

Summary
Triadin and junctin are integral sarcoplasmic reticulum membrane proteins that form a macromolecular complex with the skeletal muscle ryanodine receptor (RyR1) but their roles in skeletal muscle calcium homeostasis remain incompletely understood. Here we report that delivery of siRNAs specific for triadin or junctin into C2C12 skeletal myoblasts reduced the expression of triadin and junctin in 8-day-old myotubes by 80 and 100%, respectively. Knocking down either triadin or junctin in these cells reduced Ca2+ release induced by depolarization (10 mM KCl) by 20–25%. Unlike triadin knockdown myotubes, junctin knockdown and junctin/triadin double knockdown myotubes also had reduced Ca2+ release induced by 400 μM 4-chloro-m-cresol, 10 mM caffeine, 400 μM UTP, or 1 μM thapsigargin. Thus, knocking down junctin compromised the Ca2+ stores in the sarcoplasmic reticulum of these cells. Our subsequent studies showed that in junctin knockdown myotubes at least two sarcoplasmic reticulum proteins (RyR1 and skeletal muscle calsequestrin) were down-regulated while these proteins’ mRNA expression was not affected. The results suggest that triadin has a role in facilitating KCl depolarization-induced Ca2+ release in contrast to junctin which has a role in maintaining sarcoplasmic reticulum Ca2+ store size in C2C12 myotubes.
Keywords: C2C12 myotubes, Ryanodine receptor, Sarcoplasmic reticulum, Calcium release, Triadin, Junctin
Introduction
In the specialized triad region of skeletal muscle, the voltage-dependent L-type Ca2+ channels localized to the invaginations of the cell membrane (t-tubules) are physically coupled to the juxtaposing intracellular Ca2+ release channels/ryanodine receptors (RyR1s) in the junctional sarcoplasmic reticulum (SR). Through this physical coupling, depolarization of t-tubules sensed by the L-type Ca2+ channels provokes RyR1 to release Ca2+ stored in the SR into the cytosol, thereby initiating muscle contraction [1]. This process, known as excitation–contraction (EC) coupling, therefore requires both dihydropyridine-sensitive L-type Ca2+ channel (Cav1.1, DHPR) and RyR1. In the SR, RyR1 is also physically associated with the two integral membrane proteins junctin and triadin to form a macromolecular Ca2+ signaling complex [2-5]. However, the roles of triadin and junctin in SR Ca2+ release of skeletal muscle remain incompletely understood.
Triadin is a glycoprotein with several splice variants that have molecular weights ranging from 35 to 95 kDa [6-11]. Expressed in both heart and skeletal muscle, triadin was initially isolated from skeletal muscle as a 95-kDa splice variant linked to both the α1 subunit of L-type Ca2+ channel and RyR1 [12,13]. Triadin has a short amino-terminal cytoplasmic domain, one transmembrane spanning segment, followed by a longer C-terminal luminal domain [6]. The luminal region of triadin is enriched in clusters of alternating lysine and glutamic acid residues that interact with skeletal muscle calsequestrin (CSQ1) [14] and with negatively charged amino acids of RyR [5]. Although it has been reported that the interruption between the luminal domains of triadin and RyR1 impairs RyR1 function and SR Ca2+ release [4,15], triadin also has been reported to inhibit channel activity of RyR1 through its cytoplasmic domain by reducing channel open probability [16,17]. Consistent with the inhibitory role of triadin, over-expression of triadin in skeletal myotubes inhibited depolarization-induced SR Ca2+ release [18]. Thus, whether triadin has a positive, negative or neutral effect on skeletal muscle EC coupling remains unclear. However, in a recent study triadin-deficient mice survived to adulthood, which showed that triadin is not essential for EC coupling [19]. Skeletal myotubes from triadin-deficient mice had reduced depolarization-induced Ca2+ release that was mainly attributed to reduced Ca2+ store size.
Junctin is a smaller protein (26 kDa) with a short 21 amino acid cytoplasmic N-terminal and a single membrane-spanning domain that is followed by a longer, highly charged C-terminal domain [20]. Like triadin, junctin is also expressed in both skeletal and cardiac muscle [20,21]. Junctin-deficient mice also survived to adulthood but were susceptible to ventricular arrhythmias and sudden death due to aberrant Ca2+ homeostasis in the heart [22]. Junctin’s role in skeletal muscle EC coupling is not well characterized.
Using recombinant adeno-associated virus (rAAV) delivered siRNAs, we have previously reported the roles of SR skeletal muscle calsequestrin (CSQ1) and cardiac muscle calsequestrin (CSQ2) isoforms in stored Ca2+ release of C2C12 skeletal myotubes [23]. Using the same knockdown strategy, we assessed the roles of triadin and junctin in SR Ca2+ release induced by depolarization (10 mM KCl) in C2C12 skeletal myotubes. Here we report that knocking down either triadin or junctin in these cells reproducibly reduced SR Ca2+ release induced by depolarization by 20–25%. Moreover, the effect of knocking down both proteins is additive, as Ca2+ release induced by 10 mM KCl is decreased by ~35% in double-knockdown C2C12 myotubes. Our subsequent studies showed that triadin specifically facilitates depolarization-induced Ca2+ release but has no noticeable role in maintaining Ca2+ store size of SR. In contrast, junctin appears to maintain Ca2+ store size of SR in C2C12 myotubes, as junctin-knockdown myotubes also have reduced stored Ca2+ release provoked by pharmacological agents that do not cause membrane depolarization or activate RyR1. A preliminary report of this work has been presented in abstract form [24].
Materials and methods
Materials
Immature C2C12 skeletal muscle cells (myoblasts) derived from normal adult C3H mouse leg muscles were purchased from ATCC (Manassas, VA). Double-strand recombinant adeno-associated viral (rAAV) vector was generously provided by Dr. Douglas McCarty (University of North Carolina, Chapel Hill). Rabbit polyclonal anti-junctin raised against an amino acid region specific for junctin and C-terminal cysteine (MAEDKEAKHGGHKNGRR GC) was prepared by ProSci Incorporated (San Diego, CA). Rabbit polyclonal anti-RyR1 antibody was prepared as described previously [25]. Monoclonal anti-triadin antibody and all other primary antibodies were purchased from Affinity Bioreagents (Golden, CO). [3H]ryanodine was obtained from PerkinElmer Life Sciences. Fluo 4-AM, Alexa 488 and Alexa 647-labeled secondary antibodies were purchased from Molecular Probes (Eugene, OR) and Fura 2-AM was from TEF LABS (Austin, TX). All other chemicals were obtained from Sigma unless specified otherwise.
Construction of vector, packaging, and purification of rAAV
The sequences of the triadin and junctin siRNAs were 5′-GGAAATGCATCGACAACCATTCAAGAGATGGTTGTCGATG CATTTCC-3′ and 5′-CTGACAAGAGTTCCAAGTCTATTCAAGAGATAGACTTGGAACTCTT GTCAG-3′, respectively. BLAST searches confirmed that the selected oligonucleotide sequences were not homologous to any other genes. A control sequence of 5′-TTCTCCGAACGTGTCA CGTTTCAAGAGAACGTGACACGTTCGGAGAA-3′ was used to construct rAAV-control as a negative control. The oligonucleotides encoding the specific siRNA for junctin were inserted into pSilencer-1.0 vector (Ambion, Austin TX) downstream of the U6 promoter using ApaI and EcoRI sites. For triadin, the sequence of the oligonucleotide is near 5′ end of triadin which is homologous to the known triadin isoforms. U6 promoter-driven expression cassettes were inserted into rAAV vector (ptrs-U1a-RFP-U6) using the KpnI and NotI sites [23]. The resulting vectors are termed rAAV-triadin, rAAV-junctin and rAAV-control. Serum type 2 double strand rAAVs were produced by the triple plasmid cotransfection method and purified by ammonium sulfate precipitation and on cesium chloride gradients [26,27].
Cell culture and recombinant AAV transduction
C2C12 myoblasts were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1 × antibiotics/antimycotics. Myoblasts were seeded at a concentration of 6 × 105 cells per 10-cm plate or 1 × 105 per well in 6-well plates and cultured for 48 h to reach 100% confluence (defined as day 0 myotubes). To induce myogenic differentiation, the growth medium was changed to differentiation medium (Dulbecco’s modified Eagle’s medium supplemented with 2% horse serum and 1 × antibiotics/antimycotics) on day 0. For rAAV transduction, C2C12 myoblasts were infected with rAAV carrying the control, junctin and/or triadin siRNA silencing cassette at 1 × 104 particles/cell about 24 h after seeding as described previously [23].
Stored Ca2+ release
Stored Ca2+ release was determined using the fluorescent Ca2+ indicator dye Fluo 4-AM as described previously [23]. Briefly, 8-day-old C2C12 myotubes grown on glass coverslips were washed three times with PBS buffer and loaded with 5 μM Fluo 4-AM for 1 h at 37 °C in Krebs–Ringer–Henseleit (KRH) buffer (125 mM NaCl, 5 mM KCl, 1.2 mM KH2PO4, 6 mM glucose, 1.2 mM MgCl2, 2 mM CaCl2, and 25 mM HEPES, pH 7.4). After loading, cells were rinsed with KRH buffer to remove non-hydrolyzed fluophore and kept in KRH buffer for 30 min to complete de-esterification. Individual cells were defined as region of interest, and average fluorescence was measured using the program ImageMaster (Photon Technology International, Lawrenceville, NJ). Resting calcium levels were monitored with Fura-2 as described [23].
Depolarization-induced Ca2+ transients were provoked by addition of 10 mM KCl to Ca2+-free KRH buffer (2 mM CaCl2 was replaced by 0.5 mM EGTA). In other experiments, stored Ca2+ release was induced by 400 μM 4-chloro-m-cresol, 10 mM caffeine, 400 μM UTP, or 1 μM thapsigargin.
Immunoblot analyses
8-Day-old C2C12 myotubes grown on six-well plates were harvested, washed twice with cold phosphate-buffered saline, lysed in RIPA buffer plus protease inhibitors (Complete Mini, Roche Diagnostics), and centrifuged at 12,000 × g for 10 min to remove insoluble material. Protein concentrations were determined using BCA assay. Twenty microgram of lysates were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and probed with antibodies for skeletal muscle triadin 95, junctin, CSQ1, CSQ2, DHPR-α1 subunit of the L-type Ca2+ channel (DHPR-α1), RyR1, and SERCA1. Western blots were developed using 3,3′-diaminobenzidine or enhanced chemiluminescence and quantified using Kodak Digital Science ID Image Analysis Software.
Immunofluorescence analyses
8-Day-old C2C12 myotubes fixed with 3% paraformaldehyde were permeabilized with buffer containing 20 mM HEPES, pH 7.4, 0.3 M sucrose, 50 mM NaCl, 3 mM MgCl2 and 0.5% Triton-X100. The myotubes were then incubated with antibodies specific for triadin and junctin, followed by incubation with an appropriate Alexa 488-labeled goat anti-mouse or Alexa 647-labeled goat anti-rabbit antibody. The prepared slides were examined using a Carl Zeiss LSM 510 laser scanning confocal microscopy system attached to an Axiovert 100 inverted microscope (Carl Zeiss, Jena, Germany) as described [23]. Argon and helium/neon lasers provided the excitation light beams (488 and 633 nm, respectively) for the confocal microscopy system. The images (512 × 512 pixels) obtained with a 100 × objective (Plan-Neofluar) were recorded using LSM Image software.
Quantitative RT-PCR
C2C12 cells were harvested, and total RNA was isolated using an RNeasy mini kit per the manufacturer’s instructions (Qiagen, Valencia, CA). The gene expression levels of RyR1, RyR3, CSQ1, CSQ2 and SERCA1 were determined by quantitative RT-PCR [28]. The primer and probe sequences of RyR1 are 5′-AGAATTCTGGGGTGAACTG, 3′-AACCGCAGTGTGTAGA AATTC. Probe sequence is (FAM)AGAGAGTGAAATTCTTGAACTACTTGTCG(TAMRA). The corresponding sequences are for RyR3: 5′-CCAACTTCTTCAAAGGGCTG, 3′-GAACCTCA GGTTGTAGAAATTC and (FAM) TCAGACCAAGTTATTGCACTACCTGGC(TAMRA); for DHPR-α1: 5′-CTCATAGGGTTGCTTGCAAG, 3′-AGGGTGGAGTTGAAGTGAAC and (FAM)TCCTGCGGATGGACCTGCCGGTA(TAMRA); for CSQ1: 5′-TACGAGGCCTTCATG GAAGA, 3′-GTTTCCTCAGGGTTGATCTC and (FAM)TGACCATCCCAGACAAGCCCAACA (TAMRA); for CSQ2: 5′-AAGAGT CACCCAGATGGCTA, 3′-ATGCTCAAGTCAGGATTGTCA and (FAM)CCTAGAGATCCTGAAACAGGTTGCCC(TAMRA); and for SERCA1: 5′-TCACCA CCAACCAGATGTCA, 3′-GAGAACTCGTTCAGTGAGCA and (FAM) ATCCCCATCCA CCTTTCAATGATG (TAMRA).
[3H]ryanodine binding
Ca2+ dependent activity of RyRs was determined by a [3H]ryanodine binding assay as described [23]. Crude membrane fractions prepared from C2C12 cells were incubated for 20 h at 24 °C with 2.5 nM [3H]ryanodine in 20 mM imidazole, pH 7.0, 250 mM KCl, 150 mM sucrose, 1 mM glutathione (oxidized), protease inhibitors, and the indicated free Ca2+ concentrations. Bmax values of [3H]ryanodine binding were determined by incubating membranes for 4 h at 24 °C with a saturating concentration of [3H]ryanodine (30 nM) in 20 mM imidazole, pH 7.0, 0.6 M KCl, 0.15 M sucrose, 20 μM leupeptin, 200 μM Pefabloc, and 100 μM Ca2+.
45Ca2+ uptake
ATP-dependent 45Ca2+ uptake by C2C12 total particulate matter was determined using a filtration method. 45Ca2+ uptake was initiated by placing membranes in 0.15 M KCl, 20 mM imidazole, pH 7.0, solution containing 5 mM ATP, 8 mM Mg2+, 5 mM Koxalate (a Ca2+ precipitating agent to increase Ca2+ uptake capacity [29], 10 μM ruthenium red (to inhibit RyRs [30], 5 mM NaN3 (to inhibit mitochondrial Ca2+ uptake), 1 mM EGTA, and 45Ca2+ to yield a free Ca2+ concentration of 0.5 μM. To obtain 45Ca2+ uptake rates, aliquots were placed at 2.5, 5, and 10 min on 0.45 μm Millipore filters under vacuum and rinsed with three 3-ml volumes of ice-cold 0.175 M KCl, 5 mM imidazole, pH 7.0, solution. Radioactivity remaining with the vesicles on the filters was determined by liquid scintillation counting.
Biochemical assays and data analyses
Free Ca2+ concentrations were obtained by including in the solution the appropriate amounts of Ca2+ and EGTA as determined using the stability constants and published computer program [31]. Free Ca2+ concentrations were verified with the use of a Ca2+ selective electrode.
Results are given as mean ± S.E. Significance of differences in data (p < 0.05) were determined using Student’s t-test.
Results
Efficacies of triadin and junctin knockdown in C2C12 myotubes
The efficacies of knocking down triadin and junctin in singly and doubly infected C2C12 myotubes were first investigated by immunoblot analyses. As shown in Fig. 1A and B, the expression of junctin and triadin in 8-day-old C2C12 skeletal myotubes, as assessed by a densitometry method, was reduced by approximately 100 and 80%, respectively, by their corresponding siRNAs. Knocking down junctin did not cause a compensatory over-expression of triadin and vice versa. We also assessed the expression of these two proteins using confocal microscopy (Fig. 2). Compared to myotubes infected with rAAV vector that contained the control oligonucleotide sequences, myotubes infected with rAAV-junctin siRNA knocked down the protein to background levels. Myotubes infected with rAAV-triadin siRNA maintained a weak level of fluorescence, in agreement with an 80% reduction in protein level (Fig. 1). There were no significant differences between the resting cytosolic Ca2+ levels among the control, triadin knockdown, junctin knockdown, and double knockdown myotubes using the fluorescent Ca2+ indicator Fura-2 [23] (data not shown).
Figure 1.

Immunoblot analysis of C2C12 myotubes. (A and B) Junctin and triadin protein levels in C2C12 myotubes infected with rAAV-control (lane 1), rAAV-junctin (lane 2), rAAV-triadin (lane 3), and both rAAV-junctin and rAAV-triadin (lane 4). Immuoblots were performed on 8-day-old C2C12 myotube lysates. (C) Intensities of protein bands were normalized by comparing them to respective control bands. Data are the mean ± S.E. of three experiments. *p < 0.05 compared to controls.
Figure 2.
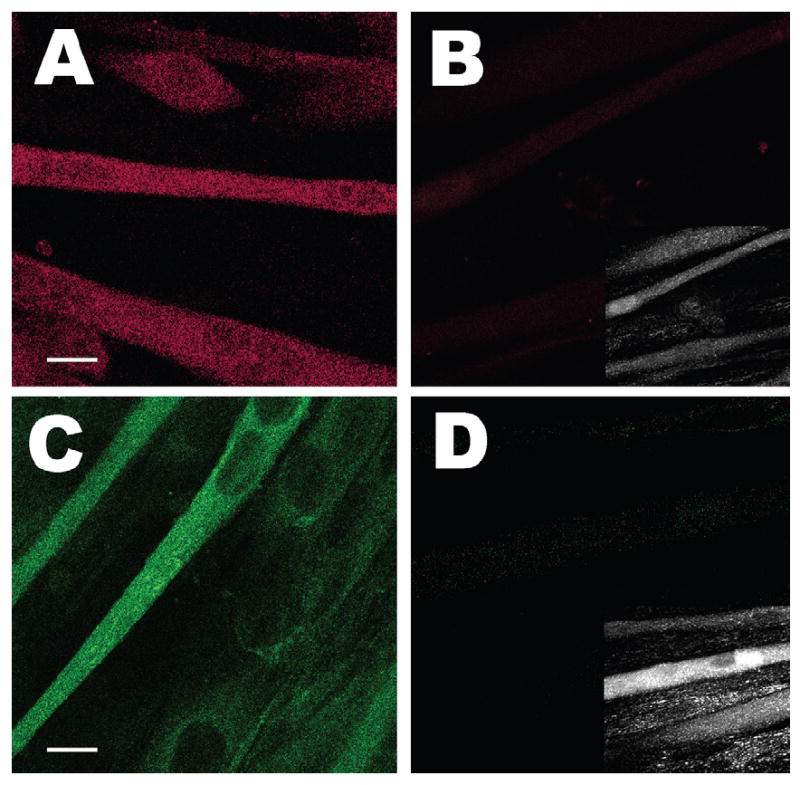
Efficacies of knocking down junctin and triadin in C2C12 myotubes. Eight-day-old control (A and C) or double knockdown (B and D) myotubes were probed with rabbit anti-junctin polyclonal antibody (A and B) or mouse anti-triadin monoclonal antibody (C and D) followed by the corresponding Alexa 647-conjugated goat-anti-rabbit or Alexa 488-conjugated goat-anti-mouse secondary antibody. The efficacies of the triadin siRNA and junctin siRNA are indicated by the nearly complete elimination of fluorescence in the double knockdown myotubes. Insets in B and D show pictures of corresponding double knockdown myotubes. Scale bars in (A) and (C): 10 μm.
Depolarization-induced stored Ca2+ release in triadin and junctin knockdown myotubes
The individual roles of triadin and junctin in EC coupling of 8-day-old C2C12 myotubes were assessed by measuring stored Ca2+ release provoked by addition of a submaximally activating KCl concentration of 10 mM in the absence of extracellular Ca2+. Stored Ca2+ release, measured as the ratio of peak fluo-4 fluorescence over baseline fluorescence ratio (F/F0), was reduced by 19 and 25% in the junctin and triadin knockdown groups, respectively (Fig. 3A and Table 1). These results suggest that a submaximally activating KCl concentration was sufficient to induce stored Ca2+ release in both triadin and junctin knockdown myotubes. Moreover, we noted that the effect of knocking down both proteins is additive of knocking down each protein individually, as stored Ca2+ release by double-knockdown myotubes exposed to 10 mM KCl was reduced by ~35% as compared to the control myotubes (Fig. 3A and Table 1).
Figure 3.
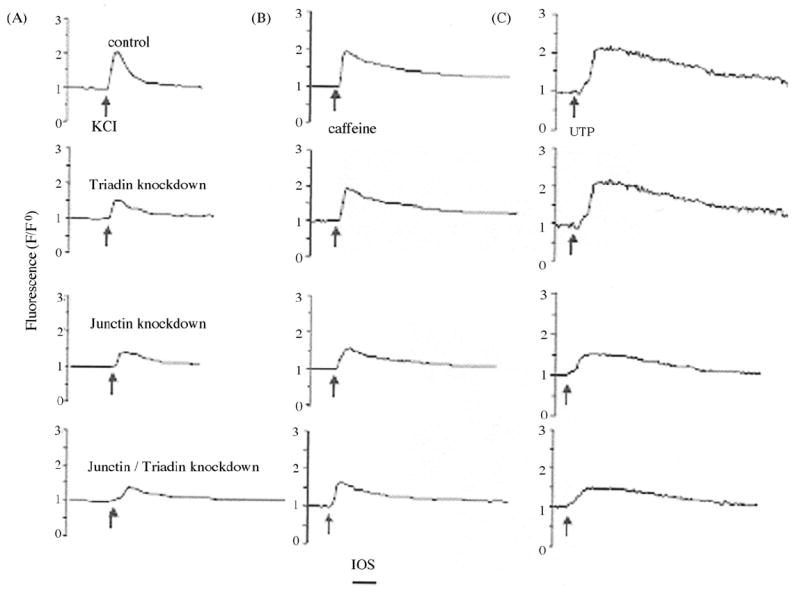
Effects of junctin and triadin knockdown on stored Ca2+ release in 8-day-old C2C12 myotubes. Depolarization (10 mM KCl) (A), caffeine (10 mM) (B), and UTP (400 μM) (C) induced Ca2+ release were determined in Ca2+-free KRH bath solutions as changes of Fluo-4 fluorescence (F/F0) in C2C12 myotubes infected with rAAV-control (top traces), rAAV-triadin (second traces), rAAV-junctin (third traces), and rAAV-junctin/triadin (bottom traces).
Table 1.
Effects of junctin/triadin knockdown on stored Ca2+ release in C2C12 myotubes
| Addition | Control | Junctin knockdown | Triadin knockdown | Junctin/triadin knockdown |
|---|---|---|---|---|
| 10 mM KCl | 1.98 ± 0.25 | 1.61 ± 0.18* | 1.49 ± 0.13* | 1.27 ± 0.08* |
| 0.4 mM 4-chloro-m-cresol | 1.88 ± 0.23 | 1.65 ± 0.17* | 2.05 ± 0.14 | 1.60 ± 0.11* |
| 10 mM caffeine | 1.83 ± 0.17 | 1.65 ± 0.14* | 1.88 ± 0.24 | 1.69 ± 0.18* |
| 1 μM thapsigargin | 1.86 ± 0.17 | 1.64 ± 0.16* | 1.93 ± 0.24 | 1.63 ± 0.14* |
| 0.4 mM UTP | 2.07 ± 0.29 | 1.46 ± 0.08* | 1.90 ± 0.16 | 1.48 ± 0.07* |
Peak values of fluorescence increases (F/F0) in C2C12 myotubes were determined in Fluo-4 loaded cells in Ca2+-free KRH buffer. Data are the mean ± S.E. of 8–17 experiments.
p < 0.05 compared to control.
Stored Ca2+ release induced by non-depolarizing agents in triadin and junctin knockdown myotubes
To determine whether triadin and/or junctin specifically facilitate KCl-depolarization induced Ca2+ release in C2C12 myotubes or have a less specific effect on stored Ca2+ release, we also exposed the four groups of C2C12 myotubes to caffeine (which activates RyR1 and RyR3), 4-chloro-m-cresol (which activates RyR1 but not RyR3 [32], or UTP (which generates inositol 1,4,5-trisphosphate and activates its receptor) in the absence of extracellular Ca2+. As shown in Fig. 3B and C and Table 1, knocking down triadin had no significant effects on the stored Ca2+ release of 8-day-old C2C12 myotubes provoked by caffeine, 4-chloro-m-cresol or UTP, thus indicating that Ca2+ store size was maintained. In contrast, junctin and junctin/triadin knockdown showed a significantly reduced Fluo-4 fluorescence signal in response to the three agents (Fig. 3B and C, Table 1). Thus, unlike triadin, junctin knockdown appeared to reduce Ca2+ store size. In support of this conclusion, thapsigargin – a SERCA inhibitor which empties Ca2+ stores independent of RyR – and inositol 1,4,5-trisphosphate receptor also provoked significantly less fluorescence responses in junctin and double knockdown myotubes as compared to control and triadin knockdown myotubes (Table 1). Taken together, data of Fig. 3 and Table 1 suggest that triadin knockdown impairs KCl-induced Ca2+ release without reducing SR Ca2+ store, whereas junctin knockdown decreases SR Ca2+ store thereby causing impaired KCl-induced Ca2+release.
Expression and function of Ca2+ handling proteins in junctin and triadin knockdown myotubes
We had previously shown that knocking down CSQ2 but not CSQ1 reduced SR Ca2+ uptake and KCl-induced Ca2+ release by affecting the expression of both RyR1 and SERCA1 in C2C12 myotubes [23]. In initial experiments, we measured the mRNA levels of RyR1, RyR3, DHPR-α1, CSQ1, CSQ2 and SERCA1 in 8-day triadin, junctin and triadin/junctin knockdown myotubes, as determined by quantitative RT-PCR. None differed substantially from those in myotubes infected with the control vector (Table 2). To determine whether the reduction in stored Ca2+ release of junctin knockdown myotubes was due to a reduction of Ca2+ handling proteins in SR, we performed immunoblot analyses of the four groups of C2C12 myotubes. CSQ1 and RyR1 were reduced by about 40 and 30% in junctin knockdown myotubes, respectively, but maintained in triadin knockdown myotubes at levels comparable to control myotubes (Fig. 4A and B). Since it is possible that the small fraction of triadin (~20%) not eliminated by our knockdown approach may still be sufficient to exert its effect, we attempted immunoprecipitation experiments to determine whether the amount of triadin interacting with RyR1 is affected by triadin knockdown. However, despite multiple attempts the relatively small amount of membrane fractions isolated from C2C12 myotubes was not sufficient for us to conclusively address this question. No significant changes of protein expression levels of SERCA1, CSQ2 or DHPR-α1 were detected among the four groups of myotubes.
Table 2.
Quantitative RT-PCR of mRNAs from junctin and triadin knockdown myotubes.
| MRNA | Control | Junctin knockdown | Triadin knockdown | Junctin/triadin knockdown |
|---|---|---|---|---|
| RyR1 | 100 | 131 ± 14 | 99 ± 7 | 103 ± 6 |
| RyR3 | 100 | 126 ± 12 | 109 ± 11 | 124 ± 13 |
| DHPR-α1 | 100 | 82 ± 23 | 107 ± 26 | 72 ± 22 |
| CSQ1 | 100 | 137 ± 29 | 68 ± 26 | 76 ± 23 |
| CSQ2 | 100 | 130 ± 22 | 105 ± 11 | 116 ± 16 |
| SERCA1 | 100 | 109 ± 10 | 97 ± 31 | 94 ± 19 |
RT-PCR analyses were performed using total RNA isolated from 8-day-old myotubes. Relative levels of gene expression as a percentage of control were determined for each gene with β-actin as reference. Data are the mean ± S.E. of 3–12 samples.
Figure 4.
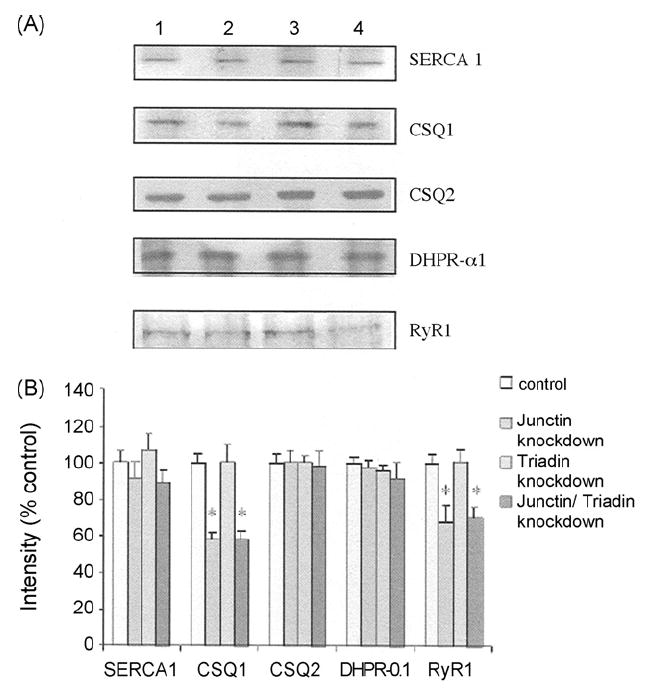
Immunoblot analysis of C2C12 myotubes. (A) Protein levels in C2C12 myotubes infected with rAAV-control (lane 1), rAAV-junctin (lane 2), rAAV-triadin (lane 3), and both rAAV-junctin and rAAV-triadin (lane 4). Immuoblots were performed on 8-day-old C2C12 myotube lysates. (B) Normalized intensities of protein bands. Data are the mean ± S.E. of three experiments. *p < 0.05 compared to controls.
In agreement with immunoblot analyses that indicate a reduction of RyR1 protein in junctin knockdown and double knockdown myotubes, the maximal [3H]ryanodine binding capacity (Bmax) of crude membrane fractions isolated from junctin knockdown (but not from triadin knockdown) myotubes was significantly reduced (Table 3). A small (not significant) decrease in Bmax of [3H]ryanodine binding was also observed in double-knockdown myotubes. We tested whether junctin and triadin knockdown altered the Ca2+ dependence of RyR1 activity, as determined by a ligand binding assay [33]. As shown in Fig. 5, knockdown of triadin, junctin and both proteins did not significantly alter the bimodal Ca2+ dependence of [3H]ryanodine binding. Although the immunoblot analyses did not suggest a reduction in SERCA1 protein in any of the four groups, we nevertheless measured 45Ca2+ uptake rates by membrane fractions isolated from each of the four groups of myotubes to determine whether a reduction in Ca2+ uptake could explain the non-specific reduction in stored Ca2+ release of junctin knockdown myotubes. The measured 45Ca2+ uptake rates (Table 3) confirmed that knocking down junctin, triadin or both proteins did not affect Ca2+ uptake in these cells that could lead to a reduction of Ca2+ storage.
Table 3.
Bmax values of [3H]ryanodine binding and 45Ca2+uptake rates of membranes isolated from control and junctin/triadin knockdown C2C12 myotubes
| Sample | Bmax of [3H]ryanodine binding (% of control) | 45Ca2+ uptake rate (% of control) |
|---|---|---|
| Control | 100 | 100 |
| Junctin knockdown | 63 ± 9* | 89 ± 12 |
| Triadin knockdown | 113 ± 54 | 107 ± 16 |
| Junctin/triadin knockdown | 77 ± 32 | 92 ± 7 |
The total number of [3H]ryanodine binding sites (Bmax) and 45Ca2+ uptake rates of crude membrane fractions were determined as described in Materials and methods. Data are mean ± S.E. of six experiments.
p < 0.05 compared to control.
Figure 5.
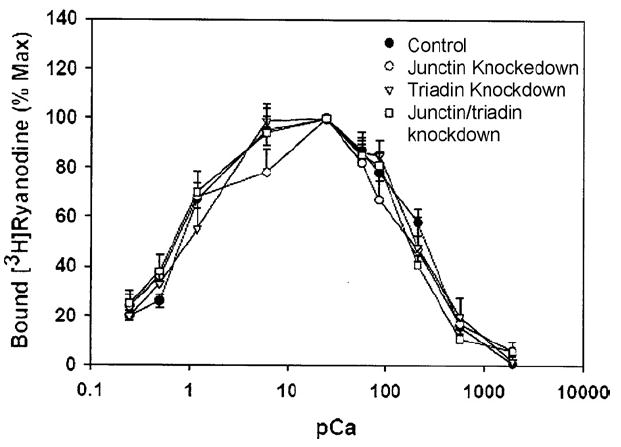
Ca2+ dependence of [3H]ryanodine binding of 8-day-old junctin and triadin knockdown C2C12 myotubes. Specific [3H]ryanodine binding was determined in 250 mM KCl, 20 mM imidazole, pH 7.0, media containing 2.5 nM [3H]ryanodine and the indicated concentrations of free Ca2+. Data are the mean ± S.E. of three to four experiments.
Discussion
To better understand the functional significance of the two ryanodine receptor associated proteins junctin and triadin in a skeletal muscle derived cell line, we used a knockdown approach that effectively reduced the two proteins by at least 80% in C2C12 myotubes (Figs. 1 and 2). We had previously shown that the transduction of rAVV by itself did not lead to alterations of other Ca2+ handling proteins in 8-day-old C2C12 myotubes and these cells released normal amounts of stored Ca2+ in response to KCl depolarization [23].
In a previous study, acute over-expression of 95-kDa triadin in skeletal myotubes inhibited depolarization-induced SR Ca2+ release [18]. These data implicated that the role of triadin could be to prevent excessive stored Ca2+ release during cell membrane depolarization. In contrast, our data (Fig. 3 and Table 1) indicate that triadin, through its physical association with RyR1 [4,15] and possibly DHPRs [12,13], actually facilitates depolarization-induced Ca2+ release in skeletal muscle. However, our data is not as dramatic as a study that showed expression of the triadin-binding deficient RyR1-D4878A/D4907A/E4908A mutant in dyspedic myotubes eliminated electrically evoked Ca2+ release [4]. Our data is more comparable to data obtained from myotubes of triadin-deficient mice, as we found that triadin knockdown myotubes exposed to KCl-induced depolarization had a reduction in stored Ca2+ release by only ~20%, as compared to control infected myotubes [19]. But in contrast to the study of skeletal myotubes from triadin-deficient mice, our triadin knockdown myotubes had preserved Ca2+ store size, as stored Ca2+ release provoked by RyR-specific agents (caffeine and 4-chloro-m-cresol) or by other agents (UTP and thapsigargin) that release stored Ca2+ via different mechanisms was not affected by triadin knockdown. The difference between our study and that of triadin-deficient mice could be due to the formation of a less well organized cell membrane and sarcoplasmic reticulum system in C2C12 myotubes compared to skeletal muscle fibers [34-36].
An alternative possibility is that the longer and complete elimination of triadin in triadin knockout mice can lead to reduced Ca2+ store size in muscle fibers.
Immunoprecipitation studies with a RyR1 mutant (D4878A/D4907A/E4908A) that lacked triadin binding showed junctin binding, which suggests that triadin and junctin bind to different sites on RyR1 [4]. Although the functional effects of over-expressing and abolishing junctin on cardiac function have been extensively studied [22,37-40], the physiological role of junctin in skeletal muscle Ca2+ release is less well understood. Because we found that junctin knockdown myotubes had reduced stored Ca2+ release provoked by KCl as well as pharmacological agents that do not affect RyR1 (UTP and thapsigargin), we conclude that knocking down junctin reduces the SR Ca2+ store in C2C12 myotubes. The expression of at least two SR proteins (RyR1 and CSQ1) was reduced in junctin knockdown myotubes. Therefore, it is conceivable that knockdown of junctin may have resulted in the disruption of the macromolecular Ca2+ signaling complex that include RyR1 and CSQ1. However, the reduction in Ca2+ stores in junctin knockdown C2C12 myotubes is unlikely due to the reduction of CSQ1, as we previously showed that knockdown of CSQ1 did not reduce the Ca2+ store in C2C12 myotubes [23]. We also note that in junctin knockdown myotubes no noticeable changes in the mRNA levels of several key Ca2+ handling proteins were observed including RyR1 and CSQ1 (Table 2). Thus, knocking down junctin may increase degradation of RyR1 and CSQ1 and possibly other SR proteins. How junctin knockdown reduces SR Ca2+ store size and the concentrations of certain SR proteins will require further studies.
The present study describes for the first time the effects of the combined knockdown of junctin and triadin. C2C12 myotubes infected with siRNAs specific to junctin and triadin exhibited reduced Ca2+ transients in response to pharmacological agents similar to those in junctin knockdown myotubes. Ca2+ transients were lower in double knockdown myotubes in response to KCl depolarization than in response to pharmacological agents in agreement with triadin knockdown myotubes. The data support the notion that knockdown of junctin decreases SR Ca2+ store size, whereas reduction in triadin decreased depolarization-induced stored Ca2+ release without affecting Ca2+ store size. Taken together, our results suggest that the two structurally related proteins do not have a redundant role in forming a macromolecular Ca2+ signaling complex in a skeletal muscle derived cell line.
Acknowledgments
The authors thank Daniel Pasek for his assistance with the biochemical assays. The work was supported by National Institutes of Health Grants AR018687 (to GM) and NHLBI081285 (to JPE).
Footnotes
Conflicts of interest None.
References
- 1.Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–729. doi: 10.1152/physrev.1997.77.3.699. [DOI] [PubMed] [Google Scholar]
- 2.Guo W, Campbell KP. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J Biol Chem. 1995;270:9027–9030. doi: 10.1074/jbc.270.16.9027. [DOI] [PubMed] [Google Scholar]
- 3.Beard NA, Laver DR, Dulhunty AF. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog Biophys Mol Biol. 2004;85:33–69. doi: 10.1016/j.pbiomolbio.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Goonasekera SA, Beard NA, Groom L, et al. Triadin binding to the C-terminal luminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J Gen Physiol. 2007;130:365–378. doi: 10.1085/jgp.200709790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee JM, Rho SH, Shin DW, et al. Negatively charged amino acids within the intraluminal loop of ryanodine receptor are involved in the interaction with triadin. J Biol Chem. 2004;279:6994–7000. doi: 10.1074/jbc.M312446200. [DOI] [PubMed] [Google Scholar]
- 6.Knudson CM, Stang KK, Moomaw CR, Slaughter CA, Campbell KP. Primary structure and topological analysis of a skeletal muscle-specific junctional sarcoplasmic reticulum glycoprotein (triadin) J Biol Chem. 1993;268:12646–12654. [PubMed] [Google Scholar]
- 7.Taske NL, Eyre HJ, RO OB, Sutherland GR, Denborough MA, Foster PS. Molecular cloning of the cDNA encoding human skeletal muscle triadin and its localisation to chromosome 6q22-6q23. Eur J Biochem. 1995;233:258–265. doi: 10.1111/j.1432-1033.1995.258_1.x. [DOI] [PubMed] [Google Scholar]
- 8.Guo W, Jorgensen AO, Campbell KP. Triadin, a linker for calsequestrin and the ryanodine receptor. Soc Gene Physiol Ser. 1996;51:19–28. [PubMed] [Google Scholar]
- 9.Kobayashi YM, Jones JR. Identification of triadin 1 as the predominant triadin isoform expressed in mammalian myocardium. J Biol Chem. 1999;274:28660–28668. doi: 10.1074/jbc.274.40.28660. [DOI] [PubMed] [Google Scholar]
- 10.Marty I, Thevenon D, Scotto C, et al. Cloning and characterization of a new isoform of skeletal muscle triadin. J Biol Chem. 2000;275:8206–8212. doi: 10.1074/jbc.275.11.8206. [DOI] [PubMed] [Google Scholar]
- 11.Hong CS, Ji JH, Kim JP, Jung DH, Kim DH. Molecular cloning and characterization of mouse cardiac triadin isoforms. Gene. 2001;278:193–199. doi: 10.1016/s0378-1119(01)00718-1. [DOI] [PubMed] [Google Scholar]
- 12.Brandt NR, Caswell AH, Wen SR, Talvenheimo JA. Molecular interactions of the junctional foot protein and dihydropyridine receptor in skeletal muscle triads. J Membr Biol. 1990;113:237–251. doi: 10.1007/BF01870075. [DOI] [PubMed] [Google Scholar]
- 13.Kim KC, Caswell AH, Talvenheimo JA, Brandt NR. Isolation of a terminal cisterna protein which may link the dihydropyridine receptor to the junctional foot protein in skeletal muscle. Biochemistry. 1990;29:9281–9289. doi: 10.1021/bi00491a025. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi YM, Alseikhan BA, Jones LR. Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged beta-strand in mediating the protein–protein interaction. J Biol Chem. 2000;275:17639–17646. doi: 10.1074/jbc.M002091200. [DOI] [PubMed] [Google Scholar]
- 15.Lee EH, Song DW, Lee JM, Meissner G, Allen PD, Kim DH. Occurrence of atypical Ca2+ transients in triadin-binding deficient-RYR1 mutants. Biochem Biophys Res Commun. 2006;351:909–914. doi: 10.1016/j.bbrc.2006.10.115. [DOI] [PubMed] [Google Scholar]
- 16.Ohkura M, Furukawa K, Fujimori H, et al. Dual regulation of the skeletal muscle ryanodine receptor by triadin and calsequestrin. Biochemistry. 1998;37:12987–12993. doi: 10.1021/bi972803d. [DOI] [PubMed] [Google Scholar]
- 17.Groh S, Marty I, Ottolia M, et al. Functional interaction of the cytoplasmic domain of triadin with the skeletal ryanodine receptor. J Biol Chem. 1999;274:12278–12283. doi: 10.1074/jbc.274.18.12278. [DOI] [PubMed] [Google Scholar]
- 18.Rezgui SS, Vassilopoulos S, Brocard J, et al. Triadin Trisk 95 overexpression blocks excitation–contraction coupling in rat skeletal myotubes. J Biol Chem. 2005;280:39302–39308. doi: 10.1074/jbc.M506566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen X, Franzini-Armstrong C, Lopez JR, et al. Triadins modulate intracellular Ca2+ homeostasis but are not essential for excitation–contraction coupling in skeletal muscle. J Biol Chem. 2007;282:37864–37874. doi: 10.1074/jbc.M705702200. [DOI] [PubMed] [Google Scholar]
- 20.Jones LR, Zhang L, Sanborn K, Jorgensen AO, Kelley J. Purification, primary structure, and immunological characterization of the 26-kDa calsequestrin binding protein (junctin) from cardiac junctional sarcoplasmic reticulum. J Biol Chem. 1995;270:30787–30796. doi: 10.1074/jbc.270.51.30787. [DOI] [PubMed] [Google Scholar]
- 21.Lim KY, Hong CS, Kim DH. cDNA cloning and characterization of human cardiac junction. Gene. 2000;255:35–42. doi: 10.1016/s0378-1119(00)00299-7. [DOI] [PubMed] [Google Scholar]
- 22.Yuan Q, Fan GC, Dong M, et al. Sarcoplasmic reticulum calcium overloading in junctin deficiency enhances cardiac contractility but increases ventricular automaticity. Circulation. 2007;115:300–309. doi: 10.1161/CIRCULATIONAHA.106.654699. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Xu L, Duan H, Pasek DA, Eu JP, Meissner G. Knocking down type 2 but not type 1 calsequestrin reduces calcium sequestration and release in C2C12 skeletal muscle myotubes. J Biol Chem. 2006;281:15572–15581. doi: 10.1074/jbc.M600090200. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Li X, Duan H, Fulton TR, Meissner G. Knockdown of sarcoplasmic reticulum triadin and junctin in C2C12 cells. Biophysical Society Meeting; Baltimore, MD. 2007. Abstract. [Google Scholar]
- 25.Du W, McMahon TJ, Zhang ZS, Stiber JA, Meissner G, Eu JP. Excitation–contraction coupling in airway smooth muscle. J Biol Chem. 2006;281:30143–30151. doi: 10.1074/jbc.M606541200. [DOI] [PubMed] [Google Scholar]
- 26.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vector in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Samulski RJ, Xiao X. Role for highly regulated rep gene expression in adeno-associated virus vector production. J Virol. 1997;71:5236–5243. doi: 10.1128/jvi.71.7.5236-5243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HS, Lee G, John SWM, Maeda N, Smithies O. Molecular phenotyping for analyzing subtle genetic effects in mice: application to an angiotensinogen gene titration. Proc Natl Acad Sci U S A. 2002;99:4602–4607. doi: 10.1073/pnas.072083799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martonosi A, Feretos R, Sarcoplasmic I. Reticulum, The uptake of Ca2+ by sarcoplasmic reticulum fragments. J Biol Chem. 1964;239:648–658. [PubMed] [Google Scholar]
- 30.Xu L, Tripathy A, Pasek DA, Meissner G. Ruthenium red modifies the cardiac and skeletal muscle Ca2+ release channels (ryanodine receptors) by multiple mechanisms. J Biol Chem. 1999;274:32680–32691. doi: 10.1074/jbc.274.46.32680. [DOI] [PubMed] [Google Scholar]
- 31.Schoenmakers TJ, Visser GJ, Flik G, Theuvenet AP. CHELATOR: an improved method for computing metal ion concentrations in physiological solutions. BioTechniques. 1992;12:870–879. [PubMed] [Google Scholar]
- 32.Fessenden JD, Wang Y, Moore RA, Chen SR, Allen PD, Pessah IN. Divergent functional properties of ryanodine receptor types 1 and 3 expressed in a myogenic cell line. Biophys J. 2000;79:2509–2525. doi: 10.1016/S0006-3495(00)76492-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meissner G. Regulation of mammalian ryanodine receptors. Front Biosci. 2002;7:d2072–d2080. doi: 10.2741/A899. [DOI] [PubMed] [Google Scholar]
- 34.Carozzi AJ, Ikonen E, Lindsay MR, Parton RG. Role of cholesterol in developing T-tubules: analogous mechanisms for T-tubule and caveolae biogenesis. Traffic. 2000;1:326–341. doi: 10.1034/j.1600-0854.2000.010406.x. [DOI] [PubMed] [Google Scholar]
- 35.Parton RG, Way M, Zorzi N, Stang E. Caveolin-3 associates with developing T-tubules during muscle differentiation. J Cell Biol. 1997;136:137–154. doi: 10.1083/jcb.136.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nori A, Valle G, Bortoloso E, Turcato F, Volpe P. Calsequestrin targeting to sarcoplasmic reticulum of skeletal muscle fibers. Am J Physiol Cell. 2006;291:C245–C253. doi: 10.1152/ajpcell.00370.2005. [DOI] [PubMed] [Google Scholar]
- 37.Hong CS, Cho MC, Kwak YG, et al. Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J. 2002;16:1310–1312. doi: 10.1096/fj.01-0908fje. [DOI] [PubMed] [Google Scholar]
- 38.Kirchhefer U, Hanske G, Jones JR. Overexpression of junctin causes adaptive changes in cardiac myocyte Ca2+ signaling. Cell Calcium. 2006;39:131–142. doi: 10.1016/j.ceca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Kirchhefer U, Neumann J, Bers DM, et al. Impaired relaxation in transgenic mice overexpressing junctin. Cardiovasc Res. 2003;59:369–379. doi: 10.1016/s0008-6363(03)00432-2. [DOI] [PubMed] [Google Scholar]
- 40.Gergs U, Berndt T, Buskase J, et al. On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am J Physiol. 2007;293:H728–H734. doi: 10.1152/ajpheart.01187.2006. [DOI] [PubMed] [Google Scholar]