A, Rost D, Tvardovskaia N, Roggenkamp A, Wiedemann A, Kirschning CJ, Aepfelbacher M, Heesemann J. Yersinia V-antigen exploits toll-like receptor 2 and CD14 for interleukin 10-mediated immunosuppression. J Exp Med. 2002;196:1017–1024. doi: 10.1084/jem.20020908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW, van Krieken JH, Hartung T, Adema G, Kullberg BJ. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol. 2004;172:3712–3718. doi: 10.4049/jimmunol.172.6.3712. [DOI] [PubMed] [Google Scholar]
- 29.Hajishengallis G, Ratti P, Harokopakis E. Peptide mapping of bacterial fimbrial epitopes interacting with pattern recognition receptors. J Biol Chem. 2005;280:38902–38913. doi: 10.1074/jbc.M507326200. [DOI] [PubMed] [Google Scholar]
- 30.Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama SI, Ratti P, Schifferle RE, Lyle EA, Triantafilou M, Triantafilou K, Yoshimura F. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- 31.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 32.Ukai T, Yumoto H, Gibson FC, 3rd, Genco CA. Macrophage-elicited osteoclastogenesis in response to bacterial stimulation requires Toll-like receptor 2-dependent tumor necrosis factor-α production. Infect Immun. 2008;76:812–819. doi: 10.1128/IAI.01241-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibson FC, 3rd, Ukai T, Genco CA. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059. doi: 10.2741/2822. [DOI] [PubMed] [Google Scholar]
- 34.Gibson FC, 3rd, Genco CA. Porphyromonas gingivalis mediated periodontal disease and atherosclerosis: disparate diseases with commonalities in pathogenesis through TLRs. Curr Pharm Des. 2007;13:3665–3675. doi: 10.2174/138161207783018554. [DOI] [PubMed] [Google Scholar]
- 35.Hajishengallis G, Shakhatreh MAK, Wang M, Liang S. Complement receptor 3 blockade Promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359–2367. doi: 10.4049/jimmunol.179.4.2359. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S-i, Yoshimura F, Demuth DR, Hajishengallis G. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- 37.Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111:927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 38.Stokes RW, Thorson LM, Speert DP. Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J Immunol. 1998;160:5514–5521. [PubMed] [Google Scholar]
- 39.Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas gingivalis fimbriae proactively modulate β2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect Immun. 2006;74:5658–5666. doi: 10.1128/IAI.00784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- 41.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, Odden AR, Shellito JE, Bagby GJ, Nelson S, Kolls JK. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hajishengallis G, Tapping RI, Martin MH, Nawar H, Lyle EA, Russell MW, Connell TD. Toll-like receptor 2 mediates cellular activation by the B subunits of type II heat-labile enterotoxins. Infect Immun. 2005;73:1343–1349. doi: 10.1128/IAI.73.3.1343-1349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boxio R, Bossenmeyer-Pourie C, Steinckwich N, Dournon C, Nusse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol. 2004;75:604–611. doi: 10.1189/jlb.0703340. [DOI] [PubMed] [Google Scholar]
- 44.Mydel P, Takahashi Y, Yumoto H, Sztukowska M, Kubica M, Gibson FC, 3rd, Kurtz DM, Jr, Travis J, Collins LV, Nguyen KA, Genco CA, Potempa J. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog. 2006;2:e76, 712–725. doi: 10.1371/journal.ppat.0020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harokopakis E, Albzreh MH, Haase EM, Scannapieco FA, Hajishengallis G. Inhibition of proinflammatory activities of major periodontal pathogens by aqueous extracts from elder flower (Sambucus nigra) J Periodontol. 2006;77:271–279. doi: 10.1902/jop.2006.050232. [DOI] [PubMed] [Google Scholar]
- 46.Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M. Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol. 1983;130:1910–1917. [PubMed] [Google Scholar]
- 47.Knapp S, von Aulock S, Leendertse M, Haslinger I, Draing C, Golenbock DT, van der Poll T. Lipoteichoic acid-induced lung inflammation depends on TLR2 and the concerted action of TLR4 and the platelet-activating factor receptor. J Immunol. 2008;180:3478–3484. doi: 10.4049/jimmunol.180.5.3478. [DOI] [PubMed] [Google Scholar]
- 48.Elbim C, Bailly S, Chollet-Martin S, Hakim J, Gougerot-Pocidalo MA. Differential priming effects of proinflammatory cytokines on human neutrophil oxidative burst in response to bacterial N-formyl peptides. Infect Immun. 1994;62:2195–2201. doi: 10.1128/iai.62.6.2195-2201.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bogdan C, Rollinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12:64–76. doi: 10.1016/s0952-7915(99)00052-7. [DOI] [PubMed] [Google Scholar]
- 50.Jadeski LC, Lala PK. Nitric oxide synthase inhibition by N(G)-nitro-L-arginine methyl ester inhibits tumor-induced angiogenesis in mammary tumors. Am J Pathol. 1999;155:1381–1390. doi: 10.1016/S0002-9440(10)65240-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balloy V, Si-Tahar M, Takeuchi O, Philippe B, Nahori MA, Tanguy M, Huerre M, Akira S, Latge JP, Chignard M. Involvement of toll-like receptor 2 in experimental invasive pulmonary aspergillosis. Infect Immun. 2005;73:5420–5425. doi: 10.1128/IAI.73.9.5420-5425.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yauch LE, Mansour MK, Shoham S, Rottman JB, Levitz SM. Involvement of CD14, toll-like receptors 2 and 4, and MyD88 in the host response to the fungal pathogen Cryptococcus neoformans in vivo. Infect Immun. 2004;72:5373–5382. doi: 10.1128/IAI.72.9.5373-5382.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Archer KA, Roy CR. MyD88-dependent responses involving toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires’ disease. Infect Immun. 2006;74:3325–3333. doi: 10.1128/IAI.02049-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fuse ET, Tateda K, Kikuchi Y, Matsumoto T, Gondaira F, Azuma A, Kudoh S, Standiford TJ, Yamaguchi K. Role of Toll-like receptor 2 in recognition of Legionella pneumophila in a murine pneumonia model. J Med Microbiol. 2007;56:305–312. doi: 10.1099/jmm.0.46913-0. [DOI] [PubMed] [Google Scholar]
- 55.Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 56.Letiembre M, Echchannaoui H, Bachmann P, Ferracin F, Nieto C, Espinosa M, Landmann R. Toll-like receptor 2 deficiency delays pneumococcal phagocytosis and impairs oxidative killing by granulocytes. Infect Immun. 2005;73:8397–8401. doi: 10.1128/IAI.73.12.8397-8401.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watanabe I, Ichiki M, Shiratsuchi A, Nakanishi Y. TLR2-mediated survival of Staphylococcus aureus in macrophages: A novel bacterial strategy against host innate immunity. J Immunol. 2007;178:4917–4925. doi: 10.4049/jimmunol.178.8.4917. [DOI] [PubMed] [Google Scholar]
- 58.Ji S, Hyun J, Park E, Lee BL, Kim KK, Choi Y. Susceptibility of various oral bacteria to antimicrobial peptides and to phagocytosis by neutrophils. J Periodontal Res. 2007;42:410–419. doi: 10.1111/j.1600-0765.2006.00962.x. [DOI] [PubMed] [Google Scholar]
- 59.Reeves EP, Lu H, Jacobs HL, Messina CGM, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 60.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 61.Yates RM, Russell DG. Phagosome maturation proceeds independently of stimulation of toll-like receptors 2 and 4. Immunity. 2005;23:409–417. doi: 10.1016/j.immuni.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 62.Gyurko R, Boustany G, Huang PL, Kantarci A, Van Dyke TE, Genco CA, Gibson FC., 3rd Mice lacking inducible nitric oxide synthase demonstrate impaired killing of Porphyromonas gingivalis. Infect Immun. 2003;71:4917–4924. doi: 10.1128/IAI.71.9.4917-4924.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steele C, Marrero L, Swain S, Harmsen AG, Zheng M, Brown GD, Gordon S, Shellito JE, Kolls JK. Alveolar macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J Exp Med. 2003;198:1677–1688. doi: 10.1084/jem.20030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shay K, Scannapieco FA, Terpenning MS, Smith BJ, Taylor GW. Nosocomial pneumonia and oral health. Spec Care Dentist. 2005;25:179–187. doi: 10.1111/j.1754-4505.2005.tb01647.x. [DOI] [PubMed] [Google Scholar]
- 66.Schroder NW, Schumann RR. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious disease. The Lancet infectious diseases. 2005;5:156–164. doi: 10.1016/S1473-3099(05)01308-3. [DOI] [PubMed] [Google Scholar]
- 67.Happel KI, Nelson S. Alcohol, immunosuppression, and the lung. Proc Am Thorac Soc. 2005;2:428–432. doi: 10.1513/pats.200507-065JS. [DOI] [PubMed] [Google Scholar]
- 68.Goral J, Kovacs EJ. In vivo ethanol exposure down-regulates TLR2−, TLR4−, and TLR9-mediated macrophage inflammatory response by limiting p38 and ERK1/2 activation. J Immunol. 2005;174:456–463. doi: 10.4049/jimmunol.174.1.456. [DOI] [PubMed] [Google Scholar]
- 69.Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol. 2004;33:147–155. doi: 10.1016/j.alcohol.2004.08.001. [DOI] [PubMed] [Google Scholar]