

Abstract
Cytomegalovirus (CMV) reactivation is a well described complication of solid organ transplantation. These studies were performed to 1.) determine if cardiac allograft transplantation of latently infected recipients results in reactivation of CMV, and 2.) determine what impact CMV might have on development of graft acceptance/tolerance. BALB/c cardiac allografts were transplanted into C57BL/6 mice with/without latent murine CMV (MCMV). Recipients were treated with gallium nitrate induction and monitored for graft survival, viral immunity, and donor reactive DTH responses. Latently infected allograft recipients had ∼80% graft loss by 100 days after transplant, compared with ∼8% graft loss in naïve recipients. PCR evaluation demonstrated that MCMV was transmitted to cardiac grafts in all latently infected recipients, and 4/8 allografts had active viral transcription compared to 0/6 isografts. Latently infected allograft recipients showed intragraft IFN-α expression consistent with MCMV reactivation, but MCMV did not appear to negatively influence regulatory gene expression. Infected allograft recipients had disruption of splenocyte DTH regulation, but recipient splenocytes remained unresponsive to donor antigen even after allograft losses. These data suggest that transplantation in an environment of latent CMV infection may reactivate virus, and that intragraft responses disrupt development of allograft acceptance.
Introduction
The ultimate goal of transplant immunology is tolerance of alloantigen without immunosuppression. Given the myriad of immune responses that occur simultaneously in response to foreign antigens, it is perhaps not surprising that achieving this goal has been quite elusive, despite decades of work. As surrogates of tolerance, we have been interested for many years in murine models of cardiac allograft acceptance (1-5) . These models allow induced graft acceptance and function without long term requirement for immunosuppression, offering the unique opportunity to study events involved with allograft acceptance outside the context of ongoing immunosuppression.
We and others have previously shown that H2d (DBA) heterotopic cardiac allografts function indefinitely in H2b (C57BL/6) recipients after transient gallium nitrate (GN) treatment (1-5). This allograft acceptance is associated with immune regulation of donor-reactive delayed type hypersensitivity (DTH) responses, which appears to be controlled by transforming growth factor beta (TGF-β) and interleukin-10 (IL-10). Perhaps even more interesting is the observation that under this immune regulation scenario, “linked non-responsiveness” occurs, so that hyporesponsiveness of allograft acceptors to alloantigen is also conferred onto third party antigens when simultaneously presented (2). Given the high incidence of infectious pathogens seen in patients following transplantation, we were therefore extremely interested to study what influence linked non-responsiveness might have upon a viral pathogen. We sought to determine if allograft recipients would develop regulated responses to virus to the detriment of the host, or conversely, develop antiviral immune responses that would disrupt development of allograft acceptance.
Our interest in effects of viral infection on transplantation tolerance is not unique, and previous work by others using murine models has shown that acute viral infection during or shortly after transplantation can interfere with allograft acceptance and tolerance (6-8). In addition, chronic infections with persistent viral shedding also appear to have detrimental effects on tolerance (9), but interestingly, acute viral infection after establishment of tolerance does not seem to influence graft survival (6, 7). Although these models are representative of acute infection with novel viral pathogens during or just after transplantation, clinically this probably occurs far less frequently than reactivation of latent infection. We therefore chose to study graft acceptance interactions with the most frequent viral pathogen encountered in clinical transplantation, latent cytomegalovirus (CMV).
Although there are several published studies evaluating the influence of CMV in graft acceptance/rejection in rodent models (reviewed in (10)), all of these studies utilize acute infection during the peritransplant period. In rat models of cardiac allotransplantation, acute infection with CMV causes allograft vasculopathy that leads to graft loss (11-14). Similarly, accelerated rejection of murine cardiac allografts following acute infection has been described using murine CMV (MCMV) models (15, 16). Nonetheless, to date there are no data that we are aware of evaluating the influence of latent CMV infection upon graft acceptance or tolerance. Previous work with MCMV in transplantation has been hampered by the fact that C57BL6 mice are highly resistant to MCMV infection (17). Fortunately, the mechanism of this resistance has recently been described (18, 19), and mutant MCMV viruses have been developed that will infect C57BL6 mice to normal titers (20). We thus combined a model of murine cardiac allograft acceptance with latent MCMV infection, transplanting latently infected recipients with MCMV naïve donor organs (D-R+), modeling a commonly encountered scenario in clinical transplantation.
In addition to evaluating influences of latent CMV upon allograft acceptance, combining these models also affords the opportunity to ask another age old question; does allogeneic stimulation or immunosuppression cause CMV reactivation after transplantation? Allogeneic stimulation was first proposed to cause CMV reactivation by Lang in 1972 either by transfusion or transplantation (21). Since then, numerous in-vitro and in-vivo studies have suggested but not proven that allogeneic stimulation can reactivate latent CMV ((22-28) reviewed in (29)). By transplanting allografts and isografts into latently infected recipients, we tested the role of allogeneic stimulation in CMV reactivation.
Methods
Mice
C57Bl/6 (H-2b) and BALB/c (H-2d) mice were obtained from Jackson Labs (Bar Harbor, MN). All mice were housed and treated in accordance with Animal Care Guidelines established by the National Institute of Health and The Ohio State University. Infected mice received 1 × 105 pfu of deletion mutant Δm157 MCMV (kind gift of Ulrich Koszinowski (20)) by intra-peritoneal injection and allowed to become latent (16 weeks) as previously published (30). Following confirmation of latency in a cohort by focused expansion assay mice underwent heterotopic cardiac transplantation. Table I summarizes cohort numbers for each time-point and treatment studied. For latent recipient experiments, latently infected C57BL/6 mice received cardiac isografts (C57BL/6, n=21) or allografts (BALB/c, n=39) and grafts were collected for analysis at day 21, day 45, or > day 100. Additionally as controls MCMV “naïve” C57BL/6 mice received cardiac allografts (BALB/c, n=56) which were collected for analysis at either day 21, day 45, or > day 100. For latent donor experiments, latently infected BALB/c donor hearts were transplanted to MCMV naïve C57BL/6 mice (n=12). Finally, MCMV pre-sensitized (but not infected) C57BL/6 recipients received MCMV-naïve BALB/c allografts 16 weeks after sensitization with 1 × 106 pfu heat killed MCMV (HK-MCMV). HK-MCMV was generated by heating Δm157 MCMV for 30 minutes at 60°C, and lack of infectivity was confirmed by plaque assay (not shown).
Table I.
Numbers of mice in each group
| Days after transplant | |||
|---|---|---|---|
| 21 | 45 | 100 | |
| D-R+ Allograft (n) | 11 | 9 | 19 |
| D-R- Allograft (n) | 9 | 15 | 32 |
| D-R+ Isograft (n) | 6 | 3 | 12 |
| D+R- Allograft (n) | 12 | ||
| D-R- HK-MCMV Allograft (n) | 10 | ||
| D-R-GN- Allograft (n) | 12 | ||
Murine Cardiac Transplantation
Heterotopic cardiac transplantation was performed as described by Corry et al (31). Briefly, native hearts from heparinized donor mice (BALB/c) were anastomosed to recipient BL6 abdominal aorta and vena cava using microsurgical techniques. Graft survival was assessed by bi-weekly trans-abdominal palpation. Gallium nitrate (GN, Ganite, Fujisawa, Deerfield, IL) was administered to all graft recipients as an initial subcutaneous bolus injection of 2.2 mg twenty-four hours prior to graft implantation, followed by 28 days of continuous delivery via subcutaneous osmotic minipumps (model 2002, Alzet Inc., Palo Alto, CA) (3).
PCR, RT-PCR and nested RT-PCR
PCR was performed as previously described (32) using QIAamp Tissue Kit (QIAGEN GmbH, Hilden, Germany) for DNA extraction, Taq DNA polymerase (GIBCO BRL), and a Perkin Elmer 9700 thermocycler (PE Applied Biosystems, Foster City, CA). MCMV mRNA detection was performed as previously described (32) using TRIzol Reagent (GIBCO BRL, Carlsbad CA) for mRNA isolation, Reverse transcription (RT) reactions were performed after DNase I treatment (GIBCO BRL) using U Super-transcriptase (GIBCO BRL). To control for DNA contamination, every sample had a concomitant parallel experiment with no RT reaction. If the first reaction yielded no visible product, a second (nested) PCR was performed using 1μl of this first PCR reaction product. Primers for glycoprotein B (GB) and β-actin were as previously described (32).
For inflammatory mediator mRNA quantitative PCR, RNA was extracted from tissues using TRIzol Reagent (GIBCO BRL, Carlsbad CA), treated with 3 U DNase I Amplification Grade (Invitrogen, Carlsbad, CA) and RT reactions were performed using SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). cDNA were amplified with SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) using Applied Biosystems 7900HT Fast Real-Time PCR System, with GAPDH serving as endogenous controls. Thermal cycling was carried out at 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Relative mediator mRNA were calculated using the 2-ΔΔCT method (33). MCMV Primers for GAPDH, IFN-α, IL-12, IL-10, TGF-β, Foxp3, and IDO were obtained from SuperArray (Frederick, MD).
Transfer DTH responses
Splenocytes isolated from graft recipients were tested using transfer DTH assays between 45 to 110 days post-transplant. For this assay, pinnae of naive B6 mice were injected using a 30-gauge insulin syringe, with 40 microliters containing 8×106 RBC depleted splenocytes from cardiac transplant recipients plus combinations of tetanus toxoid (25 limits of flocculation), heat killed MCMV virus (1×105 PFUs), subcellular donor alloantigens (100 micrograms), neutralizing TGF-β antibodies (25 micrograms) and neutralizing IL-10 antibodies (25 micrograms).Changes in ear thickness were measured both before injection and 24 hours after injection using a dial thickness gauge (Swiss Precision Instruments, Carlstadt, NJ). For reference, changes ranging 0-30 ×10−4 inches represent background swelling due to injection trauma, changes ranging 40-60 ×10−4 inches represent moderate DTH responses, and changes ranging 70-100 ×10−4 inches represent strong DTH responses. Polyclonal rabbit anti-TGF-β, goat anti-IL-10, and antiIgG antibodies were obtained from R&D Systems (Minneapolis, MN). Subcellular H2d alloantigen was prepared according to published methods of Engers et al (34) from BALB/c splenocytes. For DTH challenges, 75-125 micrograms protein of this solution was injected into murine pinnae.
Histological examination of cardiac tissue
Cardiac tissues were excised and fixed in 10% neutral buffered formalin, dehydrated in upgraded ethanol (70%, 95%, and 100%) and embedded in paraffin. For histological analysis, 3 micron sections were mounted on slides and stained with hematoxylin and eosin (H&E), Masson’s trichrome and elastic stains. The histologic slides were evaluated in blinded fashion by our transplant pathologist (TN). Graft histology was evaluated using a scoring system adapted with modifications from Russell et al (35) and the International Society of Heart and Lung Transplantation classification of cardiac allograft rejection (36). All examined histologic parameters were semi-quantitatively scored from 0-3, with 0 representing normal histology. Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue from representative MCMV-positive and negative allografts. Briefly, a double marker technique was used for simultaneous identification of Foxp3 and a surface differentiation molecule CD3. CD3 polyclonal antibody (DAKO, Carpinteria, CA) and an affinity purified rat anti-FOXP3 (clone FJK-16s) antibody (eBoioscience) were used for staining as previously described (37).
Virus recovery
In-vitro plaque assays were performed as previously described using mouse embryo fibroblasts (30). More sensitive assays of infectivity in tissue by focused expansion assay (FEA) were performed using the techniques previously described by Reddehase et al (38). Briefly, this technique is RT-PCR of lysates from spin inoculated plaque assays to detect viral transcription.
Results
Compromised allograft function in CMV positive recipients
We have previously reported that GN induces long term graft survival in ∼97% C57BL/6 mice receiving DBA/2 cardiac allografts (2). In the current study, C57BL6 recipients latently infected with MCMV (D-R+), or MCMV naïve controls (D-R-) received MCMV naïve BALB/c cardiac grafts + GN. We chose BALB/c instead of DBA mice as donors because of our extensive experience with MCMV infection this mouse strain. Figure 1A depicts Kaplan-Meier survival analyses, showing that ∼80% of MCMV latent recipients lost BALB/c cardiac allograft function by 100 days post-transplant. In contrast, significantly fewer (8%) MCMV naïve recipients lost allograft function. To control for effects of GN, latently infected C57BL6 received naïve C57BL6 cardiac isografts + GN treatment, and all cardiac isografts remained functional throughout the experiments (>100 days). Historic control data show that non-infected C57/BL6 recipients of BALB/c hearts that do not receive GN all reject their grafts by day 12 (Figure 1B, D-R-GN-).
Figure 1. Cytomegalovirus and cardiac graft survival.
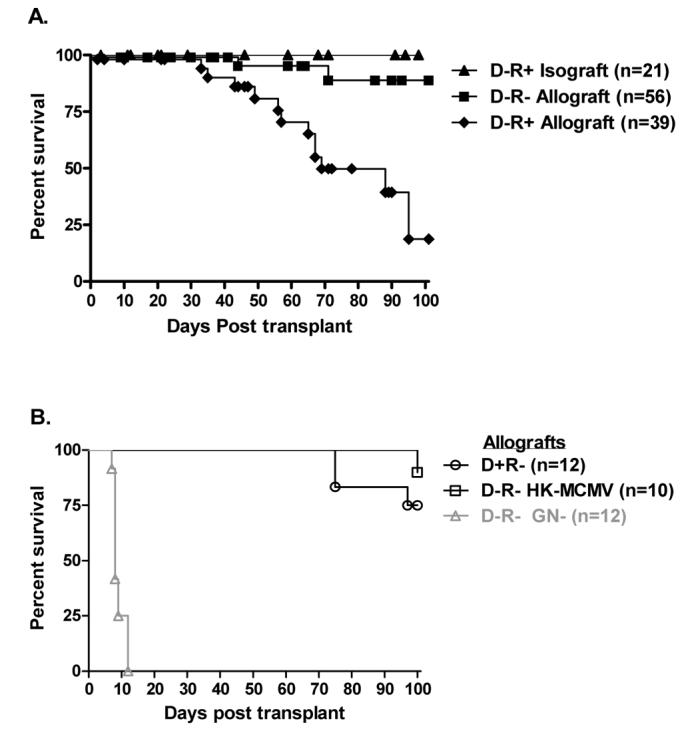
C57BL6 (H2b) mice received BALB/c (H2d) cardiac allografts or C57BL6 isografts. A. Recipients latently infected with murine cytomegalovirus (MCMV) received allografts (◆D-R+ Allograft) or isografts (▲D-R+ Isograft) and were compared with non-infected allograft recipient (■D-R- Allograft) for graft survival. Kaplan—Meier analysis shows that latently infected D-R+ allograft recipients have significantly worse graft survival than non-infected D-R-allograft recipients (21% versus 92%, p<0.0001). All D-R+ cardiac isografts remain functional beyond 100 days post-transplant. B. Latent infection of donor allografts (oD+R-) showed 100 day graft survival comparable to non-infected D-R-allografts (p=0.59). Sensitization (without infection) using heat killed MCMV (□D-R- HK-MCMV) also had no significant influence upon graft survival. Non-infected C57BL/6 mice not treated with gallium nitrate (historical controls) promptly reject their BALB/c grafts (ΔD-R- GN-). Cardiac graft function was assessed by trans-abdominal palpation.
To evaluate for possible induction of heterologous immune responses by MCMV, a cohort of allograft recipients pre-sensitized with heat killed MCMV underwent allograft transplantation. This had no impact upon allograft survival (Figure 1B), despite development of splenocytes reactive with MCMV antigen by DTH (not shown). Similarly, latent MCMV introduced via donor tissue did not influence graft acceptance (Figure 1B). Latently infected BALB/c hearts transplanted into naïve C57BL6 recipients (D+R-) showed comparable 100 day graft acceptance to non-infected D-R- allograft recipients, and significantly better acceptance than latently infected D-R+ recipients (Log-rank (Mantel Cox), p=0.59, p=0.005 respectively). This occurred despite transmission of MCMV to all naïve recipients, and development of MCMV specific splenocyte DTH responses (not shown). Together, these data suggest that introduction of an MCMV naïve graft into a latently infected recipient compromises induction of graft acceptance. Compromised graft acceptance does not appear to be due to MCMV reactive splenocytes, which develop after either pre-sensitization (without infection) or D+R-transplantation.
Histologic features of cardiac allograft damage
Accepted cardiac allografts (MCMV naïve) collected 21 and 45 days after transplant display mild leukocytic infiltration (Figure 2A&D) and scattered mild fibrosis (Figure 2G) consistent with previous results (3). In contrast, cardiac allografts from MCMV-positive recipients show significant graft injury, including infiltration of leukocytes (Figure 2B&E) and significant fibrosis (Figure 2H). This is in significant contrast to the minimal cellular infiltration or fibrosis observed in MCMV-positive isografts (Figure 2C, F, & I). Cardiac allografts from latently infected recipients had significantly worse diffuse inflammation, fibrosis, myocyte necrosis, and intimal arteritis scores (data not shown). An example of allograft arteritis associated with CMV is shown in Figure 2E. Staining for antibody deposition (C3d) showed no significant deposition for MCMV-positive recipients (data not shown). Thus our histologic evaluations are consistent with observed MCMV-associated allograft losses, and suggest a cellular and not humoral mechanism.
Figure 2. Pathologic changes in cardiac allografts of recipients latently infected with murine cytomegalovirus (MCMV).
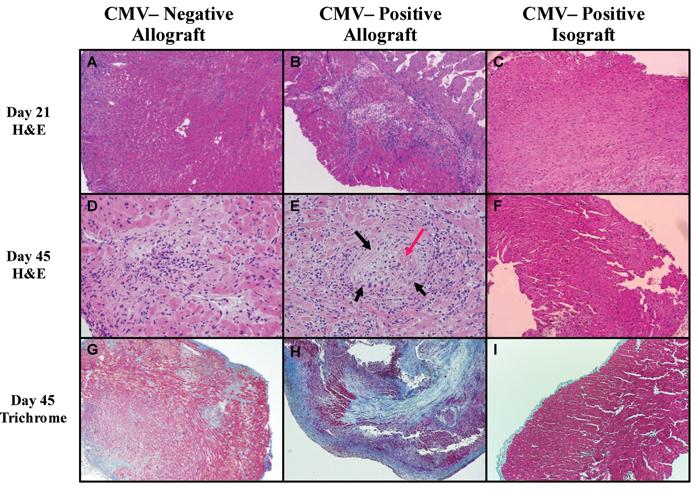
Histology in cardiac allografts (BALB H2d) from MCMV-naïve C57BL/6 recipients compared to allografts and isografts from MCMV-positive C57BL/6 recipients 21 and 45 days post-transplant. A,D&C. Representative H&E (A&D) 21 and 45 days after transplant show typical cellular infiltration associated with this model, and trichrome sections (G) show mild patchy fibrosis in CMV-naïve recipient allografts. B&E. In contrast, CMV-positive recipient allografts show prominent cellular infiltration 21 days after transplant (B), which is more pronounced by day 45 (E) and is associated with vasculitis (black arrow) with fibrin (red arrow) deposition in lumen. H. Day 45 trichrome staining shows advanced fibrosis (blue staining). C,F,G. Isografts from CMV-positive recipients show bland myocardium, with very lile cellular infiltrate at either time point, and nominal fibrosis at day 45. Specimens were formalin fixed and embedded. Magnification = 40X, except for D & E 100X.
Cardiac allograft transplantation causes CMV transcriptional reactivation
It was unknown whether transplantation would result in transmission of MCMV from recipient to graft. It was also unclear what effect GN would have upon MCMV if reactivation occurred. Finally, because previous work with this model suggested an anti-inflammatory state in accepted allografts, it was unclear what effect this would have on viral replication. Therefore, cohorts of latently infected allograft (n=9) and isograft (n=3) hearts were studied 45 days after transplant for evidence of MCMV DNA. This time point was chosen because this was when we first began to note graft losses in our model. Transplanted hearts were removed and evaluated by PCR for MCMV DNA, and all were found to be MCMV DNA positive (not shown). Transplanted hearts were also evaluated using nested RT-PCR for viral RNA and FEA, but no viral RNA (MCMV-GB) was detected at this time point (not shown). Thus, latent virus in recipients is transmitted into naïve grafts after transplantation, but it remained unclear whether this was due to viral reactivation, or simply infiltration by MCMV containing leukocytes.
We have previously reported that pro-inflammatory mediators, such as tumor necrosis factor-α or interleukin-1β, can trigger reactivation of latent CMV 21 days after stimulation (32). Because allogeneic cardiac transplantation is known to cause inflammation, with activation of numerous mediators causing stimulation of the MCMV promoter (28), we postulated that reactivation might be occurring earlier than 45 days. Accordingly, cardiac allografts (n=8) and isografts (n=6) from latently infected recipients procured 21 days post-transplantation were analyzed for active virus by focused expansion assay. MCMV transmission was confirmed in all allograft and isograft recipients by the presence of MCMV viral DNA in graft tissues (Figure 3). Although we were unable to recover live virus by plaque assay, FEA analysis shows that 4/8 latently infected allograft recipients had viral reactivation in their grafts (Figure 3A). In contrast, isografts demonstrate no evidence of MCMV reactivation (0/6) either by FEA (Figure 3B), plaque assay, or RT-PCR (not shown). Viral loads determined using quantitative real-time PCR demonstrated comparable low overall viral DNA in allografts and isografts at day 21 and 45 (not shown), confirming that MCMV was ultimately controlled. Control mice receiving GN alone (no graft) showed no evidence of viral reactivation (0/5, not shown). Thus although not all allografts have detectable MCMV replication, these data support the hypothesis that allogeneic transplant may trigger viral reactivation.
Figure 3. Molecular reactivation of latent murine cytomegalovirus (MCMV) after cardiac transplantation.
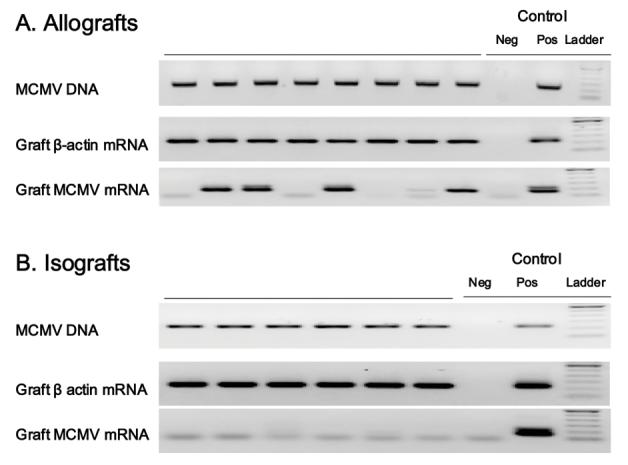
Allograft and isograft recipients latently infected with MCMV were evaluated 21 days post-transplant by focused expansion assay for MCMV glycoprotein-B (GB) mRNA transcription using nested RT-PCR. A. Allografts from latently infected recipients show definitive molecular MCMV reactivation in 4/8. B. Isografts from latently infected mice showed no evidence of molecular reactivation (0/6). Both allografts and isografts contain MCMV DNA confirming prior infection and transmission in all recipients. Each lane represents DNA or mRNA from one graft. Presence of β-actin confirms recovery of RNA and controls are technique controls. No-RT controls performed concurrently with all RT-PCR confirm absence of DNA contamination (not shown).
Disruption of Donor-reactive Immune Regulation
Although the current study utilizes a slightly different combination of BALB/c hearts (H2d) into CMV naïve or latently infected C57BL/6 recipients than previously reported (2, 4, 5), cardiac allograft acceptance in this combination is similarly associated with regulation of donor-reactive delayed type hypersensitivity (DTH) responses via TGF-β and IL-10 (Figure 4A). In addition, allograft acceptors display linked-non-responsiveness to TT when TT is mixed with donor antigen. MCMV naïve recipients manifest nominal DTH responses to MCMV antigen with or without donor alloantigens (Figure 4A).
Figure 4. Changes in immune regulation in cardiac allograft recipients latently infected with murine cytomegalovirus (MCMV).
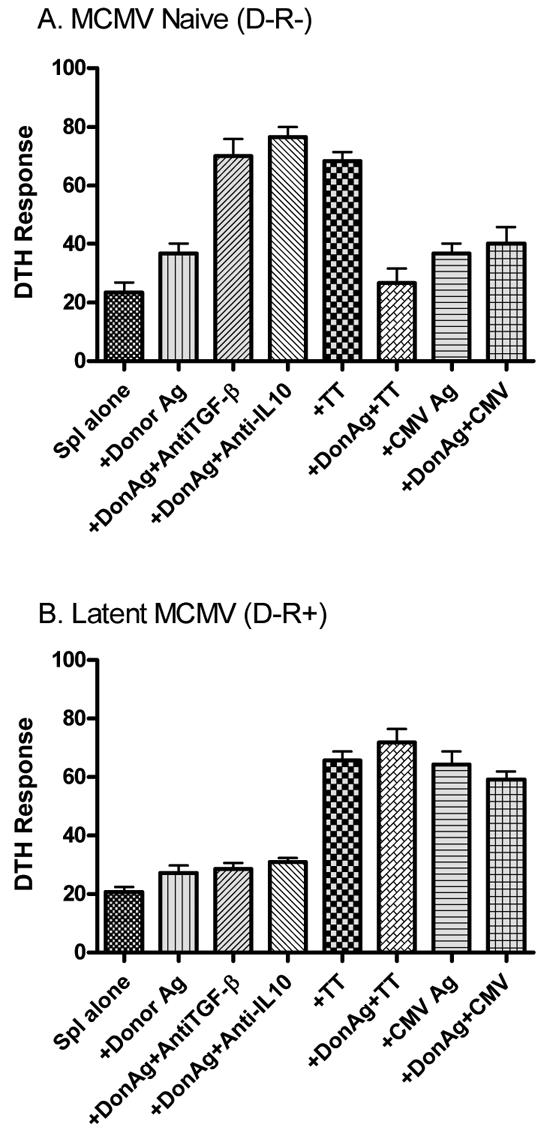
Splenocytes isolated from MCMV-naive (D-R-) and MCMV-positive (D-R+) cardiac allograft recipients between 33 and 110 days post-transplant were evaluated using trans-vivo DTH. Briefly, splenocytes (Spl) obtained from individual mice were combined with sub-cellular donor alloantigen (DonAg) alone or with polyclonal antibodies to TGF-β or IL-10, tetanus toxoid (TT) with/without DonAg, or heat inactivated MCMV with/without DonAg, and injected into pinnae of naïve C57BL/6 mice. All mice were sensitized to TT prior to evaluation. A. As previously published, MCMV-negative allograft acceptors demonstrate regulation of donor-reactive DTH responses that are restored by antibodies to TGF-β or IL-10. Further, immune regulation extends to suppress TT-reactive DTH responses when donor alloantigen is present at the DTH site, so-called “linked non-responsiveness”. As expected MCMV-naïve splenocytes did not show a full response when challenged with MCMV antigen. B. Splenocytes from recipients latently infected with MCMV no longer demonstrate TGF-β or IL-10 regulation, but interestingly remain unresponsive to donor antigen. In addition, there is disruption of linked non-responsiveness, as prominent CMV- and TT-reactive DTH responses are not suppressed when co localized with donor antigen at DTH sites. DTH responses are measured after 24 hours as the change in ear thickness (mean ± SD × 10−4 inches). Each bar represents results from 3-6 mice (D-R-) or 5 mice (D-R+).
Splenocyte DTH responses in latently infected allograft recipients were first performed around the time that cardiac allograft losses began to occur (∼day 45), and interestingly, there was negligible DTH response to donor antigen (Figure 4B). This hyporesponsiveness persisted despite disruption of TGF-β and IL-10-mediated DTH regulation and loss of linked donor DTH non-responsiveness (Figure 4B). This hyporesponsiveness was not general anergy, as latently infected recipients responded strongly to both MCMV and TT antigen (Figure 4B). To determine whether we were measuring for allo-responses too early, these DTH experiments were repeated at day >100 in a D-R+ cohort that had previously lost their grafts around day 60 after transplant. Surprisingly, this cohort showed identical results to the day 45 cohort, with splenocytes remaining hypo-responsive to donor antigen by DTH, despite absence of TGF-β and IL-10 regulation, and disruption of linked non-responsiveness (data not shown). Thus allograft losses in our model occur without demonstrable anti-donor reactive cells in spleens of recipients.
Intra-graft mediator biology
Lack of demonstrable recipient splenocyte responses to alloantigen, despite disruption of regulation suggested to us that allograft acceptance is being disrupted locally in grafts. To test for local disruption of allograft acceptance by donor-specific T-cells, we attempted to isolate graft infiltrating cells (GIC) to perform DTH testing. Despite exhaustive attempts, we were unable to isolate enough GICs from day 45-50 allografts to perform such testing. As a surrogate to GIC DTH testing, day 21 allograft or isograft recipients latently infected with MCMV and non-infected allograft recipients were tested for regulatory molecules TGF-β, IL-10, Foxp3, and IDO mRNA expression using quantitative RT-PCR. TGF-β and IL-10 were chosen for obvious reasons, and Foxp3 and IDO were chosen because we have recently shown that both might be critical to spontaneous renal allograft acceptance (37). Alternatively, allograft losses seen in MCMV-positive recipients might be consequent to an inflammatory signal induced by MCMV, and because acute MCMV infection induces IFN-α and IL-12 (39), these inflammatory cytokines were also evaluated.
As shown in Figure 5, allografts show significantly higher expression of regulatory genes Foxp3, IDO, and TGF-β compared to isografts independent of MCMV. Importantly, recipient MCMV does not appear to negatively influence graft expression of regulatory genes Foxp3, IDO, IL-10, or TGF-β, as CMV-positive allografts have significantly higher transcript levels than CMV-negative allografts. In contrast, MCMV does appear to influence inflammatory mediator expression, as CMV-positive allografts show significantly increased IFN-α mRNA compared to CMV-negative allografts. Expression of IL-12 approaches but does not reach statistical significance. In addition mRNA analyses performed on day 21 allograft and isograft recipient spleens (all CMV-positive n=3 each) showed no significant differences for any of these mediators (data not shown). Collectively, these data suggest that MCMV—positive allograft recipients display local intra-graft inflammatory and anti-inflammatory immune responses that are not observed in either MCMV-negative allografts or in MCMV-positive isografts.
Figure 5. Influence of murine cytomegalovirus (MCMV) on regulatory and inflammatory mediator mRNA in cardiac grafts.
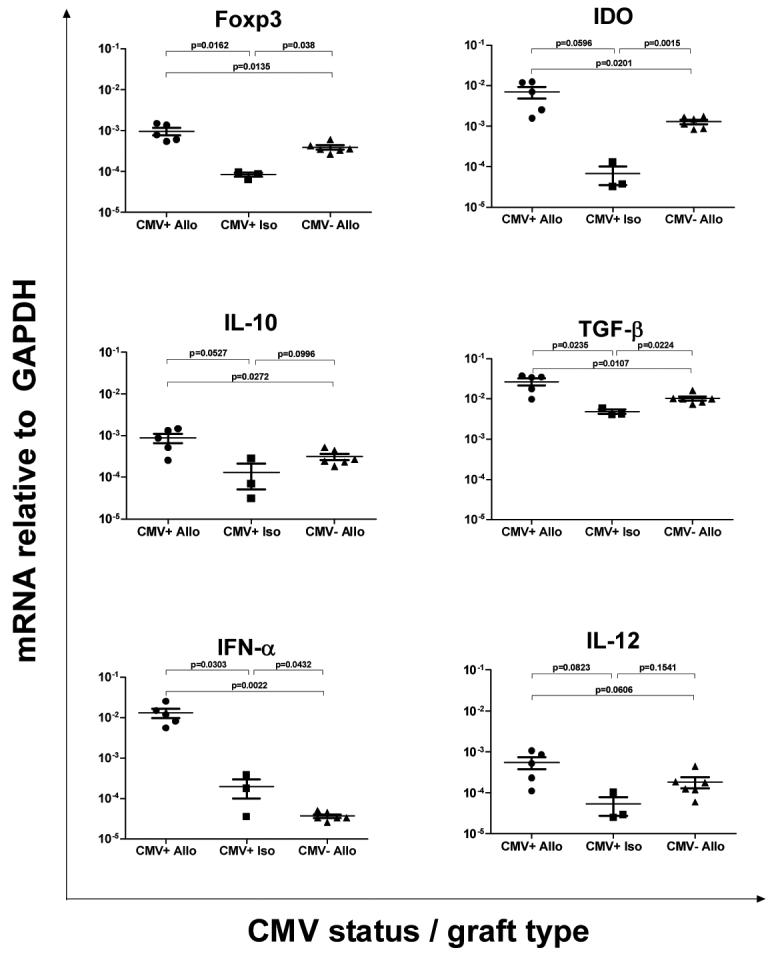
Real-time RT-PCR was performed on heterotopic cardiac grafts 21 days after transplantation for regulatory genes Foxp3, IDO, IL-10, and TGF-β, or inflammatory genes IFN-α and IL-12. Groups were allograft or isograft recipients latently infected with MCMV (CMV+ Allo or CMV+ Iso respectively) and non-infected allograft recipients (CMV- Allo). Allografts show significantly higher expression of regulatory genes Foxp3, IDO, and TGF-β compared to isografts independent of MCMV. Recipient MCMV does not appear to negatively influence graft expression of regulatory genes Foxp3, IDO, IL-10, or TGF-β, as CMV+ allografts have significantly higher transcript levels than CMV- allografts. In contrast, MCMV does appear to influence inflammatory mediator expression, as CMV+ allografts show significantly increased IFN-α mRNA compared to CMV- allografts. Each point represents mean ± standard error mRNA frequency (mRNA relative to GAPDH) from n=1 mouse (performed in duplicate).
Intragraft regulatory T-cells
Our previous work has suggested a role for regulatory T-cells in this model of cardiac allograft acceptance (4, 5, 40). To determine if MCMV was influencing development or persistence of T-regulatory cells within grafts, sections of allografts from CMV-positive and negative recipients were evaluated by immunohistochemistry for Foxp3+ cells. As shown in Figure 6, there are Foxp3+ CD3 cells in both CMV-positive and CMV-negative grafts in a similar scattered distribution (A&B). Foxp3+ CD3 cells persist in allografts at least until day 45 after transplant, and actually appear somewhat more numerous in CMV-positive allografts at this time point (Figure 6D&E). Taken together, these data confirm the inflammatory mediator data, suggesting that allograft losses observed in MCMV-positive recipients is not consequent to loss of T-regulatory cells in these grafts.
Figure 6. Foxp3 staining of cardiac allografts.

Immunohistochemical staining was performed on representative sections from CMV-positive and negative allografts 21 and 45 days after transplantation. A&D. Sections from CMV-negative allografts show numerous CD3+ cells (brown) with scattered Foxp3+ cells (blue). B&E. CMV-positive allografts show a similar staining pattern at day 21, but have more numerous Foxp3+ cells at day 45 after transplant. C&F. 40X views of CMV-positive grafts, clearly showing persistence of Foxp3+ T-cells (blue).
Discussion
This study demonstrates that presence of MCMV in recipients clearly impairs allograft acceptance after MHC-mismatched heterotopic cardiac transplantation. It is not entirely surprising that CMV influences graft acceptance in our model, given the well known relationship between rejection and CMV reactivation in patients (41-43). Since Lopez et al first suggested that reactivation of CMV might trigger graft rejection (44), it has become clearer clinically that CMV reactivation might have a causal influence, as prophylaxis of CMV seems to reduce the incidence of acute rejection (45).
The obvious question is what causes allograft losses in recipients latently infected with CMV in our model? Murine models of allograft tolerance are associated with development of metastable regulatory mechanisms that might be quite sensitive to disruptive forces (4, 5). Possible disruptive forces include non-donor-antigen-specific antiviral immune responses following viral reactivation, which could alter either the inflammatory or regulatory milieu within transplanted grafts. Alternatively, donor-antigen-specific responses might develop either from MCMV infection (heterologous immunity) or following loss of regulation induced by MCMV.
Perhaps the simplest explanation for disruption of allograft acceptance in our model is that MCMV reactivated by allogeneic stimulation acutely infects naive allografts, with subsequent local antiviral immune responses causing graft losses, and the preponderance of our data support this hypothesis. We have recently shown using a comparable MHC mismatched skin model (H2b donor, H2d recipient) that allografts, but not isografts, will reactivate latent MCMV 21 days after grafting (MF Forster, AA Bickerstaff, CH Cook, manuscript in preparation). In the current study, viral DNA are detectable in all grafts from latently infected recipients (Figure 3 and not shown), but MCMV is harbored in leukocytes (46), and could simply travel as a passenger during leukocytic infiltration (Figure 2). Focused expansion assay of infectivity is considered the most sensitive method to detect viral activity (38), and using this method reactivation was detected in 4/8 allografts from D-R+ allograft recipients 21 days after transplantation (Figure 3). It is possible that reactivation occurs slightly before or after this 21 day time point and is being missed, or that inherently low cardiac titers coupled with active antiviral immune responses exceed our abilities of detection. Host immune response could be the most sensitive indicator of viral activity (47), thus the most compelling support of reactivation is the association of elevated IFN-α mRNA levels with CMV-positive allografts (Figure 5). Acute MCMV infection is known to cause type-I interferon release, and interestingly this response is mediated by plasmacytoid dendritic cells (39, 48-50), which have also been implicated in various models of tolerance (51-53). Thus although not demonstrated with absolute certainty, we feel that data from our skin model taken together with data reported here support the hypothesis that reactivated virus acutely infects naïve allografts causing type-I interferon expression.
Not only does IFN-α expression support the hypothesis of MCMV reactivation from latency, but it also suggests a possible mechanism of graft loss. Exogenous type I interferons have been linked with clinical allograft rejection (54-57), accelerated cardiac rejection in rodents (58), and recently disruption of tolerance and graft acceptance in several different models (59-61). If MCMV is reactivating and acutely infecting naïve allografts, it becomes similar to other acute viral infections that induce IFN-α and disrupt allograft acceptance, such as lymphocytic choriomeningitis virus and Pichinde virus (6-8, 62-64). IFN-α directly stimulates murine NK-cells to expand, become cytotoxic, and release both TNF-α and IFN-γ (49, 65, 66), as well as non-specifically stimulate bystander T cells (67). In addition, virally activated NK-cells can lyse allogeneic cells (68, 69) as well as disrupt T-regulatory cell function (70). To more conclusively demonstrate that induced IFN-α is the mechanism which causes allograft losses in our model, we considered using IFN receptor knockout mice as recipients, but unfortunately, MCMV infection is lethal in these mice precluding such experiments (71). Thus although not definitively proven, our data and those of others are certainly consistent with the hypothesis that type I interferon induction by MCMV might contribute to our observed graft losses.
Loss of DTH regulation and linked non-responsiveness, with concomitant eventual viral control in MCMV-positive allograft acceptors suggests that in this model, anti-viral responses trump or subvert the “tolerogenic” forces that would normally lead to allograft acceptance. Surprisingly, despite loss of regulation, allograft recipient splenocytes remain unresponsive to donor antigen even months after graft loss. Lack of systemic donor antigen reactivity suggests to us that allograft acceptance is disrupted locally within grafts, either specifically or non-specifically. DTH studies using graft infiltrating cells might have answered the specificity question, but these studies were technically not possible to perform (see Results). Up-regulation of intragraft “tolerogenic” mediators, including Foxp3 and IDO in CMV-positive recipients suggests that MCMV does not disrupt intragraft accumulation of regulatory T-cells, and this was confirmed by immunohistochemistry. It is also important to note that when MCMV is introduced with the graft (D+R-), there is no significant influence on graft acceptance, despite viral transmission to the recipient. Taken together, these data suggest that introduction of CMV into naïve allografts tips the balance of local tolerogenic forces toward inflammation, causing non-donor specific graft loss. Encouragingly, the non-TGF-β/IL-10 mechanism mediating splenocyte DTH hyporesponsiveness to donor antigen in the current report appears to be fairly robust, persisting despite graft loss. This suggests that if CMV reactivation or the IFN-α response to it could be controlled, that induced allograft acceptance might be restored.
Virus induced heterologous immunity has been shown in numerous investigations to have significant influences upon graft acceptance and tolerance induction, although MCMV remains relatively unstudied. Importantly, MCMV infection has been shown to induce T cells reactive with H2d antigen (72), which is relevant to our model that utilizes H2d donor to H2b recipients previously infected with MCMV. While pre-sensitization with heat killed virus might not completely replicate the immune response to actual infection, HK-MCMV recipient sensitization did induce splenocytes reactive to MCMV. Nonetheless, these cells appear insufficient to cause allograft loss in our model (Figure 1B). Similarly recipient sensitization to MCMV following D+R- allograft transplantation did not influence graft acceptance. Recent work by others has suggested that induction of TCR-Vβ4 CD8 T-cells can influence tolerance induction (73). Although we did not specifically test for H2d reactive TCR-Vβ4 CD8 T-cells, these cells have not been strongly associated with MCMV infection (74-76), and more importantly there was no functional evidence of these cells in our splenocyte DTH testing (Figure 4B). Thus it does not appear that MCMV induced donor specific heterologous immune responses contribute to disruption of graft acceptance in our model.
In conclusion, it is clear that prior CMV infection in cardiac allograft recipients has significant impact on induction of allograft acceptance. Our data support the hypothesis of latent CMV reactivation by allogeneic stimulation, with subsequent infection of naïve grafts causing non-donor-specific immune responses and graft loss. Independent of whether CMV is reactivated, previous recipient CMV infection is associated with an intragraft type-I interferon response that has been shown by others to cause graft loss. Because CMV is so prevalent, at first blush these data seem discouraging to potential development of tolerance strategies in patients. Nonetheless, it is exciting that in our model splenocyte DTH assays demonstrate unresponsiveness to donor antigen despite disruption of regulation. Together these findings are consistent with graft losses occurring consequent to local anti-viral immune responses taking out already vulnerable grafts as “innocent bystanders”. Given the prevalence of CMV in the population, we suspect that tolerance induction strategies will need to directly address influences of CMV or the associated type-I interferon antiviral responses to achieve success.
Acknowledgments
The authors would like to thank Kelly Nye, Christina Sass, Jonathan Campbell, and Bree Heestand for their technical assistance with these studies. We also thank Patricia Della Pelle for her assistance with the immunohistochemistry.
Supported by NIH RO1 AI053094 (CHC) and a grant from the Roche Organ Transplant Foundation (RBC)
References
- 1.Orosz CG, Wakely E, Sedmak DD, Bergese SD, VanBuskirk AM. Prolonged murine cardiac allograft acceptance: characteristics of persistent active alloimmunity after treatment with gallium nitrate versus anti-CD4 monoclonal antibody. Transplantation. 1997;63(8):1109–1117. doi: 10.1097/00007890-199704270-00010. [DOI] [PubMed] [Google Scholar]
- 2.VanBuskirk AM, Wakely ME, Sirak JH, Orosz CG. Patterns of allosensitization in allograft recipients: long-term cardiac allograft acceptance is associated with active alloantibody production in conjunction with active inhibition of alloreactive delayed-type hypersensitivity. Transplantation. 1998;65(8):1115–1123. doi: 10.1097/00007890-199804270-00017. [DOI] [PubMed] [Google Scholar]
- 3.Orosz CG, Wakely E, Bergese SD, VanBuskirk AM, Ferguson RM, Mullet D, et al. Prevention of Murine Cardiac Allograft Rejection with Gallium Nitrate: Comparison with Anti-CD4 Monoclonal Antibody 1. Transplantation. 1996;61(5):783–791. doi: 10.1097/00007890-199603150-00019. [DOI] [PubMed] [Google Scholar]
- 4.Bickerstaff AA, VanBuskirk AM, Wakely E, Orosz CG. Transforming growth factor-beta and interleukin-10 subvert alloreactive delayed type hypersensitivity in cardiac allograft acceptor mice. Transplantation. 2000;69(7):1517–1520. doi: 10.1097/00007890-200004150-00055. [DOI] [PubMed] [Google Scholar]
- 5.Bickerstaff AA, Xia D, Pelletier RP, Orosz CG. Mechanisms of graft acceptance: evidence that plasminogen activator controls donor-reactive delayed-type hypersensitivity responses in cardiac allograft acceptor mice. J Immunol. 2000;164(10):5132–5139. doi: 10.4049/jimmunol.164.10.5132. [DOI] [PubMed] [Google Scholar]
- 6.Williams MA, Tan JT, Adams AB, Durham MM, Shirasugi N, Whitmire JK, et al. Characterization of Virus-Mediated Inhibition of Mixed Chimerism and Allospecific Tolerance. J Immunol. 2001;167(9):4987–4995. doi: 10.4049/jimmunol.167.9.4987. [DOI] [PubMed] [Google Scholar]
- 7.Welsh RM, Markees TG, Woda BA, Daniels KA, Brehm MA, Mordes JP, et al. Virus-Induced Abrogation of Transplantation Tolerance Induced by Donor-Specific Transfusion and Anti-CD154 Antibody. J Virol. 2000;74(5):2210–2218. doi: 10.1128/jvi.74.5.2210-2218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehl S, Hombach J, Aichele P, Rulicke T, Odermatt B, Hengartner H, et al. Viral and Bacterial Infections Interfere with Peripheral Tolerance Induction and Activate CD8+ T Cells to Cause Immunopathology. J Exp Med. 1998;187(5):763–774. doi: 10.1084/jem.187.5.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams MA, Onami TM, Adams AB, Durham MM, Pearson TC, Ahmed R, et al. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade-based regimen. J Immunol. 2002;169(10):5387–5391. doi: 10.4049/jimmunol.169.10.5387. [DOI] [PubMed] [Google Scholar]
- 10.Streblow DN, Orloff SL, Nelson JA. Acceleration of allograft failure by cytomegalovirus. Curr Opin Immunol. 2007;19(5):577–582. doi: 10.1016/j.coi.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemstrom K, Koskinen P, Krogerus L, Daemen M, Bruggeman C, Hayry P. Cytomegalovirus Antigen Expression, Endothelial Cell Proliferation, and Intimal Thickening in Rat Cardiac Allografts After Cytomegalovirus Infection. Circulation. 1995;92(9):2594–2604. doi: 10.1161/01.cir.92.9.2594. [DOI] [PubMed] [Google Scholar]
- 12.Lemstrom K, Sihvola R, Bruggeman C, Hayry P, Koskinen P. Cytomegalovirus infection-enhanced cardiac allograft vasculopathy is abolished by DHPG prophylaxis in the rat. Circulation. 1997;95(12):2614–2616. doi: 10.1161/01.cir.95.12.2614. [DOI] [PubMed] [Google Scholar]
- 13.Orloff SL, Streblow DN, Soderberg-Naucler C, Yin Q, Kreklywich C, Corless CL, et al. Elimination of donor-specific alloreactivity prevents cytomegalovirus-accelerated chronic rejection in rat small bowel and heart transplants. Transplantation. 2002;73(5):679–688. doi: 10.1097/00007890-200203150-00005. [DOI] [PubMed] [Google Scholar]
- 14.Streblow DN, Kreklywich C, Yin Q, De La Melena VT, Corless CL, Smith PA, et al. Cytomegalovirus-Mediated Upregulation of Chemokine Expression Correlates with the Acceleration of Chronic Rejection in Rat Heart Transplants. J Virol. 2003;77(3):2182–2194. doi: 10.1128/JVI.77.3.2182-2194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlquist JF, Shelby J, Shao YL, Greenwood JH, Hammond ME, Anderson JL. Accelerated rejection of murine cardiac allografts by murine cytomegalovirus-infected recipients. Lack of haplotype specificity. J Clin Invest. 1993;91(6):2602–2608. doi: 10.1172/JCI116499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao YL, Shelby J, Hisatake G, Kern ER, Nelson EW, Gay WA.Accelerated cardiac allograft rejection in murine cytomegalovirus-infected C3H recipients Transplant Proc 199123(1 Pt 1) 129–130. [PubMed] [Google Scholar]
- 17.Allan JE, Shellam GR. Genetic control of murine cytomegalovirus infection: virus titres in resistant and susceptible strains of mice. Arch Virol. 1984;81(12):139–150. doi: 10.1007/BF01309303. [DOI] [PubMed] [Google Scholar]
- 18.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct Recognition of Cytomegalovirus by Activating and Inhibitory NK Cell Receptors. Science. 2002;296(5571):1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 19.Brown MG, Dokun AO, Heusel JW, Smith HRC, Beckman DL, Blattenberger EA, et al. Vital Involvement of a Natural Killer Cell Activation Receptor in Resistance to Viral Infection. Science. 2001;292(5518):934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- 20.Bubic I, Wagner M, Krmpoti A, Saulig T, Kim S, Yokoyama WM, et al. Gain of Virulence Caused by Loss of a Gene in Murine Cytomegalovirus. J Virol. 2004;78(14):7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lang DJ. Cytomegalovirus infections in organ transplantation and post transfusion. An hypothesis. Archiv fur die Gesamte Virusforschung. 1972;37(4):365–377. doi: 10.1007/BF01241460. [DOI] [PubMed] [Google Scholar]
- 22.Olding LB, Kingsbury DT, Oldstone MB. Pathogenesis of cytomegalovirus infection. Distribution of viral products, immune complexes and autoimmunity during latent murine infection. J Gen Virol. 1976;33(2):267–280. doi: 10.1099/0022-1317-33-2-267. [DOI] [PubMed] [Google Scholar]
- 23.Olding LB, Jensen FC, Oldstone MB. Pathogenesis of of cytomegalovirus infection. I. Activation of virus from bone marrow-derived lymphocytes by in vitro allogenic reaction. J Exp Med. 1975;141(3):561–572. doi: 10.1084/jem.141.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soderberg-Naucler C, Fish KN, Nelson JA. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell. 1997;91(1):119–126. doi: 10.1016/s0092-8674(01)80014-3. [DOI] [PubMed] [Google Scholar]
- 25.Guedes MIMC, Risdahl JM, Wiseman B, Molitor TW. Reactivation of Porcine Cytomegalovirus through Allogeneic Stimulation. J Clin Microbiol. 2004;42(4):1756–1758. doi: 10.1128/JCM.42.4.1756-1758.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheung KS, Lang DJ. Transmission and activation of cytomegalovirus with blood transfusion: a mouse model. J Infect Dis. 1977;135(5):841–845. doi: 10.1093/infdis/135.5.841. [DOI] [PubMed] [Google Scholar]
- 27.Schmader KE, Rahija R, Porter KR, Daley G, Hamilton JD. Aging and reactivation of latent murine cytomegalovirus. J Infect Dis. 1992;166(6):1403–1407. doi: 10.1093/infdis/166.6.1403. [DOI] [PubMed] [Google Scholar]
- 28.Hummel M, Zhang Z, Yan S, DePlaen I, Golia P, Varghese T, et al. Allogeneic transplantation induces expression of cytomegalovirus immediate-early genes in vivo: a model for reactivation from latency. J Virol. 2001;75(10):4814–4822. doi: 10.1128/JVI.75.10.4814-4822.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hummel M, Abecassis MM. A model for reactivation of CMV from latency. J Clin Virol. 2002;25(Suppl 2):S123–136. doi: 10.1016/s1386-6532(02)00088-4. [DOI] [PubMed] [Google Scholar]
- 30.Cook C, Zhang X, McGuinness B, Lahm M, Sedmak D, Ferguson R. Intra-abdominal Bacterial Infection Reactivates Latent Pulmonary Cytomegalovirus in Immunocompetent Mice. J Infect Dis. 2002;185:1395–1400. doi: 10.1086/340508. [DOI] [PubMed] [Google Scholar]
- 31.Corry RJ, Winn HJ, Russell PS. Heart transplantation in congenic strains of mice. Transplant Proc. 1973;5(1):733–735. [PubMed] [Google Scholar]
- 32.Cook CH, Trgovcich J, Zimmerman PD, Zhang Y, Sedmak DD. Lipopolysaccharide, Tumor Necrosis Factor Alpha, or Interleukin-1{beta} Triggers Reactivation of Latent Cytomegalovirus in Immunocompetent Mice. J Virol. 2006;80(18):9151–9158. doi: 10.1128/JVI.00216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-deltadeltaCt Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Engers HD, Thomas K, Cerottini JC, Brunner KT. Generation of cytotoxic T lymphocytes in vitro. V. Response of normal and immune spleen cells to subcellular alloantigens. J Immunol. 1975;115(2):356–360. [PubMed] [Google Scholar]
- 35.Russell PS, Chase CM, Winn HJ, Colvin RB. Coronary atherosclerosis in transplanted mouse hearts. II. Importance of humoral immunity. J Immunol. 1994;152(10):5135–5141. [PubMed] [Google Scholar]
- 36.Billingham ME, Cary NR, Hammond ME, Kemnitz J, Marboe C, McCallister HA, et al. A working formulation for the standardization of nomenclature in the diagnosis of heart and lung rejection: Heart Rejection Study Group. The International Society for Heart Transplantation. J Heart Transplant. 1990;9(6):587–593. [PubMed] [Google Scholar]
- 37.Cook CH, Bickerstaff AA, Wang J-J, Nadasdy T, Della Pelle P, Colvin RB, et al. Spontaneous Renal Allograft Acceptance Associated with “Regulatory” Dendritic Cells and IDO. J Immunol. 2008;180(5):3103–3112. doi: 10.4049/jimmunol.180.5.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurz SK, Rapp M, Steffens HP, Grzimek NK, Schmalz S, Reddehase MJ. Focal transcriptional activity of murine cytomegalovirus during latency in the lungs. J Virol. 1999;73(1):482–494. doi: 10.1128/jvi.73.1.482-494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, et al. Interferon {alpha}/{beta} and Interleukin 12 Responses to Viral Infections: Pathways Regulating Dendritic Cell Cytokine Expression In Vivo. J Exp Med. 2002;195(4):517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orosz CG, Bickerstaff AA, Adams P, Hennessey P, Pelletier RP. Evidence that a similar range of alloimmune responses can develop in murine and human allograft recipients. Transplant Immunology. 2002;9(24):143–147. doi: 10.1016/s0966-3274(02)00035-7. [DOI] [PubMed] [Google Scholar]
- 41.Rubin RH. Impact of cytomegalovirus infection on organ transplant recipients. Rev Infect Dis. 1990;12(Suppl 7):S754–766. doi: 10.1093/clinids/12.supplement_7.s754. [DOI] [PubMed] [Google Scholar]
- 42.Sagedal S, Nordal KP, Hartmann A, Sund S, Scott H, Degre M, et al. The Impact of Cytomegalovirus Infection and Disease on Rejection Episodes in Renal Allograft Recipients. American Journal of Transplantation. 2002;2(9):850–856. doi: 10.1034/j.1600-6143.2002.20907.x. [DOI] [PubMed] [Google Scholar]
- 43.McLaughlin K, Wu C, Fick G, Muirhead N, Hollomby D, Jevnikar A. Cytomegalovirus seromismatching increases the risk of acute renal allograft rejection. Transplantation. 2002;74(6):813–816. doi: 10.1097/00007890-200209270-00014. [DOI] [PubMed] [Google Scholar]
- 44.Lopez C, Simmons RL, Mauer SM, Najarian JS, Good RA. Association of renal allograft rejection with virus infections. The American Journal of Medicine. 1974;56(3):280–289. doi: 10.1016/0002-9343(74)90609-3. [DOI] [PubMed] [Google Scholar]
- 45.Potena L, Holweg CT, Chin C, Luikart H, Weisshaar D, Narasimhan B, et al. Acute rejection and cardiac allograft vascular disease is reduced by suppression of subclinical cytomegalovirus infection. Transplantation. 2006;82(3):398–405. doi: 10.1097/01.tp.0000229039.87735.76. [DOI] [PubMed] [Google Scholar]
- 46.Mitchell BM, Leung A, Stevens JG. Murine cytomegalovirus DNA in peripheral blood of latently infected mice is detectable only in monocytes and polymorphonuclear leukocytes. Virology. 1996;223(1):198–207. doi: 10.1006/viro.1996.0468. [DOI] [PubMed] [Google Scholar]
- 47.von Muller L, Klemm A, Durmus N, Weiss M, Suger-Wiedeck H, Schneider M, et al. Cellular Immunity and Active Human Cytomegalovirus Infection in Patients with Septic Shock. J Infect Dis. 2007;196(9):1288–1295. doi: 10.1086/522429. [DOI] [PubMed] [Google Scholar]
- 48.Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2(12):1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 49.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, et al. Dendritic Cell Responses to Early Murine Cytomegalovirus Infection: Subset Functional Specialization and Differential Regulation by Interferon {alpha}/{beta} J Exp Med. 2003;197(7):885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zucchini N, Bessou G, Robbins SH, Chasson L, Raper A, Crocker PR, et al. Individual plasmacytoid dendritic cells are major contributors to the production of multiple innate cytokines in an organ-specific manner during viral infection. Int Immunol. 2008;20(1):45–56. doi: 10.1093/intimm/dxm119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts.[see comment]. Nature Immunology 200676652–662. [DOI] [PubMed] [Google Scholar]
- 52.Sharma MD, Baban B, Chandler P, Hou D-Y, Singh N, Yagita H, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007:JCI31911. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abe M, Wang Z, de Creus A, Thomson AW. Plasmacytoid Dendritic Cell Precursors Induce Allogeneic T-Cell Hyporesponsiveness and Prolong Heart Graft Survival. American Journal of Transplantation. 2005;5(8):1808–1819. doi: 10.1111/j.1600-6143.2005.00954.x. [DOI] [PubMed] [Google Scholar]
- 54.Kramer P, ten Kate FW, Bijnen AB, Jeekel J, Weimar W. Recombinant leucocyte interferon A induces steroid-resistant acute vascular rejection episodes in renal transplant recipients. Lancet. 1984;1(8384):989–990. doi: 10.1016/s0140-6736(84)92327-4. [DOI] [PubMed] [Google Scholar]
- 55.Kovarik J, Mayer G, Pohanka E, Schwarz M, Traindl O, Graf H, et al. Adverse effect of low-dose prophylactic human recombinant leukocyte interferon-alpha treatment in renal transplant recipients. Cytomegalovirus infection prophylaxis leading to an increased incidence of irreversible rejections. Transplantation. 1988;45(2):402–405. doi: 10.1097/00007890-198802000-00031. [DOI] [PubMed] [Google Scholar]
- 56.Takahara S, Kakimoto K, Kokado Y, Kameoka H, Ishibashi M, Kawada S, et al. Interferon alpha therapy for chronic active hepatitis type C after renal transplantation and allograft rejection. Int Urol Nephrol. 1995;27(4):479–485. doi: 10.1007/BF02550087. [DOI] [PubMed] [Google Scholar]
- 57.Rostaing L, Izopet J, Baron E, Duffaut M, Puel J, Durand D. Treatment of chronic hepatitis C with recombinant interferon alpha in kidney transplant recipients. Transplantation. 1995;59(10):1426–1431. doi: 10.1097/00007890-199505270-00012. [DOI] [PubMed] [Google Scholar]
- 58.Slater AD, Klein JB, Sonnenfeld G, Ogden LL, 2nd, Gray LA., Jr The effects of interferon-alpha/beta in a model of rat heart transplantation. J Heart Lung Transplant. 1992;11(5):975–978. [PubMed] [Google Scholar]
- 59.Wang T, Chen L, Ahmed E, Ma L, Yin D, Zhou P, et al. Prevention of Allograft Tolerance by Bacterial Infection with Listeria monocytogenes. J Immunol. 2008;180(9):5991–5999. doi: 10.4049/jimmunol.180.9.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thornley TB, Brehm MA, Markees TG, Shultz LD, Mordes JP, Welsh RM, et al. TLR Agonists Abrogate Costimulation Blockade-Induced Prolongation of Skin Allografts. J Immunol. 2006;176(3):1561–1570. doi: 10.4049/jimmunol.176.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thornley TB, Phillips NE, Beaudette-Zlatanova BC, Markees TG, Bahl K, Brehm MA, et al. Type 1 IFN Mediates Cross-Talk between Innate and Adaptive Immunity That Abrogates Transplantation Tolerance. J Immunol. 2007;179(10):6620–6629. doi: 10.4049/jimmunol.179.10.6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Merigan TC, Oldstone MBA, Welsh RM. Interferon production during lymphocytic choriomeningitis virus infection of nude and normal mice. Nature. 1977;268(5615):67–68. doi: 10.1038/268067a0. [DOI] [PubMed] [Google Scholar]
- 63.Ou R, Zhou S, Huang L, Moskophidis D. Critical Role for Alpha/Beta and Gamma Interferons in Persistence of Lymphocytic Choriomeningitis Virus by Clonal Exhaustion of Cytotoxic T Cells. J Virol. 2001;75(18):8407–8423. doi: 10.1128/JVI.75.18.8407-8423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Molomut N, Padnos M, Papperman TW, Pevear DC, Pfau CJ. Immune recognition of tumor cells in mice infected with Pichinde virus. Cancer Immunology, Immunotherapy. 1984;17(1):56–61. doi: 10.1007/BF00205498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei X-q, Liew FY, et al. Coordinated and Distinct Roles for IFN-{alpha}{beta}, IL-12, and IL-15 Regulation of NK Cell Responses to Viral Infection. J Immunol. 2002;169(8):4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- 66.Orange J, Biron C. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J Immunol. 1996;156(3):1138–1142. [PubMed] [Google Scholar]
- 67.Tough DF, Borrow P, Sprent J. Induction of Bystander T Cell Proliferation by Viruses and Type I Interferon in Vivo. Science. 1996;272(5270):1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 68.Welsh JRM. Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J Exp Med. 1978;148(1):163–181. doi: 10.1084/jem.148.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct Visualization of Cross-Reactive Effector and Memory Allo-Specific CD8 T Cells Generated in Response to Viral Infections. J Immunol. 2003;170(8):4077–4086. doi: 10.4049/jimmunol.170.8.4077. [DOI] [PubMed] [Google Scholar]
- 70.Sahasrabudhe DM. Inhibition of suppressor T lymphocytes by murine interferon beta. J Exp Med. 1987;166(5):1573–1578. doi: 10.1084/jem.166.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salazar-Mather TP, Lewis CA, Biron CA. Type I interferons regulate inflammatory cell trafficking and macrophage inflammatory protein 1alpha delivery to the liver. J Clin Invest. 2002;110(3):321–330. doi: 10.1172/JCI15376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang HY, Dundon PL, Nahill SR, Welsh RM. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. 1989;142(5):1710–1718. [PubMed] [Google Scholar]
- 73.Stapler D, Lee ED, Selvaraj SA, Evans AG, Kean LS, Speck SH, et al. Expansion of Effector Memory TCR V{beta}4+CD8+ T Cells Is Associated with Latent Infection-Mediated Resistance to Transplantation Tolerance. J Immunol. 2008;180(5):3190–3200. doi: 10.4049/jimmunol.180.5.3190. [DOI] [PubMed] [Google Scholar]
- 74.Flano E, Woodland DL, Blackman MA. A Mouse Model for Infectious Mononucleosis. Immunologic Research. 2002;25(3):201–218. doi: 10.1385/IR:25:3:201. [DOI] [PubMed] [Google Scholar]
- 75.Karrer U, Sierro S, Wagner M, Oxenius A, Hengel H, Koszinowski UH, et al. Memory Inflation: Continuous Accumulation of Antiviral CD8+ T Cells Over Time. J Immunol. 2003;170(4):2022–2029. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- 76.Pahl-Seibert M-F, Juelch M, Podlech J, Thomas D, Deegen P, Reddehase MJ, et al. Highly Protective In Vivo Function of Cytomegalovirus IE1 Epitope-Specific Memory CD8 T Cells Purified by T-Cell Receptor-Based Cell Sorting. J Virol. 2005;79(9):5400–5413. doi: 10.1128/JVI.79.9.5400-5413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]