

Abstract
Differentiation of antigen specific T cells into IFN-γ producers is essential for protective immunity to intracellular pathogens. In addition to stimulation through the TCR and costimulatory molecules, IFN-γ production is thought to require other inflammatory cytokines. Two such inflammatory cytokines are IL-12 and type-I IFN (IFN-I), which have been shown to have a role in priming naïve T cells IFN-γ production in vitro. However, their role in priming antigen specific T cells for IFN-γ production during experimental infection in vivo is less clear. Herein, we examine the requirements for IL-12 and IFN-I, either individually or in combination, for priming antigen specific T cells for IFN-γ production after Listeria monocytogenes infection. Surprisingly, neither individual nor combined defects in IL-12 or IFN-I signaling altered IFN-γ production by antigen specific CD8 T cells after Lm infection. In contrast, individual defects in either IL-12 or IFN-I signaling conferred partial (∼50%) reductions, while combined deficiency in both IL-12 and IFN-I signaling conferred more dramatic (75−95%) reductions in IFN-γ production by antigen specific CD4 T cells after Lm infection. The additive effects of IL-12 and IFN-I signaling on IFN-γ production by CD4 T cells were further demonstrated by adoptive transfer of transgenic IFN-IR+/+ and IFN-IR−/− CD4 T cells into normal and IL-12-deficient mice, and infection with recombinant Lm. These results demonstrate an important dichotomy between the signals required for priming IFN-γ production by CD4 and CD8 T cells in response to bacterial infection.
Keywords: Bacterial infection, T cells, cell differentiation
INTRODUCTION
IFN-γ plays an important protective role in both innate and adaptive immunity to infection with viruses, bacteria, and other intracellular pathogens. Antigen specific IFN-γ-producing T cells are the hallmark of protective adaptive immunity to this group of pathogens. Inflammatory cytokines produced early during infection are believed to play an important role in the differentiation of naïve T cells into IFN-γ producing cells. Two well-characterized inflammatory cytokines that can influence the differentiation of T cells are IL-12 and type-I IFN (IFN-I) (1-5). Most of the studies examining the signals required for T cell activation and priming for IFN-γ production were conducted in vitro by stimulating T cells with plastic beads coated with MHC ligands + peptide in the presence of exogenously added cytokines (1-3). These experiments have clearly demonstrated the direct effects of these cytokines on isolated T cells in vitro, but do not address the role of these cytokines in vivo, and especially in the context of infection when a myriad of other cytokines and immune stimulatory pathways are activated. Nevertheless, understanding the role and relative contributions of IL-12 and IFN-I in T cell activation during in vivo infection is important because of their known direct effects on T cell IFN-γ production and augmentation or inhibition of the other cytokine during certain experimental infections (6, 7).
Previous studies using infection and vaccination models have limited their analysis to either IFN-I or IL-12. For example, IFN-I receptor-deficient (IFN-IR−/−) mice can generate IFN-γ producing CD8 T cells after LCMV infection (7, 8). But, since more IL-12 is produced after LCMV infection in IFN-IR−/− mice, these results suggest that IL-12 can substitute for the lack of IFN-I signaling in these mice (7). For IL-12-deficient mice, infection with various viral or other intracellular pathogens, unlike model antigens administered with adjuvants, show that IL-12 is not required for IFN-γ production by pathogen specific T cells (7, 9-12). Yet in these infection models where IFN-γ is produced by antigen specific T cells in the absence of IL-12, the role of IFN-I in bypassing the requirement for IL-12 has not been clearly demonstrated. Given that IFN-I may be compensating for the lack of IL-12, and vice versa, assessing the combined deficiency of IFN-I and IL-12 is critical to understanding the role of IFN-I and IL-12 plays in priming T cells for IFN-γ production during infection, in vivo. To address this, we used a well-characterized mouse model of infection with the Gram-positive bacterium Listeria monocytogenes (Lm) in which both immune mediators of innate host resistance and generation of antigen specific T cells in response to bacterial infection can be characterized (13). Following primary Lm infection, both IL-12 and IFN-I are produced (14-17) and antigen specific CD4 and CD8 T cells expand and produce IFN-γ. In this study, we examined the individual and additive roles of IL-12 and IFN-IR signaling in priming CD8 and CD4 T cells for IFN-γ production after Lm infection.
MATERIALS AND METHODS
Mice
C57Bl/6 and Thy1.1 mice were obtained from The Jackson Laboratory. IL-12β-deficient (IL12P40−/−) (11), and IL-12α-deficient (IL12P35−/−) (7), backcrossed to B6 mice for 11 generations, were purchased from The Jackson Laboratory. IFN-I receptor-deficient (IFN-IR−/−) mice (18) were backcrossed to B6 mice for 12 generations prior to use (8). IFN-IR−/−IL12P40−/− double-deficient mice were obtained by intercrossing IL-12P40−/− with IFN-IR−/− mice. B6-Tg(TcrLCMV)1Aox, LCMV-GP61−80-specific CD4 TCR transgenic mice (SMARTA) (19) were obtained from Dr. C. Surh (The Scripps Research Institute, La Jolla, CA) via Dr. M.J. Bevan (University of Washington, Seattle, WA) and intercrossed with Thy1.1 mice or IFN-IR−/− (Thy1.2+) mice to generate SMARTA transgenic IFN-IR+/+ (Thy1.1+) and SMARTA transgenic IFN-IR−/− (Thy1.1+Thy1.2+) mice, respectively. All mice were housed in a specific pathogen free facility at the University of Washington and experiments performed under IACUC approved protocols.
Infections with Listeria and LCMV
The recombinant Lm strains Lm-OVA and Lm-GP61−80 were kindly provided by Dr. Hao Shen (University of Pennsylvania). Lm-OVA ΔactA was constructed from Lm-OVA using homologous recombination after cloning ∼500 base-pair fragments of the Lm actA locus into the HindIII/Kpn1 sites of the temperature sensitive plasmid pKSV7 with the following primers: upstream flanking region, forward primer 5’ aagcttgcagcgaccgatagcgaag 3’, reverse primer 5’ gaattccgctgcgctatccgatgg 3’, downstream flanking region, forward primer 5’ gaattcgttaagtccaaaggtatcg 3’, reverse primer 5’ ggtacctaaagagaacacgccaatag 3’ (underlined sequences indicate introduced restriction sites). Lm were grown to early log phase (OD600 0.1) in brain heart infusion media (Becton Dickinson Company) at 37°C, washed in saline, and diluted in 200 μl final volume. For endogenous responses, 106 Lm-OVA ΔactA were injected intravenously into mice. For adoptive transfer experiments, 105 Lm-GP61−80 were injected intraperitoneally into mice. LCMV strain Armstrong was plaque purified, grown in baby hamster kidney cells, and titered on Vero cells. 2 × 105 PFU LCMV diluted in 200 μl final volume was injected intravenously into mice.
Reagents, Abs, in vitro cultures, adoptive transfer and cell staining
For in vivo depletion, 1.0 mg of purified anti-mouse IL-12P40 (clone C17.8) or rat IgG2a isotype control (Bioexpress) was injected into mice 1 day prior to infection as described previously (7). For in vitro culture, splenocytes were plated into 96-well round bottom plates (5 × 106 cells/ml), and either stained directly with tetramer, or stimulated with the indicated peptides (10−6 M) for 5 hours (intracellular cytokine staining) or 72 hours (culture supernatants) as described (8, 20). For intracellular cytokine staining, monensin (BD GolgiStop reagent) was added to cell culture prior to peptide stimulation. For adoptive transfer of SMARTA cells, CD4 T cells were purified by negative selection from SMARTA transgenic IFN-IR+/+ (Thy1.1+) or SMARTA transgenic IFN-IR−/− (Thy1.1+Thy1.2+) mice using CD4 T cell isolation kits (R&D Systems). These cells were transferred intravenously into recipient mice one day prior to Lm infection and contained 1×105 SMARTA transgenic IFN-IR+/+ (Thy1.1+) and 1×105 SMARTA transgenic IFN-IR−/− (Thy1.1+Thy1.2+) CD4 T cells per recipient mouse. The concentration of IL-12P40 and IL-12P70 in serum, and IFN-γ, IL-4, and IL-13 in splenocyte culture supernatants was determined by ELISA using reagents from R&D Systems.
Statistics
The differences in percentages and numbers of stimulated splenocytes, percentage of cytokine producing transgenic cells, and cytokine concentrations in culture supernatants between groups of mice were evaluated by using the Student's t test with P < 0.05 taken as statistically significant.
RESULTS
Generation of antigen-specific CD8 T cells in the absence of IFN-I receptor after Lm infection
The role of IFN-I on the adaptive T cell response to bacterial infection is unknown. IFN-I receptor-deficient (IFN-IR−/−) compared with control mice are more resistant to primary Lm infection (15-17). This may be due to a reduction in IFN-I mediated lymphocyte apoptosis in the first few days after infection in IFN-IR−/− mice (21). The increased resistance of IFN-IR−/− mice to Lm infection is in sharp contrast to the protective role that IFN-I plays in both innate and adaptive immunity during viral infection (22). Thus, we sought to examine the contribution of IFN-I in the adaptive T cell response after Lm infection, and directly compare the relative importance of IFN-I for priming antigen specific T cells after Lm compared with LCMV infection in either normal or IFN-IR−/− mice. Consistent with previous studies in our laboratory (8), the expansion of LCMV-specific CD8 T cells was dramatically reduced in IFN-IR−/− mice compared with B6 control mice confirming the important role of IFN-I for expansion of CD8 T cells in response to LCMV infection (Figure 1). In contrast following infection with Lm-OVA, expansion of OVA-specific CD8 T cells occurred with the same magnitude in both IFN-IR−/− and B6 mice (Figure 1). These results suggest that while IFN-I plays an important role for the optimal expansion of CD8 T cell response to LCMV infection, it is not required for expansion of antigen-specific CD8 T cells in response to Lm infection.
Figure 1.
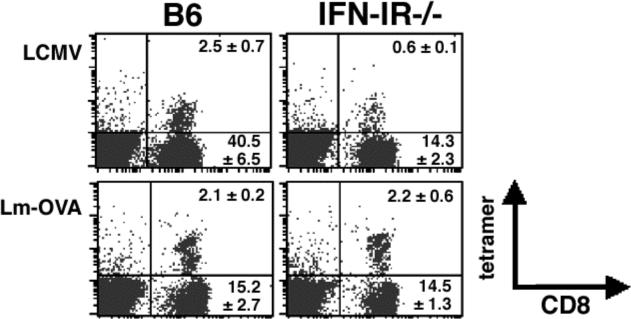
Antigen-specific CD8 T cell expansion after LCMV or Lm-OVA infection. Mice were infected with LCMV Armstrong (2×105 PFUs) or Lm-OVA (104 CFUs) and splenocytes were analyzed by LCMV-GP33−41 and OVA257−264 tetramer and cell surface staining day 8 after infection. The number in each quadrant indicates the percentage ± standard error of gated cells in each quadrant from 3−4 mice per group, and is representative of two independent experiments.
Increased production of IL-12 in IFN-I receptor-deficient mice
For CD8 T cells cultured in vitro, either IL-12 of IFN-I can provide in addition to TCR and costimulation, a third signal allowing for proliferation and IFN-γ production (2). Since Lm infection in vivo triggers the production of both IL-12 and IFN-I (16, 17, 23), we hypothesized that IL-12 produced early after Lm infection may overcome the requirement for IFN-I signaling and allow for the apparent normal expansion of antigen-specific CD8 T cells in IFN-IR−/− compared with B6 mice after Lm infection. Consistent with this hypothesis, beginning at 8 hours after Lm-OVA infection, increased concentrations of both IL-12P40 and IL-12P70 were present in B6 and IFN-IR−/− mice; and by 24 hours after infection, maximal levels of IL-12P40 and IL-12P70 are observed (Figure 2A,B). Of note, the serum concentration of both IL-12P40 and IL-12P70 was ∼ 5-fold higher in IFN-IR−/− compared with B6 mice. In contrast following LCMV infection, only minimal amounts of IL-12P40 and no IL-12P70 could be detected in either B6 or IFN-IR−/− mice (Figure 2A,B).
Figure 2.
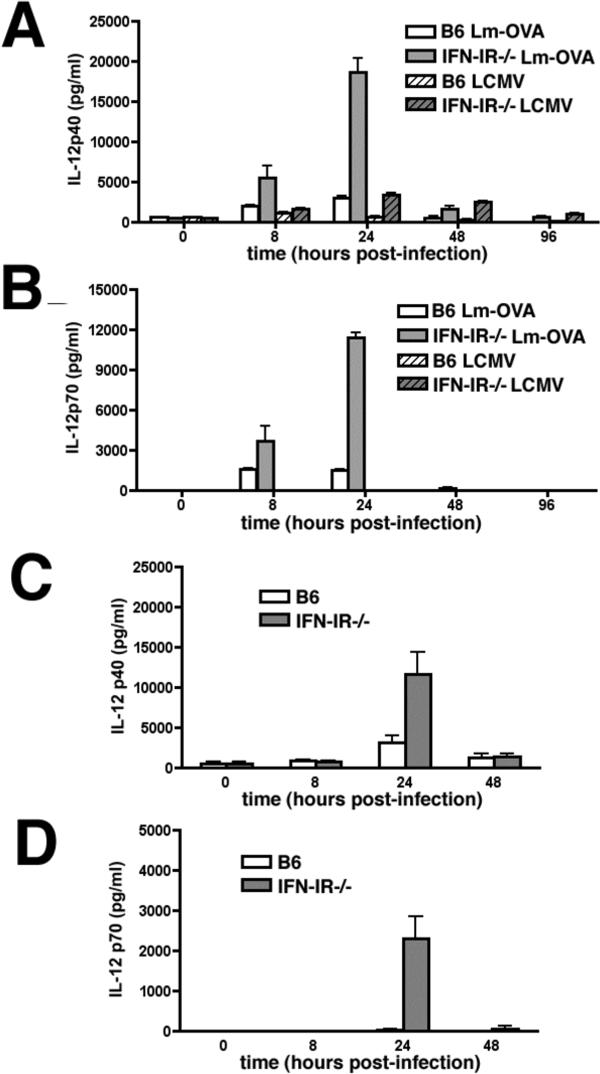
Mean serum concentration of IL-12P40 (A,C) or IL-12P70 (B,D) within the first 96 hours after infection with Lm-OVA (104 CFUs), LCMV (2×105 PFUs) (A, B) or Lm-OVA ΔactA (106 CFUs ) (C,D) in B6 or IFN-IR−/− mice. Each data point represents 3−4 mice per group, and is representative of at least two independent experiments. Bar, standard error.
Relative roles of IL-12 and IFN-I for IFN-γ production by antigen-specific T cells during Listeria infection
To determine the relative contribution of IL-12 produced in response to Lm infection on the antigen-specific adaptive T cell response in normal and IFN-IR−/− mice, we examined the effects of IL-12 depletion on the expansion and priming of CD8 and CD4 T cells for IFN-γ production in IFN-IR−/− compared with B6 mice. Since IL-12 plays a protective role, and IFN-I plays a detrimental role early in the course of WT Lm infection (10, 15-17, 24), a comparison of the adaptive T cell responses in mice that lack of each of these cytokines (or cytokine receptors) is complicated by potential differences in Lm antigen load. To bypass this potential limitation, we examined the immune response to the attenuated Lm mutant containing a targeted deficiency in actA instead of WT Lm. Within an infected cell, unipolar expression of ActA by Lm facilitates and is required for bacterium to recruit host cell actin into organized tails allowing for intracellular spread leading to a productive infection (25). We and others have demonstrated that infection with the ΔactA Lm mutant is able to prime Lm-specific CD8 and CD4 T cells, and yet is non-lethal even at relatively high inocula (up to 106 CFUs) in mice deficient in components of innate immunity such as MyD88, IFN-γ, or TNF-α receptor normally essential for protection from WT Lm infection (20, 26, 27). To verify that the absence of either IL-12 or IFN-IR signaling does not significantly alter the Lm load following infection with Lm-OVA ΔactA, we examined the number of bacteria in the spleen of IFN-IR−/−, IL-12P40−/−, and control mice following infection with 106 CFUs of Lm-OVA ΔactA (Figure 3). For Lm infection, the amount of antigen 24 hours after infection as reflected by the amount of live bacteria is an important determinant of the magnitude of the ensuing T cell response since administration of antimicrobials before this time point abrogates the response and administration of antimicrobials after this time point has no effect (28). For either IFN-IR−/− or IL-12P40−/− mice compared with B6 mice, no differences in Lm CFUs could be detected in the spleen 24 hours after infection with 106 CFUs of Lm-OVA ΔactA; and by day 8 after infection no Lm could be recovered (Figure 3). Moreover, consistent with what we observe for Lm-OVA infection, infection with this inocula of Lm-OVA ΔactA triggered the production of IL-12P40 and IL-12P70 in both B6 and IFN-IR−/− mice with similar kinetics, and conferred a similar reciprocal increase in IL-12 serum concentration in IFN-IR−/− compared with B6 mice (Figure 2C,D). Therefore to overcome potential limitations related to differences in antigen load following infection in mice with increased resistance (IFN-IR−/− mice) or increased susceptibility (IL-12-deficient mice) to WT Lm infection, all subsequent experiments analyzing the endogenous adaptive T cell response were performed with the ΔactA mutant in Lm.
Figure 3.
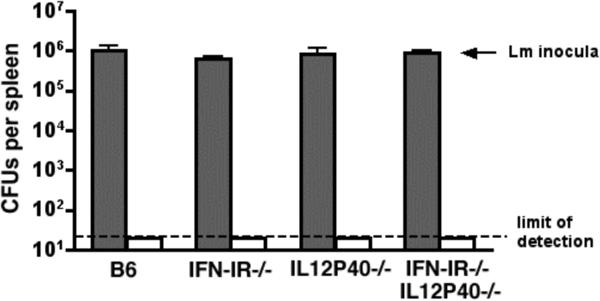
Number of recoverable bacteria per mouse spleen 24 hours (shaded bars) and 8 days (clear bars) after infection with 106 Lm-OVA ΔactA in B6, IFN-IR−/−, IL12P40−/−, and IFN-I−/−IL12P40−/− mice. Each bar represents 4−6 mice per group, and is representative of two independent experiments. Bar, standard error.
To examine the relative contribution of IL-12 on the antigen specific T cell response after Lm infection, IFN-IR−/− and B6 were treated with the anti-mouse IL-12P40 antibody (hybridoma clone C17.8) or an isotype control antibody one day prior to infection with Lm-OVA ΔactA. Eight days after infection, similar expansion of OVA-specific CD8 T cells and priming of OVA-specific CD8 T cells to produce IFN-γ could be detected in B6, B6 depleted of IL-12, IFN-IR−/− mice, or IFN-IR−/− depleted of IL-12 (Figure 4A-D). These data suggest that during Lm infection, neither IL-12 nor IFN-I is required for expansion or priming antigen-specific CD8 T cells for IFN-γ production. In contrast, examination of IFN-γ production by antigen-specific CD4 T cells in these same mice revealed important and additive roles for IL-12 and IFN-I. In either IFN-IR−/− mice or B6 mice depleted of IL-12 compared with B6 control mice, there was a modest (∼50%), but significant (P < 0.05) reduction in both percentage and absolute number of IFN-γ producing CD4 T cells following stimulation with the LLO189−201 peptide (Figure 4E,F). However for IFN-IR−/− mice depleted of IL-12 compared with B6 mice treated with control antibody, there was a substantial reduction (∼75−80% reduction, P < 0.05) in both the percentage and number of IFN-γ producing CD4 T cells. To determine if these differences in percentage and absolute numbers of IFN-γ producing CD4 T cells (and the lack of differences for CD8 T cells) reflect differences in the total amount of IFN-γ produced by these cells, we measured the concentration of cytokine in culture supernatants of cells from B6 mice, B6 mice depleted of IL-12, IFN-IR−/− mice, IFN-IR−/− depleted of IL-12 after stimulation with the MHC class II (LLO189−201) or MHC class I (OVA257−264) peptides (Figure 4G). Following stimulation with the MHC class II, LLO189−201 peptide, there was an ∼70% reduction in concentration of IFN-γ in splenocytes from IFN-IR−/− mice or B6 mice depleted of IL-12 compared with splenocytes from B6 control mice (P < 0.05 for each group compared with B6), and a 90% reduction (P < 0.05) in IFN-γ production by splenocytes from IFN-IR−/− depleted of IL-12 compared with splenocytes from B6 control mice. For these same splenocytes, stimulation with the MHC class I, OVA257−264 peptide revealed no differences in IFN-γ production by splenocytes from IFN-IR−/− mice, B6 mice depleted of IL-12, or IFN-IR−/− mice depleted of IL-12 compared with splenocytes from B6 control mice (Figure 4G). Furthermore, the reduction in IFN-γ production by CD4 T cells in IFN-IR−/− mice, B6 mice depleted of IL-12, or IFN-IR−/− mice depleted of IL-12 was not associated with a reciprocal increase in antigen-specific IL-4 or IL-13 production (data not shown).
Figure 4.
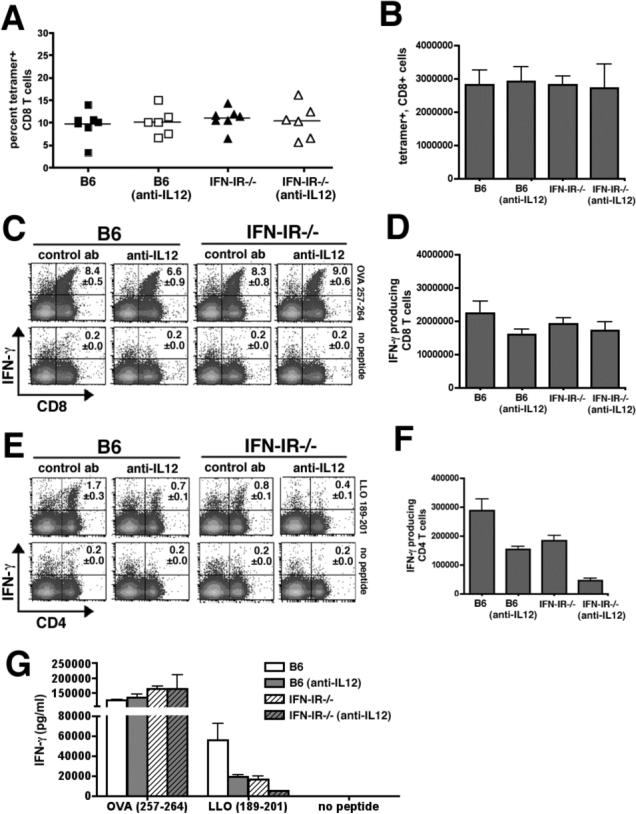
Expansion and IFN-γ production by CD8 and CD4 T cells day 8 after infection with 106 Lm-OVA ΔactA in B6 mice, B6 mice depleted of IL-12, IFN-IR−/− mice, or IFN-IR−/− depleted of IL-12. Percent (A) and total number (B) of OVA257−264 tetramer positive CD8 T cells per mouse spleen, and FACS plots demonstrating percent (C) and total number (D) of IFN-γ producing CD8 T cells per spleen after stimulation with OVA257−264 peptide or no peptide. FACS plots demonstrating percent (E) and total number (F) of IFN-γ producing CD4 T cells per spleen after stimulation with LLO189−201 peptide or no peptide. G. Concentration of IFN-γ in splenocytes culture supernatants 72 hours after stimulation with OVA257−264 peptide (MHC class I), LLO189−201 peptide (MHC class II), or no peptide determined by ELISA. Each data point represents 6−8 mice per experimental group from two independent experiments. Bar, one standard error.
In additional experiments we sought to more stringently test the role of IL-12 for priming CD4 and CD8 T cells for IFN-γ production. For these studies, the antigen specific immune response to Lm infection in IL-12-deficient mice (resulting from targeted deficiency in either the IL-12P40 or IL-12P35 subunits) and mice deficient in both IL-12P40 and IFN-IR (IFN-IR−/−IL12P40−/−) generated by intercrossing IL12P40−/− mice with IFN-IR−/− mice was examined. Combined deficiency in both IFN-IR and IL-12P40 did not affect Lm antigen load as there was no difference in the number of bacteria recovered in the spleen 24 hours after infection with 106 CFUs of Lm-OVA ΔactA in IFN-IR−/−IL12P40−/− compared with IFN-IR−/−, IL12P40−/−, or B6 mice (Figure 3). Consistent with results using IL-12 depleting antibody, examination of the antigen specific CD8 T cell response day 8 after infection with Lm-OVA ΔactA revealed similar percentages of IFN-γ producing CD8 T cells, absolute numbers of IFN-γ producing CD8 T cells, or accumulation of IFN-γ in splenocyte culture supernatants after stimulation with OVA257−264 peptide between B6, IFN-IR−/−, IL12P40−/−, IL12P35−/−, or IFN-IR−/−IL12P40−/− mice (Figure 5A,B,F). In these experiments, we also examined the CD8 T cell response by stimulation with a subdominant MHC class I peptide within LLO, LLO296−304 (29, 30). Although the percentage and total number of CD8 T cells that respond to the LLO296−304 peptide is much lower than response to OVA257−264, no differences were observed in either percentage or absolute number of CD8 T cells that produce IFN-γ in B6, IFN-IR−/−, IL12P40−/−, IL12P35−/−, or IFN-IR−/−IL12P40−/− mice to this subdominant antigen (Figure 5A,C). These data demonstrate that in response to Lm infection, neither IL-12 nor IFN-I signaling is required for priming antigen specific CD8 T cells for IFN-γ production.
Figure 5.

IFN-γ production by CD8 and CD4 T cells day 8 after infection with 106 Lm-OVA ΔactA in B6 mice, IFN-IR−/− mice, IL12P40−/−, IL12P35−/−, or IFN-IR−/−IL12P40−/− mice. FACS plots demonstrating percent (A) and total number (B,C) of IFN-γ producing CD8 T cells per spleen after stimulation with the MHC class I peptides: OVA257−264, LLO296−304, or no peptide control. FACS plots demonstrating percent (D) and total number (E) of IFN-γ producing CD4 T cells per spleen after stimulation with LLO189−201 peptide or no peptide. G. Concentration of IFN-γ in splenocytes culture supernatants 72 hours after stimulation with OVA257−264 peptide (MHC class I), LLO189−201 peptide (MHC class II), or no peptide as determined by ELISA. Each data point represents 4−6 mice per experimental group from two independent experiments. Bar, one standard error.
In these same mice infected with Lm-OVA ΔactA, we examined the antigen-specific CD4 T cell response. Consistent with results from these previous studies using IL-12 depleting antibody, IL12P40−/−, IL12P35−/−, and IFN-IR−/− mice compared with B6 mice each had partial (40−60%) reductions in percentage and absolute numbers of IFN-γ producing CD4 T cells after stimulation with the LLO189−201 peptide, while combined deficiency of both IL-12 and IFN-IR signaling in IFN-IR−/−IL12P40−/− mice revealed in a dramatic (95%) reduction in both percentage and absolute numbers of IFN-γ producing CD4 T cells (Figure 5D,E). Taken together, these data demonstrate that during Lm infection, IL-12 and IFN-I each have important and together have additive roles for priming IFN-γ production by CD4 T cells while neither IL-12 nor IFN-I are required for priming CD8 T cells for IFN-γ production.
Additive roles for IL-12 and IFN-I in priming transgenic CD4 T cells for IFN-γ production
IFN-I can be produced and exert biological responses in multiple cell types, and thereby influence the T cell response to infection directly by signaling on T cells or indirectly through stimulation of other cell types (8, 31, 32). Therefore, a comparison of CD4 T cell responses in Lm infected B6 and IFN-IR−/− mice does not specifically address the role of IFN-I signaling in antigenic specific T cells. To examine if the modest reductions in percentage and numbers of IFN-γ producing CD4 T cells in IFN-IR−/− mice, and the more dramatic reductions in IFN-γ production by CD4 T cells in mice with combined defects in both IL-12 and IFN-IR is due to defects in IFN-IR signaling on CD4 T cells, we measured expansion and IFN-γ production by IFN-IR+/+ and IFN-IR−/− TCR transgenic CD4 T cells after adoptive transfer into normal or IL-12-deficient mice. For these experiments, we utilized CD4 SMARTA cells specific for LCMV GP61−80, and transferred an equal number of IFN-IR+/+ SMARTA and IFN-IR−/− SMARTA cells into either B6 or IL12P35−/− mice. These mice were then infected with a recombinant strain of Lm expressing the MHC class II LCMV antigen, Lm-GP61−80. As expected, for cells transferred into B6 or IL-12P35−/− mice that were uninfected, SMARTA donor cells were recovered at a relatively low frequency (∼5−10% of transferred cells) (Figure 6A). By day 5 post infection, both IFN-IR+/+ and IFN-IR−/− SMARTA cells expanded ∼10−20-fold in both B6 and IL12P35−/− recipient mice, and at day eight post-infection, the numbers of SMARTA cells had undergone significant contraction in both B6 and IL12P35−/− mice. At the peak of expansion, the response of SMARTA donor T cell population to stimulation with the GP61−80 peptide was examined in splenocytes from B6 and IL12P35−/− mice. In both B6 and IL12P35−/− mice, there was only a modest reduction in the percentage of IFN-γ producing SMARTA IFN-IR+/+ compared with IFN-IR−/− cells (Figure 6B), however there was a more dramatic (37±6 compared with 14±4, P < 0.05) reduction in IFN-γ production for SMARTA IFN-IR+/+ cells in B6 mice compared with IFN-IR−/− cells in IL12P35−/− mice. Importantly, the reduction in IFN-γ production by IFN-IR−/− SMARTA cells in either IL12P35−/− or B6 mice compared with IFN-IR+/+ cells in B6 mice is not due to general defects in cell-activation because under the same stimulation conditions, there were no differences in CD40 ligand expression by these cells (Figure 6B). Taken together, these results demonstrate that during Lm infection, IFN-I acting directly on CD4 T cells primes them for IFN-γ production, and IFN-I and IL-12 synergize to prime CD4 T cells for maximal IFN-γ production.
Figure 6.
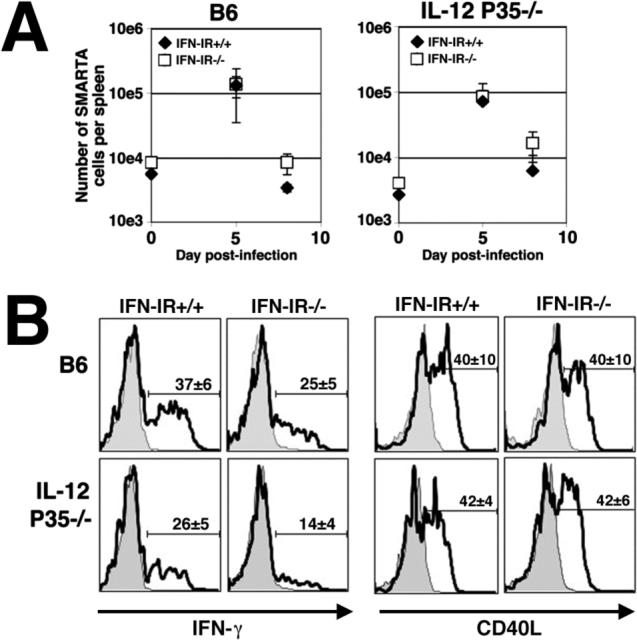
A. Number of SMARTA CD4 T cells (IFN-IR+/+, Thy1.1+ closed diamond; IFN-IR−/−, Thy1.1+Thy1.2+, open square) in congenic (Thy1.2+) B6 or IL-12 deficient (IL12P35−/−) recipient mice prior to infection (day 0), and day 5 and 8 after infection with rLm-GP61−80. B. IFN-γ production and CD40 ligand (CD40L) expression by IFN-IR+/+ (Thy1.1) or IFN-IR−/− (Thy1.1, Thy1.2) SMARTA transgenic cells day 5 after infection with rLm-GP61−80 in B6 or IL-12-deficient (IL12P35−/−) mice after in vitro stimulation with GP61−80 peptide (line histogram) or no peptide (filled histogram). The numbers in each histogram represent the mean ± standard error for three mice per experimental group, and are representative of three independent experiments with similar results. Bar, one standard error.
DISCUSSION
In this study we utilized the well-characterized Lm infection model in mice to examine the individual and combined roles of IFN-I and IL-12 in priming antigen specific T cells for IFN-γ production after in vivo infection. We first compared the number and percentage of antigen-specific IFN-γ producing CD8 and CD4 T cells in Lm infected B6 mice, IFN-IR−/− mice, B6 mice depleted of IL-12, and IFN-IR−/− mice depleted of IL-12. These experiments showed that the percentage and number of IFN-γ producing antigen specific CD8 T cells is not substantially affected by individual or combined defects in IL-12 or IFN-IR signaling. In contrast, both the percentage and number of IFN-γ producing CD4 T cells were partially reduced (∼50%) in both IFN-IR−/− and B6 mice treated with anti-IL-12 antibody, and substantially reduced (∼90%) in IFN-IR−/− mice treated with anti-IL-12 antibody. These results with IL-12 depleting antibody in normal and IFN-IR−/− mice were confirmed using mice with targeted deficiency in IL-12, and in mice deficient with targeted deficiency in both IL-12 and IFN-IR. We then performed further experiments to examine the direct action of IFN-I on antigen specific CD4 T cells by comparing the response of adoptively transferred IFN-IR+/+ versus IFN-IR−/− TCR transgenic CD4 T cells in Lm infected B6 or IL-12 deficient mice. These experiments demonstrated that in the presence of IL-12, IFN-I stimulation of CD4 T cells contributes to IFN-γ production only partially; while in the absence of IL-12, IFN-I stimulation of CD4 T cells plays a more significant role in IFN-γ production. Together these results demonstrate that during Lm infection, IL-12 and IFN-I act synergistically for priming CD4 T cells for IFN-γ production, while neither IFN-I nor IL-12 are required for priming CD8 T cells for IFN-γ production.
The precise mechanisms that govern the differentiation of CD4 T cells into IFN-γ producing Th1 or IL-4/IL-13 producing Th2 cells after infection in vivo are incompletely understood. IL-12 is essential for IFN-γ production and Th1 differentiation by CD4 T cells in the context of immunization with dead antigens (e.g., KLH or toxoplasma extract) and some infections (e.g., Lm, Mycobacterium avium, Leishmania major,) while for other infections (e.g., LCMV) a deficiency of IL-12 appears to have little or no effect on IFN-γ production by antigen-specific CD4 T cells (9-12, 33). Herein we demonstrate that this alternative IL-12 independent pathway for IFN-γ production by antigen specific CD4 T cells during Lm infection is mediated by IFN-I. To our knowledge, this is the first report demonstrating functional redundancy between IL-12 and IFN-I in priming IFN-γ production and Th1 differentiation of CD4 T cells in response to any infection or immunization. Importantly, in the absence of IL-12 and IFN-I signaling, Lm specific CD4 T cells do not have a reciprocal increase in production of the Th2 cytokines IL-4 or IL-13. These data suggest for Lm infection despite the absence signals that prime CD4 T cells to undergo a Th1 differentiation pathway, additional Th2 “promoting” signals are necessary for priming antigen-specific CD4 T cells to produce cytokines such as IL-4 and IL-13.
The reciprocal regulation of IL-12 and IFN-I and their functional redundancy in priming CD8 T cell for IFN-γ production has been previously described after infection with LCMV (7). In this study, depletion of IL-12 or IFN-I, or targeted deficiency in IL-12 or IFN-IR signaling reciprocally increased the concentration of the other cytokine. Therefore depletion of either IL-12 or IFN-I alone had little or no effect, whereas combined ablation of both IL-12 and IFN-I resulted in dramatic reductions in IFN-γ production by LCMV specific CD8 T cells. Overall, the data we present here are consistent with their conclusions that IL-12 and IFN-I play functionally redundant roles for priming CD8 T cells during LCMV infection. However, our finding that IFN-IR−/− mice (backcrossed extensively to B6) compared with B6 mice have reduced CD8 T cell expansion after LCMV infection is inconsistent with their finding of a relatively normal CD8 T cell response in IFN-IR−/− mice on a 129s/v background compared with 129s/v mice. The reasons for this difference are unclear, but may relate to mouse strain specific differences in IL-12 production following LCMV infection. Indeed, in our hands, even after infection with a ∼10-fold increased inocula of virus used in our present study, we could not detect biologically active IL-12P70 production in response to LCMV infection in either B6 or IFN-IR−/− mice. Thus, the dramatic reduction in LCMV-specific CD8 T cells in IFN-IR−/− compared with B6 mice that we describe is most likely due to the lack of biologically active IL-12 produced in response to LCMV infection in these mice.
Stimulation of naïve CD8 T cells in vitro with artificial antigen presenting cells coated with TCR + peptide and costimulation molecules does not trigger proliferation or IFN-γ production; but under these same conditions, addition of either IL-12 or IFN-I results in both proliferation and IFN-γ production (2, 34, 35). Our finding that antigen-specific CD8 T cells have no defects in expansion or IFN-γ production in the absence of both IL-12 and IFN-IR suggest that Lm infection triggers other cytokines or immune pathways that can provide a functionally redundant third signal for priming CD8. A likely candidate for this cytokine is IL-18 since it can trigger activation and IFN-γ production by T cells in the absence of IL-12 in other contexts (36-38). However, when tested in vitro with artificial APCs coated with TCR and costimulation molecules, IL-18 could not restore activation of naïve CD8 T cells (2). We are currently investigating whether this reflects inherent differences between priming during in vivo infection and culture in vitro, and if priming T cells in the absence of IL-12 and/or IFN-IR will impart a functional defect in their ability to confer protective immunity in response to WT Lm rechallenge.
Acknowledgements
We thank Drs. C.B. Wilson, M. Mathis, T. Kollmann, and A. Hajjar for helpful discussions and critical reading of the manuscript, Drs. M.J. Bevan and C. Surh for providing SMARTA mice, and Dr. H. Shen for providing recombinant Lm strains. This work was supported in part by the National Institutes of Health grants 1K08 HD051584 (to SSW), 1R01AI053146 and R21AI051386 (to KMK) and by funds from the Washington National Primate Center and the University of Washington Department of Immunology. CHD is supported by Cancer Research Institute's training grant.
Footnotes
Publisher's Disclaimer: This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org
REFERENCES
- 1.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 2.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 3.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 4.Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol. 1997;159:591–598. [PubMed] [Google Scholar]
- 5.Van Parijs L, Perez VL, Biuckians A, Maki RG, London CA, Abbas AK. Role of interleukin 12 and costimulators in T cell anergy in vivo. J Exp Med. 1997;186:1119–1128. doi: 10.1084/jem.186.7.1119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, Trinchieri G, Caux C, Garrone P. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201:1435–1446. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cousens LP, Peterson R, Hsu S, Dorner A, Altman JD, Ahmed R, Biron CA. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J Exp Med. 1999;189:1315–1328. doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jankovic D, Kullberg MC, Hieny S, Caspar P, Collazo CM, Sher A. In the absence of IL-12, CD4(+) T cell responses to intracellular pathogens fail to default to a Th2 pattern and are host protective in an IL-10(−/−) setting. Immunity. 2002;16:429–439. doi: 10.1016/s1074-7613(02)00278-9. [DOI] [PubMed] [Google Scholar]
- 10.Brombacher F, Dorfmuller A, Magram J, Dai WJ, Kohler G, Wunderlin A, Palmer-Lehmann K, Gately MK, Alber G. IL-12 is dispensable for innate and adaptive immunity against low doses of Listeria monocytogenes. Int Immunol. 1999;11:325–332. doi: 10.1093/intimm/11.3.325. [DOI] [PubMed] [Google Scholar]
- 11.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, Stewart C, Sarmiento U, Faherty DA, Gately MK. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 12.Oxenius A, Karrer U, Zinkernagel RM, Hengartner H. IL-12 is not required for induction of type 1 cytokine responses in viral infections. J Immunol. 1999;162:965–973. [PubMed] [Google Scholar]
- 13.Busch DH, Pamer EG. T lymphocyte dynamics during Listeria monocytogenes infection. Immunol Lett. 1999;65:93–98. doi: 10.1016/s0165-2478(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 14.Stockinger S, Reutterer B, Schaljo B, Schellack C, Brunner S, Materna T, Yamamoto M, Akira S, Taniguchi T, Murray PJ, Muller M, Decker T. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol. 2004;173:7416–7425. doi: 10.4049/jimmunol.173.12.7416. [DOI] [PubMed] [Google Scholar]
- 15.O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 19.Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus-specific MHC-class II-restricted TCR-transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol. 1998;28:390–400. doi: 10.1002/(SICI)1521-4141(199801)28:01<390::AID-IMMU390>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 20.Way SS, Kollmann TR, Hajjar AM, Wilson CB. Cutting edge: protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J Immunol. 2003;171:533–537. doi: 10.4049/jimmunol.171.2.533. [DOI] [PubMed] [Google Scholar]
- 21.Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med. 2006;203:933–940. doi: 10.1084/jem.20060045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 23.Seki E, Tsutsui H, Tsuji NM, Hayashi N, Adachi K, Nakano H, Futatsugi-Yumikura S, Takeuchi O, Hoshino K, Akira S, Fujimoto J, Nakanishi K. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J Immunol. 2002;169:3863–3868. doi: 10.4049/jimmunol.169.7.3863. [DOI] [PubMed] [Google Scholar]
- 24.Tripp CS, Gately MK, Hakimi J, Ling P, Unanue ER. Neutralization of IL-12 decreases resistance to Listeria in SCID and C.B-17 mice. Reversal by IFN-gamma. J Immunol. 1994;152:1883–1887. [PubMed] [Google Scholar]
- 25.Brundage RA, Smith GA, Camilli A, Theriot JA, Portnoy DA. Expression and phosphorylation of the Listeria monocytogenes ActA protein in mammalian cells. Proc Natl Acad Sci U S A. 1993;90:11890–11894. doi: 10.1073/pnas.90.24.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White DW, Badovinac VP, Fan X, Harty JT. Adaptive immunity against Listeria monocytogenes in the absence of type I tumor necrosis factor receptor p55. Infect Immun. 2000;68:4470–4476. doi: 10.1128/iai.68.8.4470-4476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 28.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–6839. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 29.Bahjat KS, Liu W, Lemmens EE, Schoenberger SP, Portnoy DA, Dubensky TW, Jr., Brockstedt DG. Cytosolic Entry Controls CD8+-T-Cell Potency during Bacterial Infection. Infect Immun. 2006;74:6387–6397. doi: 10.1128/IAI.01088-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geginat G, Schenk S, Skoberne M, Goebel W, Hof H. A novel approach of direct ex vivo epitope mapping identifies dominant and subdominant CD4 and CD8 T cell epitopes from Listeria monocytogenes. J Immunol. 2001;166:1877–1884. doi: 10.4049/jimmunol.166.3.1877. [DOI] [PubMed] [Google Scholar]
- 31.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 32.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 33.Mattner F, Magram J, Ferrante J, Launois P, Di Padova K, Behin R, Gately MK, Louis JA, Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- 34.Schmidt CS, Mescher MF. Peptide antigen priming of naive, but not memory, CD8 T cells requires a third signal that can be provided by IL-12. J Immunol. 2002;168:5521–5529. doi: 10.4049/jimmunol.168.11.5521. [DOI] [PubMed] [Google Scholar]
- 35.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, Akira S, Nakanishi K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 37.Neighbors M, Xu X, Barrat FJ, Ruuls SR, Churakova T, Debets R, Bazan JF, Kastelein RA, Abrams JS, O'Garra A. A critical role for interleukin 18 in primary and memory effector responses to Listeria monocytogenes that extends beyond its effects on Interferon gamma production. J Exp Med. 2001;194:343–354. doi: 10.1084/jem.194.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller U, Kohler G, Mossmann H, Schaub GA, Alber G, Di Santo JP, Brombacher F, Holscher C. IL-12-independent IFN-gamma production by T cells in experimental Chagas’ disease is mediated by IL-18. J Immunol. 2001;167:3346–3353. doi: 10.4049/jimmunol.167.6.3346. [DOI] [PubMed] [Google Scholar]