

Abstract
Microglial activation is one of the earliest and most prominent features of nearly all CNS neuropathologies often occurring prior to other indicators of overt neuropathology. Whether microglial activation in seemingly healthy CNS tissue during the early stages of several is a response to early stages of neuronal or glial distress or an early sign of microglial dysfunction causing subsequent neurodegeneration is unknown. Here we characterize and discuss how changes in the CNS microenvironment (neuronal activity/viability, glial activation) lead to specific forms of microglial activation. Specifically, we examine the potential role that TREM-2 expressing microglia may play in regulating the effector function of autoreactive T cell responses. Thus, we suggest that ubiquitous suppression of microglial activation during CNS inflammatory disorders rather than targeted manipulation of microglial activation, may in the end be maladaptive leading to incomplete remission of symptoms.
Keywords: Multiple sclerosis, Alzheimer's disease, Neurodegeneration, Autoimmunity, EAE, Facial axotomy, Pattern recognition receptors, Dendritic cells, Antigen-presentation, Antigen-presenting cell
1. Introduction
Microglia are the tissue macrophage of the central nervous system (CNS) and are found in all regions of the CNS and spinal cord (Streit, 2000; Aloisi, 2001; Carson, 2002; Rezaie and Male, 2002; Raivich, 2005). As suggested by their name, microglia are small cells. As shown in Fig. 1, fine processes extend out in all directions from the very small cell body of a microglia. Static pictures such as these have helped promote the concept that microglia are inactive in the healthy brain, and do not become active until environmental cues (pathogens and/or local cellular damage) stimulate the cells to change their morphology, as evidenced by increase in the size of the cell body and microglial processes. This idea of inactive immobile cells as the phenotype of microglia in the healthy brain was dramatically overturned in two recent studies. In these studies, live imaging of microglia in adult murine CNS tissue dramatically demonstrated that the cell bodies of the cells tended to stay in one place. Strikingly, the cell processes extending from the microglia were constantly in motion, suggesting constant monitoring of signals from the many cells in their environment (Davalos et al., 2005; Nimmerjahn et al., 2005). Furthermore, the slightly enlarged tips of the process extensions suggested that microglia were constantly engulfing material from their environment. As with other tissue macrophages, it is likely these cells are playing dual roles as sentinel cells and as “clean-up” cells helping to maintain the integrity of a physiologically active organ. These recent studies have triggered a renewed interest in defining the role of microglia!
Fig. 1.
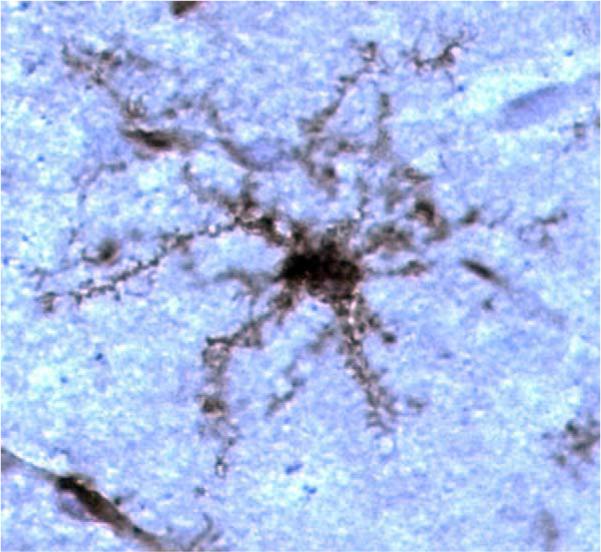
Microglia in adult murine CNS visualized with tomato lectin.
As early as the 1930s, Rio Hortega developed a staining method to label these cells and speculated on their roles in the CNS as brain macrophage (Rezaie and Male, 2002). Today, after approximately 75 years of additional research, the scientific community is still debating about the function of microglia in the healthy, injured and diseased CNS. On one hand, a very large literature exists that conclusively demonstrates the ability of activated microglia to produce large quantities of neurotoxic molecules. In vitro and in vivo experiments have also implicated activated microglia as promoting maladaptive autoreactive T cell responses. Furthermore, treating mice with agents such as minocycline that decrease microglial activation, does partially ameliorate clinical symptoms of rodent models of CNS inflammation and/or neurodegeneration (Zemke and Majid, 2004; Cai et al., 2006; Familian et al., 2006; Nikodemova et al., 2006). On the other hand, a smaller body of literature demonstrates the neuroprotective potential of microglia. Several studies have directly demonstrated the potential for activated microglia to be directly neuroprotective by their production of growth factors or to be indirectly protective by their production of immunosuppressive molecules (Polazzi et al., 2001; Polazzi and Contestabile, 2002; Minghetti, 2004; Streit, 2005). In addition, recent data suggest that microglial interactions with CD4+ T cells may be essential for the development and/or maintenance of neuroprotective T cell responses (Byram et al., 2004).
The obvious question arises of how to integrate these two apparently opposing lines of data. In part the inability of the scientific community to come to a consensus about the consequences of microglial activation for CNS function may be due to at least four factors:
The high degree of functional plasticity observed when studying microglia.
The inherent difficulty of examining microglial function in vivo.
The inability to distinguish acutely infiltrating peripheral macrophages from long-term CNS resident microglia when examining CNS tissue sections, and.
The artificially induced propensity of model systems to respond to insult and pathogenic signals with either balanced, well-regulated responses or disproportionate, dysregulated responses.
In this article we discuss these issues and try to reconcile the apparently conflicting data regarding microglial function. We will examine the extent microglia are a tissue-specific type of macrophage specialized for the CNS, and the extent that their phenotype and function are regulated by neurons and glia. Specifically, we discuss how the context of microglial activation may be the key indicator of whether microglia contribute toward directing infiltrating immune cells toward neuroprotective versus neurodestructive effector functions. To address these issues, first, we need to discuss what is an appropriate and predictive model to define microglial function.
2. Are cultured microglia predictive models of microglial function in vivo?
Microglia comprise between 5 and 15% of the resident cell population of the CNS, but current methods for isolating these cells acutely from the CNS are labor intensive and notorious for their relatively low yields (Streit, 2000; Aloisi, 2001; Carson, 2002; Rezaie and Male, 2002; Raivich, 2005). Thus, we and others have often used the more easily prepared primary microglia derived from mixed glial cultures established from neonatal mice and rats (Carson et al., 1998). However, as noted by numerous groups, we have found that these cultured microglia differ both in gene expression and in function from microglia acutely isolated from adult rodent CNS.
Specifically, in our initial efforts to generate a molecular definition of microglia, we performed a comprehensive comparison of neonatal cultured microglial and peritoneal macrophage gene expression (Schmid et al., 2002; Carson et al., 2004). We found several molecules such as the chemokine CXCL14 and protease nexin 1 (PN-1) that were expressed at higher levels in cultured microglia than in peritoneal macrophages and that were regulated by 24 h treatment with LPS/IFNg (Fig. 2). However, when we examined the expression of these molecules in vivo by in situ hybridization analysis, we found that neurons not microglia were the primary expressers of CXCL14 and astrocytes and not microglia were the primary expressers of PN-1. Thus, one of the primary outcomes of our initial gene expression studies was the demonstration that cultured microglia express molecules that are either not expressed or that are expressed at only very low levels (below detection) by microglia in vivo.
Fig. 2.
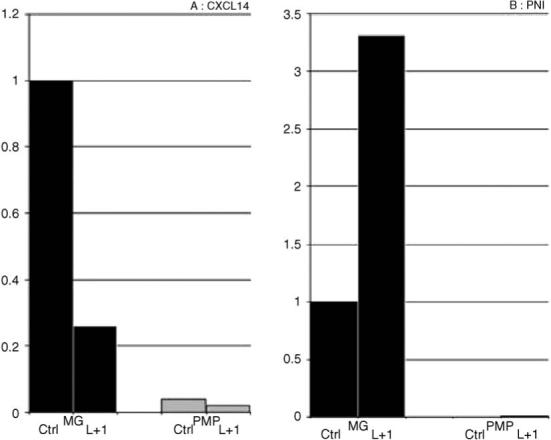
Real-time PCR analysis of CXCL14 (A) and protease-nexin 1 (B) in cultured microglia and peritoneal macrophages, untreated or LPS/IFNgamma treated as previously described in Schmid et al. (2002). CXCL14 primers: (forward primer: 5′-ATAAGGGGTTTTGTATTTGTCCAT; reverse primer: 5′-CATGCTCACTGTTCCTCCCA; amplification product: 76 bp), PN-1 primers: (forward primer: 5′-CTGGGATGCTGTAGTGAAGGAT; reverse primer: 5′-CCTGGAAAGTCACACATATCAACA; amplification product: 163 bp).
3. Are cultured microglia still useful models of microglia function in vivo?
Streit and colleagues have often noted that much of the data demonstrating the neurotoxic potential of microglia are from experiments using cultured microglia (Streit, 2005). Our molecular description of the differences between cultured and in vivo microglia also indicate that conclusions based on cultured microglia should be interpreted cautiously. Another complication in determining appropriate models has been revealed by several research groups, but also by our molecular studies: microglial heterogeneity in the rodent CNS (Schmid et al., 2002). Not surprisingly, we found that perhaps due to the homogenous environment of the culture dish, microglia in vitro display a much more homogenous phenotype.
Despite these observations, the great difficulty in isolating sufficiently large numbers of cells for RNA isolation and/or functional assays preclude routine use of adult microglia by most labs. Lack of access to mice and appropriate instrumentation will also prevent many labs from using live imaging techniques to monitor microglial activity within the living animal. Therefore, it is likely that a large body of microglial research will continue to rely on cultured forms of microglia. However, when interpreting data derived from cultured microglia, we must be aware that we are studying a cell type that is designed to monitor the health and activity of the neurons and glia in its environment! Cultured microglia, even when cultured in the presence of mixed glial cells are usually deprived of neuronal signals.
4. Is there evidence that neuronal signals do actively regulate microglial phenotype?
The most obvious answer to this question comes from a neuronal:microglial co-culture model. In this system, microglial expression of MHC class II was inhibited when neurons were electrically active, but was rapidly induced when electrical activity was suppressed (Neumann, 2001). More recently, several neuronally expressed molecules have been found to regulate the baseline activation status of microglia in vivo. For example, in vivo, CNS neurons express CD200, while microglia (and other macrophages) express the CD200 receptor (Hoek et al., 2000). When CD200 expression was deleted in knock-out mice, microglia displayed an activated phenotype (elevated expression of CD45, MHC class II, complement receptor 3) in the absence of pathogenic stimuli and displayed a more rapid and robust activation in the presence of cellular damage or pathogenic stimuli.
These data indicate the need to carefully interpret experiments using cultured microglia. Cultured microglia experiment must be considered as models of microglia activated after neuronal signals are lost and when glial proliferation is occurring! This is not how these studies are usually interpreted, but may indicate why results from in vitro assays are not always predictive of in vivo events. It is likely that culture models may have greater relevance for stroke and other neurological disorders with large neuronal lesions and reactive gliosis.
5. Do parenchymal microglia differ from other macrophage populations?
When examining histological sections of CNS pathologies, microglia and macrophages are often considered together as one population of microglia/macrophages. Indeed, activated microglia do express all of the common macrophage markers, including iba1, Fc receptor, CD11b, F4/80, CD14 and CD45 preventing an easy distinction between microglia and acutely infiltrating macrophages (Sedgwick et al., 1991; Becher et al., 1996; Carson et al., 1998; Aloisi, 2001; Popovich and Hickey, 2001). However, despite expressing many of the same molecules, several recent studies clearly illustrate that microglia and macrophages are phenotypically distinct cell types and are not fully interchangeable in their functions.
Most notably, microglia and peripheral macrophage populations appear to differ in the rate that they are replaced by bone marrow stem cells: parenchymal microglia are only slowly or rarely replaced by bone marrow stem cells, while all other macrophage populations tend to be replaced 100% within days to months (Matsumoto and Fujiwara, 1987; Hickey and Kimura, 1988; Schilling et al., 2003). This is tissue dependent, with the fastest rate of turnover being evident in the blood, and slower rates observed in the skin (∼20−30 days) and in the heart (∼3−6 months). These determinations of the lifespan and turnover of macrophages in tissues has primarily relied on the use of irradiation bone marrow chimeric mice in which bone marrow of a recipient animal is killed by lethal irradiation and replaced by donor bone marrow expressing markers such as green fluorescent protein (GFP). We have recently reviewed this literature (Carson et al., 2004). Therefore, here we will merely note the following. In most of these types of studies, small numbers of donor derived (i.e. GFP+) cells with a microglia morphology and expressing macrophage markers can be found in the otherwise unmanipulated adult rodent CNS (Matsumoto and Fujiwara, 1987; Hickey and Kimura, 1988; Schilling et al., 2003). These cells do not appear with equal frequency or with equal kinetics in all brain regions. Rather when care is taken to note whether these cells have truly left the vasculature, donor derived cells are found primarily associated with cerebral vasculature (i.e. perivascular macrophages), within the leptomeninges and the cerebellum. In other brain regions such as the cerebral cortex, caudoputamen and hippocampal formation, donor derived GFP+ parenchymal microglia were never found (Matsumoto and Fujiwara, 1987; Hickey and Kimura, 1988). For humans, a small number of bone marrow transplant studies in which donor and recipient were gender mismatched suggest that these data derived from rodent studies apply to human physiology (Unger et al., 1993; Bauer et al., 2001).
In sum, the primary finding from these irradiation bone marrow chimeric studies is the demonstration that the CNS is populated by two very different types of myeloid cells: a parenchymal population slowly or only rarely replenished by bone marrow stem cells and a primarily perivascular, meningeal and cerebellum associated population that is rapidly and frequently replaced by bone marrow stem cells. The regional differences in the relative proportions of these two populations is quite likely to have significant ramifications for CNS:immune system interactions and the propensity of different regions to develop autoimmune disease.
Of particular interest is the speculation that these two populations have different abilities to leave the CNS and migrate to the draining cervical lymph nodes. Previously we have shown that following injection of dye-labeled peripheral antigen-presenting cells (dendritic cells and macrophages) directly into the CNS, we could detect these cells in the T cell zones of the cervical lymph nodes (Carson et al., 1999). By contrast, following injection of dye-labeled cultured microglia into the CNS, we were unable to detect microglia in the cervical lymph nodes. These data suggest that at best microglia are several orders of magnitude less efficient at migrating out of the CNS than other macrophage populations, and at worst microglia may not be able to leave the CNS at all!
6. What are the consequences of this potential differential ability to migrate to the draining lymph nodes?
The primary initiation of T cell responses to antigen does not occur within tissues (Lo et al., 1999). The initial activation of T cell responses occurs when tissue macrophages/immature dendritic cells migrate to the lymph nodes, after having phagocytosed material (cellular debris, pathogens) from within their starting tissue (i.e. pancreas, liver, thyroid). Immature dendritic cells and macrophages are able to proteolytically process this material (a process referred to as antigen-processing) and place this processed material within the binding cleft of MHC class I and MHC class II. The antigen-loaded MHC is then transported to the cell surface. Upon arrival in the lymph nodes, these cells can then present the antigen to the T cell receptors on either CD8 T cells (antigen presented within the binding cleft of MHC class I) or CD4 T cells (antigen presented within the binding cleft of MHC class II). This antigen-presentation event is an essential rate-limiting step for T cell activation. Without an antigen-presenting cell, T cells cannot detect their target antigens because the T cell receptor is incapable of binding antigen when it is not presented within MHC. Thus, If microglia cannot migrate to the cervical lymph nodes, unlike other macrophages populations they will be unable to initiate primary immune responses against antigens located within the CNS. If confirmed by other measures, this feature of microglial biology would be consistent with the well-described phenomenon referred to as immune privilege: the ability of foreign material (grafts, pathogens) to reside in the CNS parenchyma without initiating a destructive proinflammatory T cell response.
7. Are the same microglia there at all times?
When do microglia first appear within the CNS? Although there is a bit of a controversy of precisely when microglia populate the CNS, microglia can clearly be detected early in the development of the CNS. In rodents, they are clearly apparent as early as embryonic day 15, and can be detected by transgenic methodologies as early as embryonic day 10.5 (Hirasawa et al., 2005). However, around birth, “fountains of microglia” become apparent in the CNS. By histology, the implication is that these microglia are entering from the periphery. This would suggest that the CNS is populated by waves of myeloid populations. Surprisingly, the potential consequences for multiple origins of parenchymal microglia has only rarely been explored and has not been confirmed by more than histological methods!
8. Molecular markers of parenchymal microglia versus peripheral macrophage populations
The holy grail of neuroimmunology would be to identify molecular markers that distinguish microglia from acutely infiltrating macrophage populations. As yet, no such markers have bee identified. However, microglia and macrophages do express significantly different levels of key regulatory molecules indicating that these two cell types do indeed exist in two different stable physiological states. Sedgwick and his colleagues were the first to discover that in contrast to acutely infiltrating macrophages and lymphocytes, microglia expressed much lower levels of CD45 (Sedgwick et al., 1991; Renno et al., 1995; Carson et al., 1998). In response to chronic pathology or robust in vivo inflammatory signals, the CD45 levels on both microglia and other macrophages does increase. However even after activation, microglial levels of CD45 tend to remain at an intermediate level between that of unactivated microglia and of mature macrophages, although there is some overlap in the expression levels of CD45 between the various myeloid populations (Sedgwick et al., 1991; Renno et al., 1995; Carson et al., 1998).
Subsequent to the seminal studies by Sedgwick and colleagues, we compared CD45 expression among microglia and other leukocyte populations as a function of development. Surprisingly, when comparing embryonic day 15, birth, and adulthood, we found that only immune cells from embryonic day 15 fetal liver expressed the same low levels of CD45 as adult parenchymal microglia (Carson et al., 1998). From this observation, we suggested that microglia were relatively unique as they were maintained as a relatively immature macrophage population in the adult. Recent studies by Finson and colleagues following microglial expression CD34, an early marker of immune cell development supports our earlier speculation (Ladeby et al., 2005).
9. What is the consequence of the differential expression of CD45?
CD45 (also called leukocyte common antigen) is a protein tyrosine phosphatase expressed by all nucleated cells of hemopoetic origin (Sedgwick et al., 1991; Renno et al., 1995; Carson et al., 1998). In peripheral macrophages, CD45 functions as both an activating signal and as a Janus kinase (JAK) tyrosine phosphatase that negatively regulates cytokine receptor signaling involved in the differentiation, proliferation, and antiviral immunity of hematopoietic cells (Irie-Sasaki et al., 2003). CD45 is also a receptor for CD22, a molecule first described as being expressed on B cells. More recently, CNS neurons have been found to express high levels of CD22 (Mott et al., 2004). Taken together these observations suggest that CD22 expressing neurons would be more efficient at inhibiting macrophage activation than microglia activation for the simple reason that macrophages express a order of magnitude higher levels of CD45 than microglia! This in turn suggests that activated microglia may be performing different functions that are perhaps less destructive or even more beneficial than those performed by macrophages!
10. Is there functional evidence for differential functions of activated microglia and macrophages in vivo?
Microglia and macrophages cannot be distinguished histologically (reliable measurements of CD45 need to be made by flow cytometric cell analysis of cell suspensions). Therefore, several studies seeking to differentiate the functions of resident microglia from acutely infiltrating cells have relied on either irradiation bone marrow chimeric mice or on selective depletion of peripheral immune cells using various detergent depletion regimens.
Using bone marrow chimeric animals, Popovich and colleagues illustrated that microglia and blood-derived macrophages were each recruited to different portions of a lesion caused by traumatic injury to the spinal cord (Popovich and Hickey, 2001; Popovich et al., 2003). In this model, macrophage accumulation followed microglial activation. Strikingly, regions with the most severe neurodegeneration and subsequent necrosis contained blood-derived macrophages, while spared white matter regions were adjacent to accumulations of activated microglia. Depletion of peripheral macrophages by C12MDP liposome treatment dramatically decreased macrophage recruitment to the injured spinal cord and dramatically decreased the area of spinal cord necrosis. These data strongly suggest that microglia and acutely infiltrating macrophages play distinct roles and that microglia may be playing less destructive or even actively neuroprotective roles.
11. Can microglial activation be directly neuroprotective?
One of the earliest microglial responses to neuronal injury is the induction of MHC class II (Streit, 2000; Aloisi, 2001; Carson, 2002; Rezaie and Male, 2002; Raivich, 2005). This induction of MHC class II on microglia has been frequently postulated to promote maladaptive neurotoxic proinflammatory T cells response. We previously mentioned that microglia were unlikely to migrate to the cervical lymph nodes in sufficiently large numbers to initiate T cell response. However T cells activated by other antigen-presenting cells outside the CNS will non-specifically enter tissues searching for their target antigen (Medzhitov and Janeway, 1998). Antigen-inexperienced (naiäve) T cells can also be recruited non-specifically to the CNS by chemoattractant signals produced by damaged and stressed neurons, glia and/or endothelium (Krakowski and Owens, 2000). Within the CNS, MHC-expressing microglia can present antigen and thus, inform T cells that their target antigen is in the local vicinity (Byram et al., 2004). If T cell activation within the CNS is maladaptive, it is unclear why the frequent induction of MHC class II does not lead to destructive autoimmune responses on a more frequent basis. One clue to this puzzle is suggested by recent studies using the well-characterized facial axotomy model of neurodegeneration (Byram et al., 2004).
Sanders and Jones had previously shown that motoneuron degeneration following facial axotomy is associated with microglial activation and CNS-infiltration of macrophages and T cells (Serpe et al., 1999). Surprisingly, Sanders and Jones observed that this inflammatory response served to limit motoneuron degeneration (Raivich et al., 1998; Serpe et al., 1999). Again, using irradiation bone marrow chimeric mice, mice were generated in which either only microglia or only peripheral immune cells could act as antigen-presenting cells (Byram et al., 2004). Not surprisingly, when only microglia could express MHC class II and present antigen, microglia alone were unable to initiate the protective CD4+ T cell response. Conversely, peripheral macrophage also by themselves were insufficient to elicit the protective immune response, even though GFP+, blood-derived cells could be detected within the CNS. However, by serially transferring T cells from one chimeric mouse model to the next, it was found that microglia and peripheral immune cells each played a different function during different stages of T cell activation. Macrophages were required to initiate the response the CD4+ T cell mediated neuroprotection of motoneurons. However, once T cell activation was initiated by peripheral macrophages, protection of motoneurons following facial axotomy was absolutely dependent on antigen-presentation by microglia activated by local neurodegenerative signals (Byram et al., 2004).
12. How do we explain these results with those observed in proinflammatory EAE studies?
The spinal cord injury and facial axotomy studies suggest that microglia play essential roles in minimizing neurodegeneration and maintaining neuronal integrity. In contrast, several studies suggest the reverse. Here we also present two studies using bone marrow chimeric mice that demonstrate specific microglial contributions to T cell mediated neuropathology. Experimentally induced autoimmune encephalomyelitis (EAE) is an animal model for multiple sclerosis (Bauer et al., 1995). In two separate studies, Becher and colleagues generated bone marrow chimeric mice in which only parenchymal microglia or hematogenous macrophages express the proinflammatory cytokine IL-23 (Becher et al., 2003) and chimeric mice in which microglia could not express CD40, the receptor for CD154 (Becher et al., 2001). Becher and colleagues showed that when microglia could not express IL-23 or CD40 the severity of clinical symptoms of MOG-EAE was drastically reduced. Additionally, while there was little impact on the degree of inflammation occurring within the CNS, the T cell cytokine profile shifted from a proinflammatory Th1 response to a protective Th2 response. Thus even in the presence of CNS-infiltrating macrophages, interrupting the ability of microglia to support proinflammatory T cell responses is sufficient to alter the course of the disease.
These studies clearly demonstrate the potential for microglia and macrophages to play significantly different roles in neurodegenerative disease. Therefore, it is crucial to directly examine their differential ability to alter neuronal function and viability and whether changes in the CNS microenvironment shifts microglial functions from neuroprotective to proinflammatory/neurodestructive.
13. Hints from other models
The development of T cell effector function is entirely dependent on the context of activation (Medzhitov and Janeway, 1998; Lo et al., 1999). Namely what types of co-stimulatory factors does the antigen-presenting cell express (CD40, B7, etc.)? In the absence of co-stimulation, antigen-presentation can promote the development T cell inactivation (anergy) and possibly the development of immunosuppressive regulatory T cell function. When considering EAE studies, it is important to realize that destructive autoimmunity requires either repetitive active immunization of the antigen (i.e. myelin protein) in the presence of strong adjuvants (complete Freund's adjuvant, heat killed mycobacterium tuberculosis and pertussis toxin) or the transfer of activated T cells already strongly differentiated into a proinflammatory Th1 phenotype. By contrast, in the facial axotomy model, no adjuvant was used, and the only activating stimulus was mechanical injury followed by Wallerian degeneration. The former EAE associated treatment leads to systemic activation of T cells and antigen-presenting cells throughout the body and to systemic changes in chemokine expression and vascular function. By contrast the systemic effects of axotomy are modest at best as overt systemic signs of activation are not detected and the only signs of inflammation are very local to the affected motoneuronal tract.
It is also important to note that the same stimuli that induce microglia expression of MHC also prime and promote microglial production of nitric oxide and prostaglandins (Minghetti, 2004). Although these molecules are often characterized as neurotoxic these molecules also suppress expression of MHC expression by CNS-infiltrating antigen-presenting cells and directly inhibit T cell activation. The relevance of these pathways for immunosuppression are demonstrated by the increased severity of EAE in mice deficient in NO production (Willenborg et al., 1999).
However, since these molecules have demonstrated neurotoxic effects, the amount of NO/prostaglandin production will also determine the relative beneficial:deleterious ratio of microglial activation. In this light it is important to note the dose of the pathogenic insults used in various published studies when analyzing outcomes of microglial activation. For example, a 1 mg/kg dose of intracerebrally injected LPS induces permanent CNS neurodegeneration and white matter pallor, while the same treatment with an approximately 25,000-fold lower dose results in a rapid, robust but entirely self-resolving CNS inflammation (Schmid et al., 2002). Similarly, in vitro high does of LPS induce activation induced cell death, while low does do not!
14. Molecular cues: TREM-2
While the activation state of the peripheral immune system is clearly important, the studies above also suggest that the activation status of microglia may play a significant or contributing role in determining beneficial versus detrimental outcome. To try to define microglial activation in molecular terms, we have performed surveys of gene expression in unactivated and activated microglia isolated from adult murine CNS. To determine which molecular profiles were associated with general microglial activation, or with beneficial versus detrimental outcomes we have also analyzed spatial and kinetic expression of a subset of molecule identified from our molecular screens in contrasting models of acute but resolving versus chronic progressive models of CNS inflammation (Schmid et al., 2002; Carson et al., 2004).
In our first set of studies, we have initially focused on the role of a new family of putative activating receptors: the triggering receptors expressed on myeloid cells (TREMs) and their closely related putatively inhibitory receptors: TREM-like transcripts (TLTs) (Schmid et al., 2002; Carson et al., 2004). In our initial observations, we discovered that subsets of microglia in vivo and nearly all microglia in vitro express one member of this new receptor family, TREM-2. Colonna and colleagues had recently implicated TREM-2 in promoting antigen-presenting cell function of immature dendritic cells, while Daws and his colleagues had implicated TREM-2 in regulating macrophage production of NO (Bouchon et al., 2001; Daws et al., 2001, 2003). In addition, positional cloning studies revealed that humans lacking a functional TREM-2 signaling develop an early onset dementia (Paloneva et al., 2001). Lastly, our lab has found that microglial expression of TREM-2, but not any other member of the TREM family is induced in response to nearly any neurodegenerative signal (Thrash et al., unpublished results). The one striking exception is the reduction in TREM-2 expression observed following intracerebral injection of LPS or LPS plus IFNgamma. However, in our model and with the low LPS does used, we also do not observe neuronal distress or degeneration. Therefore, TREM-2 appeared as a potential candidate for regulating the key beneficial versus detrimental activation states of microglia.
15. What does TREM-2 do?
Since TREM-2 is an orphan receptor, to define the consequences of TREM-2 mediated activation, Colonna used antibodies against human TREM-2 to cross-link this receptor on the surface of immature human dendritic cells (Bouchon et al., 2001). Using this method, Colonna and colleagues found that TREM-2 mediated activation triggered dendritic cell expression of molecules required for antigen-presentation to CD4+ T cells (including MHC class II, CD40, B7.1, B7.2, CCR7), without decreasing expression of molecules required to capture antigen from the environment and process the protein into antigenic peptides.
To test this hypothesis that TREM-2 could promote the differentiation of microglia into more potent antigen-presenting cells, and to monitor TREM-2 protein expression, we have also used antibodies against the extracellular domains of murine TREM-2. Using TREM-2 antisera, we confirmed that TREM-2 triggered activation caused the BV-2 microglial cell line to upregulate the expression of some molecules required for antigen presentation, specifically MHC class II. In contrast to what Colonna and colleagues observed with human dendritic cells, TREM-2 triggered activation failed to induce cultured microglia to upregulate expression of CD40 (the receptor for a molecule expressed by activated T cells: CD40 ligand) (Bouchon et al., 2001). T cells directly induce their antigen-presenting cells to make cytokines (IL-12/IL23) via CD40 ligand:CD40 interactions that serve to drive and support proinflammatory T cell function. Therefore, our data imply that: (a) TREM-2 triggers different activation cascades in microglia than in other TREM-2 expressing peripheral immune cells and (b) the TREM-2 positive microglia are likely to drive a different T cell effector function than that of other peripheral antigen-presenting cells. More recent data from Daws and colleagues has highlighted the potential importance of TREM-2 expression in the CNS (Daws et al., 2003). Performing a widescale survey of cells and small molecules, they have defined an endogenous binding activity (a putative ligand?) on astrocytes. Taken together these data suggest that astrocytes may be able to directly regulate the antigen-presenting function of TREM-2 expressing microglia and macrophages.
We have also discovered a novel non-membrane bound splice variant of TREM-2 (svTREM-2) with the potential to be a soluble “decoy” receptor for the endogenous ligand (Fig. 3) (Schmid et al., 2002; Carson et al., 2004). In both unactivated microglia and unactivated macrophages, the ratio of membrane bound TREM to svTREM-2 expression is ∼9:1. Upon LPS/IFNg induced activation, microglia and macrophages differentially regulate the relative ratio of these two forms of TREM-2. In activated microglia, the ratio of membrane bound to sv forms becomes 12:1, while it becomes 6:1 in activated macrophages. Thus activated microglia (in vivo and in vitro) differ from peripheral macrophage populations in that they preferentially are able to bind TREM-2 ligands (rather than block them with a soluble decoy receptor).
Fig. 3.
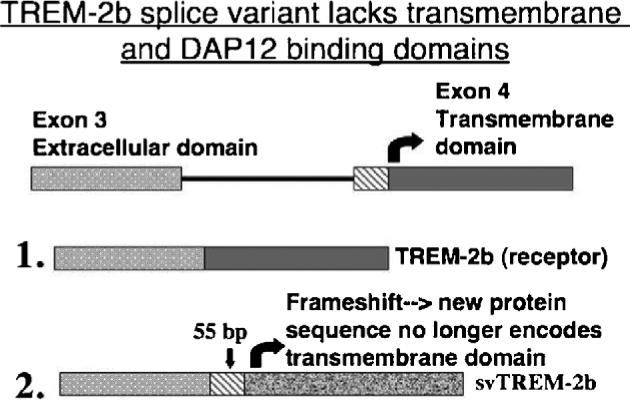
Alternative splicing of TREM-2 generates a non-receptor form of the molecule with potential to serve as a “decoy” receptor.
Recently, there has been a bit of controversy concerning whether other CNS cells (oligodendrocytes and neurons) express TREM-2 (Kiialainen et al., 2005). In our studies it may be possible that we only detected the most prominent expression of TREM-2 on microglia. One cautionary note should also be raised. The studies detecting TREM-2 on neurons and oligodendrocytes primarily used antisera. However, the presence of a potentially soluble form of TREM-2 (svTREM-2) could be a confounding factor causing cells associated with microglial secreted svTREM-2 to be identified as TREM-2 expressing cells. Lastly, TREM-2 belongs to the family of receptors lacking an intracellular signaling tail and is entirely dependent on the presence of DAP12 to mediate TREM-2 induced intracellular signaling. To date, DAP12 expression has not been described in neurons. Thus it is unlikely that even if TREM-2 is expressed by neurons that it is functionally active.
16. Summary and conclusions
Taken together, we have presented evidence from several labs including our own indicating that microglia are indeed a CNS-specific type of macrophage. Whether the outcome of microglial activation is likely to lead to T cell mediated neuroprotection or neurodegeneration is clearly dependent on the context of activation. We suggest that current strategies aimed at completely suppressing microglial function and T cell activation may only partially ameliorate clinical symptoms of neurodegeneration. However, since these same strategies will also inhibit microglia and T cell functions aimed at promoting CNS repair and regeneration, these strategies may ultimately lead to maladaptive outcomes.
References
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Bauer J, Rauschka H, Lassmann H. Inflammation in the nervous system: the human perspective. Glia. 2001;36:235–243. doi: 10.1002/glia.1112. [DOI] [PubMed] [Google Scholar]
- Bauer J, Huitinga I, Zhao W, Lassmann H, Hickey WF, Dijkstra CD. The role of macrophages, perivascular cells, and microglial cells in the pathogenesis of experimental autoimmune encephalomyelitis. Glia. 1995;15:437–446. doi: 10.1002/glia.440150407. [DOI] [PubMed] [Google Scholar]
- Becher B, Fedorowicz V, Antel JP. Regulation of CD14 expression on human adult central nervous system-derived microglia. J. Neurosci. Res. 1996;45:375–381. doi: 10.1002/(SICI)1097-4547(19960815)45:4<375::AID-JNR6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Noelle RJ. IL-23 produced by CNS-resident cells controls T cell encephalitogenicity during the effector phase of experimental autoimmune encephalomyelitis. J. Clin. Invest. 2003;112:1186–1191. doi: 10.1172/JCI19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ. The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J. Exp. Med. 2001;193:967–974. doi: 10.1084/jem.193.8.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchon A, Hernandez-Munain C, Cella M, Colonna M. A dap12-mediated pathway regulates expression of cc chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, Deboy CA, Serpe CJ, Sanders VM, Jones KJ. CD4+ T cell-mediated neuroprotection requires dual compartment antigen presentation. J. Neurosci. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137:425–435. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Carson MJ. Microglia as liaisons between the immune and central nervous systems: functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Thrash JC, Lo D. Analysis of microglial gene expression: identifying targets for CNS neurodegenerative and autoimmune disease. Am. J. Pharmacogenomics. 2004;4:321–330. doi: 10.2165/00129785-200404050-00005. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Disproportionate recruitment of CD8+ T cells into the central nervous system by professional antigen-presenting cells. Am. J. Pathol. 1999;154:481–494. doi: 10.1016/S0002-9440(10)65294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Daws MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 2001;31:783–791. doi: 10.1002/1521-4141(200103)31:3<783::aid-immu783>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE. Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 2003;171:594–599. doi: 10.4049/jimmunol.171.2.594. [DOI] [PubMed] [Google Scholar]
- Familian A, Boshuizen RS, Eikelenboom P, Veerhuis R. Inhibitory effect of minocycline on amyloid beta fibril formation and human microglial activation. Glia. 2006;53:233–240. doi: 10.1002/glia.20268. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J. Neurosci. Res. 2005;81:357–362. doi: 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Irie-Sasaki J, Sasaki T, Penninger JM. CD45 regulated signaling pathways. Curr. Top. Med. Chem. 2003;3:783–796. doi: 10.2174/1568026033452339. [DOI] [PubMed] [Google Scholar]
- Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol. Dis. 2005;18:314–322. doi: 10.1016/j.nbd.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Krakowski ML, Owens T. Naive T lymphocytes traffic to inflamed central nervous system, but require antigen recognition for activation. Eur. J. Immunol. 2000;30:1002–1009. doi: 10.1002/(SICI)1521-4141(200004)30:4<1002::AID-IMMU1002>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Dalmau I, Gregersen R, Garcia-Ovejero D, Babcock A, Owens T, Finsen B. Proliferating resident microglia express the stem cell antigen CD34 in response to acute neural injury. Glia. 2005;50:121–131. doi: 10.1002/glia.20159. [DOI] [PubMed] [Google Scholar]
- Lo D, Feng LL, Li L, Carson MJ, Crowley M, Pauza M, Nguyen A, Reilly CR. Integrating innate and adaptive immunity in the whole animal. Immunol. Rev. 1999;169:225–239. doi: 10.1111/j.1600-065x.1999.tb01318.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Fujiwara M. Absence of donor-type major histocompatibility complex class I antigen-bearing microglia in the rat central nervous system of radiation bone marrow chimeras. J. Neuroimmunol. 1987;17:71–82. doi: 10.1016/0165-5728(87)90032-4. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr. Innate immune recognition and control of adaptive immune responses. Semin. Immunol. 1998;10:351–353. doi: 10.1006/smim.1998.0136. [DOI] [PubMed] [Google Scholar]
- Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004;63:901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- Mott RT, Ait-Ghezala G, Town T, Mori T, Vendrame M, Zeng J, Ehrhart J, Mullan M, Tan J. Neuronal expression of CD22: novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia. 2004;46:369–379. doi: 10.1002/glia.20009. [DOI] [PubMed] [Google Scholar]
- Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and Ikappa-Balpha degradation in a stimulus-specific manner in microglia. J. Neurochem. 2006;96:314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. 2001;56:1552–1558. doi: 10.1212/wnl.56.11.1552. [DOI] [PubMed] [Google Scholar]
- Polazzi E, Contestabile A. Reciprocal interactions between microglia and neurons: from survival to neuropathology. Rev. Neurosci. 2002;13:221–242. doi: 10.1515/revneuro.2002.13.3.221. [DOI] [PubMed] [Google Scholar]
- Polazzi E, Gianni T, Contestabile A. Microglial cells protect cerebellar granule neurons from apoptosis: evidence for reciprocal signaling. Glia. 2001;36:271–280. doi: 10.1002/glia.1115. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Hickey WF. Bone marrow chimeric rats reveal the unique distribution of resident and recruited macrophages in the contused rat spinal cord. J. Neuropathol. Exp. Neurol. 2001;60:676–685. doi: 10.1093/jnen/60.7.676. [DOI] [PubMed] [Google Scholar]
- Popovich PG, van Rooijen N, Hickey WF, Preidis G, McGaughy V. Hematogenous macrophages express CD8 and distribute to regions of lesion cavitation after spinal cord injury. Exp. Neurol. 2003;182:275–287. doi: 10.1016/s0014-4886(03)00120-1. [DOI] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CU, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J. Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J. Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Rezaie P, Male D. Mesoglia & microglia—a historical review of the concept of mononuclear phagocytes within the central nervous system. J. Hist. Neurosci. 2002;11:325–374. doi: 10.1076/jhin.11.4.325.8531. [DOI] [PubMed] [Google Scholar]
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2003;183:25–33. doi: 10.1016/s0014-4886(03)00082-7. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hil-bush BS, Sutcliffe JG, Carson MJ. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002;83:1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl. Acad. Sci. U.S.A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J. Neurosci. 1999;19 doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglial response to brain injury: a brief synopsis. Toxicol. Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res. Brain Res. Rev. 2005;48:234–239. doi: 10.1016/j.brainresrev.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Unger ER, Sung JH, Manivel JC, Chenggis ML, Blazar BR, Krivit W. Male donor-derived cells in the brains of female sex-mismatched bone marrow transplant recipients: a Y-chromosome specific in situ hybridization study. J. Neuropathol. Exp. Neurol. 1993;52:460–470. doi: 10.1097/00005072-199309000-00004. [DOI] [PubMed] [Google Scholar]
- Willenborg DO, Staykova MA, Cowden WB. Our shifting understanding of the role of nitric oxide in autoimmune encephalomyelitis: a review. J. Neuroimmunol. 1999;100:21–35. doi: 10.1016/s0165-5728(99)00212-x. [DOI] [PubMed] [Google Scholar]
- Zemke D, Majid A. The potential of minocycline for neuroprotection in human neurologic disease. Clin. Neuropharmacol. 2004;27:293–298. doi: 10.1097/01.wnf.0000150867.98887.3e. [DOI] [PubMed] [Google Scholar]