

Abstract
The majority of gastrointestinal stromal tumors (GISTs) are characterized by oncogenic gain-of-function mutations in the receptor tyrosine kinase (RTK) c-KIT with a minority in PDGFRα. Therapy for GISTs has been revolutionized by the use of the selective tyrosine kinase inhibitor imatinib mesylate (IM). For the subset (∼10-15%) of GISTs that lack oncogenic mutations in these receptors, the genetic changes driving tumorigenesis are unknown. We recently reported that the gene encoding the insulin-like growth factor 1 receptor (IGF-1R) is amplified in a subset of GISTs, and the IGF-1R protein is over-expressed in wild-type and pediatric GISTs. In this report we present a more complete picture of the involvement of components of the insulin-like growth factor-signaling pathway in the pathogenesis of GISTs. We also discuss how the IGF pathway may provide additional molecular targets for the treatment of GISTs that respond poorly to IM therapy.
Keywords: GISTs, insulin growth factor, imatinib mesylate, tyrosine kinase inhibitors, NVP-AEW541
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gut, with an estimated annual incidence of 3,300-6,000 cases in the United States.1 Clinically, diagnosis of GIST is typically confirmed by immunohistochemical staining of CD117, which recognizes the 145 kDa transmembrane glycoprotein KIT.2 The coding gene, c-KIT, is the normal cellular homologue of a viral oncogene (v-Kit, Hardy Zuckerman 4 feline sarcoma viral oncogene homologue). The majority of primary GISTs contain gain-of-function mutations in KIT (∼70%) or in the related RTK PDGFRα (∼10%).3-8 KIT mutations in GISTs cluster in exon 11, encoding the juxtamembrane domain of the receptor, and in exon 9 encoding the extracellular domain, with less frequent mutations in exon 13 encoding the ATP binding site and exon 17 encoding the activation loop; mutations in PDGFRα are found in homologous sequences in exons 12 and 18. These mutations lead to constitutive receptor auto-phosphorylation and activation of downstream signaling pathways (PI3-kinase/AKT pathways, MAP kinase, STAT)6, 9 that influence cell survival and proliferation, neoplastic transformation and malignant progression.10-13 The discovery of oncogenic RTK mutations in GISTs has led to the successful application of the selective tyrosine kinase inhibitor imatinib mesylate (IM) in the treatment of metastatic and/or unresectable GISTs.14, 15 However, a significant subset of GISTs lack mutations in KIT and PDGFRα, and these “wild-type” (WT) GISTs, along with certain mutant GISTs (i.e. exon 9 and 17 KIT mutants) are more refractory to IM treatment than exon 11 mutants.1, 16-18 Only ∼28% of wild type GISTs, and 38% of exon 9 mutants, show an objective response to imatinib therapy, as compared to 71% of exon 11 KIT mutants, and these patients exhibit shorter time-to-progression and decreased overall survival benefit than those with “IM-sensitive” tumors.15 This seems to be true as well for the subset of GISTs that arise in pediatric patients, which as a class are more likely to be wild-type for both receptors.19 A current focus for the field then is to search for genetic determinants of GIST pathogenesis, other than KIT or PDRGR-α, that may be addressed by molecularly targeted therapeutics.
Several recent papers suggest that members of the insulin-like growth factor (IGF) system may play a role in GIST pathogenesis.20-23 These studies coincide with the development of drugs targeting this pathway, some of which are already in the clinic for other cancers. The IGF signaling system has been implicated in a variety of cancers, and the reader is directed to recent reviews on the potential involvement of the IGF pathway in tumorigenesis.24-27 The IGF system consists of the receptors IGF-1R and IGF-2R, two ligands (growth factors IGF-1 and IGF-2), and six binding proteins (IGF-BP1-6) (Figure 1). IGF-1, produced mainly in the liver, circulates as an endocrine hormone, but it is also synthesized in target tissues where it works in a paracrine/autocrine fashion. Binding of IGF-1 (or IGF-2) to the type 2 membrane tyrosine kinase IGF-1R leads to receptor activation through auto-phosphorylation of intracellular tyrosine residues, to which adaptor proteins such as the insulin receptor substrates (IRS) 1-4 and Shc dock. This leads to activation of downstream signaling cascades (PI3-kinase/AKT/mTOR, RAF-1/MEK/MAPK) triggering protein synthesis, as well as anti-apoptotic and proliferative pathways (see Figure 1 and ref. 26). IGF-2R lacks the tyrosine kinase domain and acts as a scavenger for IGF-2, regulating its interaction with IGF-1R. The binding proteins, IGF-BP1-6, also produced in the liver, circulate and bind to the IGFs with varying affinities, thus regulating the interaction of the IGFs with their receptors. The system is somewhat more complex, as hybrid receptors exist consisting of heterodimers of IGF-1R and insulin receptor (IR) monomers; these hybrid receptors can be activated by IGF-1, IGF-2, and to a lesser extent by insulin itself. While insulin and the insulin receptor serve to maintain glucose homeostasis, the IGF system plays a vital role in normal growth and development.
Figure 1. The insulin-like growth factor system and its potential role in tumorigenesis.

The IGF system ligands IGF-1, IGF-2, and insulin (Ins) circulate and interact with binding proteins (IGF-BP) or bind to the membrane-bound receptors IGF-1R, IGF-2R, the insulin receptor itself (IR) or hybrid receptors (IGF-1R/IR). Ligand binding results in receptor autophosphorylation and activation of downstream signaling pathways leading to cell proliferation, survival, and metastasis.
Various lines of evidence suggest that the IGF system may play a role in cancer cell growth and metastasis. Numerous studies focusing mainly on IGF-1, IGF-2 and the IGF-1R have demonstrated that these genes are over-expressed as compared to normal tissue in cancer cells, including brain, breast, GI tract, ovary, prostate and others. IGF-2 over-expression is of special interest in sarcomas, where activation of the normally imprinted maternal allele, a common epigenetic modification first described in Wilms tumors28, leads to an increase in IGF-2 expression and is associated with increased cell proliferation and tumor risk in rhabdomyosarcoma as well as other cancers.29 Multiple epidemiological studies have shown a correlation between high serum levels of IGFs and the risk for various cancers, and studies in some cancers make the case for an association between high IGF levels and tumor progression (for a summary see ref. 26). The case for the IGF type 1 receptor is not as clear, where for example, in breast cancer studies there are conflicting results concerning the prognostic and clinical importance of IGF-1R levels.30-33 The observed correlation between outcome and IGF component expression levels seen in these studies may result from the ability of the IGF pathway to influence metastatic potential. In vitro studies suggest that at the molecular level the IGF system may be able to interact with other signaling pathways to promote processes such as vascularization (through VEGF in crosstalk with the hypoxia response pathway), as well as extra-cellular matrix remodeling and tumor cell invasiveness through interactions with metalloproteinases or the urokinase plasminogen activation system.34-39 Animal models as well support the role of the IGF system in cancer growth. Forced over-expression with the use of an IGF-1 transgene promotes prostate cancer progression in TRAMP mice40, while in rats increased IGF-1 levels resulting from exposure to high levels of sex hormones leads to progression to adenocarcinoma of the prostate.41 On the other hand, animal models with decreased levels of activation of the IGF pathway (for example lit/lit, dw/dw, LID mice) have a lower cancer incidence when challenged with carcinogenic inducers such as dimethylbenz[α]anthracene and diethylnitrosamine.42, 43 These models indicate that forced expression or activation of the IGF system can indeed influence tumorigenesis in cancer cells and animal models, but understanding the role of IGF signaling in specific cancers will depend on a thorough understanding of the interactions between all the components of the pathway within the cellular mileau.
Expression of IGF Signaling Components in GISTs
Interest in the IGF pathway in our group began with the discovery of low-level copy number gain (3 to 8 copies per cell) in the IGF-1R locus using Fluorescent In Situ Hybridization (FISH) analysis of WT and mutant GIST samples.21 Genomic quantitative PCR (qPCR) analysis was used to confirm amplification (3-4 copies) of the gene in a subset of GISTs, including both mutant and WT. In our study, amplification of IGF-1R was more frequently detected in WT samples (7 of 10) as compared to c-KIT or PDGFRα-mutated samples (5 of 18). Immunoblot analysis of 17 tumor samples (15 mutant, 2 WT) revealed the protein was present and phosphorylated in all samples, but was overexpressed 10- to 30-fold in WT GISTs compared with mutant GISTs. Since that report, we have examined IGF-1R protein expression in additional GIST tumor samples and found that IGF-1R was similarly overexpressed in 75% of WT samples (Figure 2). We also were able to detect IGF-1R amplification and over-expression in a tumor specimen from a pediatric GIST patient, the latter observation in agreement with a previous study.44 Pediatric GISTs are an intriguing subset of GISTs that are found almost exclusively in females with a multifocal gastric presentation, and generally lack detectable mutations in c-KIT and PDGFRα.44 In a molecular characterization of a panel of pediatric GISTs compared to adult WT and adult gastric GISTs, Agaram and colleagues found over-expression of a number of genes including IGF-1R and the IGF-122, suggesting that this pathway may be further up-regulated in pediatric as compared to adult WT GISTs. In addition, IGF-2 was determined to be one of the top genes to discriminate GISTs from other sarcomas.45
Figure 2. IGF-1R expression in GIST biopsies.

Immunoblot assays of 27 consecutive fresh-frozen GIST biopsies using an anti-IGF-1R antibody. 100 µg of WCE from each sample was subjected to immunoblotting. c-KIT or PDGFRα genotype for each GIST is listed below (+ or −). Asterisks indicate WT GISTs.
We have since extended our analysis to the other described members of the IGF pathway, including the IGF-2R, the two insulin-like growth factors, and the six binding proteins. Table 1 summarizes a qPCR analysis of these genes performed on eight (8) c-KIT- and PDGFRα-mutation negative fresh-frozen GIST specimens, including one pediatric WT, and thirteen (13) mutated samples. We find that IGF-1R mRNA is over-expressed > 17 to 19-fold in WT versus mutant GISTs (with a P value = 0.0013 using HPRT as RNA control), which is in agreement with our previous immunoblot assays.21 IGF-2R mRNA is also expressed at higher levels in the WT samples analyzed (∼ 2-fold, P = 0.0268). In contrast, IGF-1 mRNA is expressed at very low levels in these samples, with little difference between WT and mutant samples. IGF-2 transcript was expressed at much higher levels than IGF-1 in virtually all samples and > 6 fold higher in mutants than in WT, although without statistical correlation. Interestingly, the levels of IGF-2 transcript detected in GISTs was significantly higher relative to the other IGF pathway transcripts, such as IGF-1R and IGF-2R suggesting that IGF-2 protein may be rapidly degraded and/or metabolized in GISTs (data not shown). Interestingly, a number of reports in the literature have implicated abnormally high plasma levels of IGF-2 with occurrences of non-hyperinsulinemic hypoglycemia in some GIST patients (see ref. 23 and references within). Our qPCR data and IHC data (see below) are in agreement with the suggestion that overexpression of IGF-2 transcript may lead to abnormal protein accumulation impacting glucose homeostasis.
Table 1.
Relative expression of IGF system genes in WT v. Mutant GISTsa
| IGF Pathway Gene | Fold difference in expression, WT/mutant (range) | |
|---|---|---|
| v. HPRT control | v. b-actin control | |
| IGF-1R | 17.6 (3.84 - 81.0)b | 19.2 (2.27 - 162)c |
| IGF-2R | 2.11 (1.03 - 4.31)c | 1.98 (0.39 - 10.0) |
| IGF-1 | 1.92 (0.68 - 5.41) | 2.09 (0.16 - 28.0) |
| IGF-2 | 0.14 (0.016 - 1.22) | 0.15 (0.010 - 2.32) |
| IGF-BP1 | 1.25 (0.56 - 2.82) | 0.45 (0.19 - 1.04) |
| IGF-BP2 | 1.83 (0.60 - 5.59) | 1.88 (0.12 - 28.79) |
| IGF-BP3 | 1.98 (0.82 - 4.79) | 2.04 (0.16 - 26.4) |
| IGF-BP4 | 1.67 (0.76 - 3.71) | 1.73 (0.21 - 14.4) |
| IGF-BP5 | 5.46 (1.29 - 23.1) | 5.63 (0.32 - 99.8) |
| IGF-BP6 | 0.62 (0.18 - 2.06) | 1.75 (0.014 - 214) |
RNA expression levels were normalized to HPRT or b-actin RNA. RNA extracted from imatinib-naïve flash-frozen GIST specimens was subjected to quantitative RT-PCR analysis using TaqMan gene expression assays on an ABI PRISM 7900 HT Sequence Detection System, Applied Biosystems, Foster, CA.
P value < 0.005 as determined by Student T test.
P value < 0.05 as determined by Student T test.
In a recent report examining the expression of IGF-1 and IGF-2 in a set of 94 GISTs, Braconi and colleagues were able to correlate the intensity of staining of both growth factors with tumor parameters such as mitotic index and risk potential, and low IGF-1 and IGF-2 expression was found to be associated with a better trend in disease-free survival.23 Interestingly, we failed to detect significant levels of IGF-1 by IHC in the 34 GIST specimens using either the polyclonal rabbit anti-IGF1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) used in the aforementioned study or a monoclonal anti-IGF-1 antibody (R&D System Company), although both antibodies were able to detect IGF-1 on sarcoma tissue microarrays (Figure 3 and data not shown). Therefore, our IGF-1 IHC data are consistent with low qPCR levels detected by qRTPCR in our specimens. Consistent with the Braconi study, we were able to detect IGF-2 in most of our samples (Figure 4). For these samples we attempted to correlate our staining intensities with mutation status or with malignant risk potential46, but neither parameter was significantly correlated with IGF-2 staining intensity, albeit in a relatively small sample set as compared to the previous study. Furthermore, our study did not have clinical outcome data so we could not confirm or reject their observations. We also performed IHC for IGF-1R expression in a larger sample set of GIST samples (n = 34, 22 mutant, 12 WT) and confirmed that IGF-1R is significantly overexpressed in WT GISTs as compared to mutant GISTs (P = 0.0084, two-sided Fisher's exact test). In addition, a strong correlation was observed between weak IGF-1R and weak IGF-2 expression by IHC (P = 0.0002).
Figure 3. IGF-1 expression in various sarcomas.

IHC staining was performed as described in ref. 21 on tissue from a leiomyosarcoma, liposarcoma, WT GIST and mutant GIST. The primary IGF-1 antibody was used at a 1:100 dilution and was purchased from Santa Cruz Biotechnology Inc. Top two panels show positive IGF-1 staining in the leiomyosarcoma and liposarcoma, whereas the two bottom panels show no IGF-1 expression in both GIST samples (WT and mutant). Pictures were taken with 200x magnification.
Figure 4. Expression of IGF components in WT and mutant GISTs by immunohstochemistry (IHC).
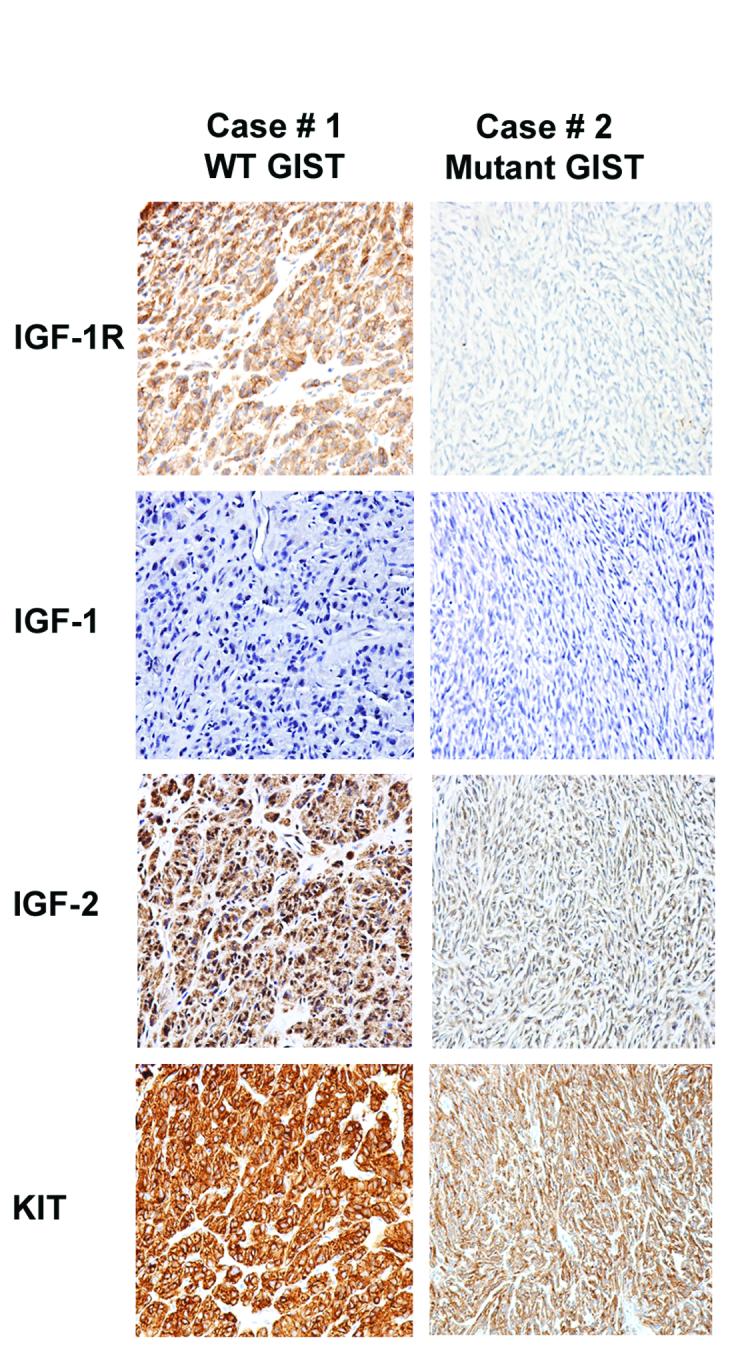
IGF-1R, IGF-1, IGF-2 and KIT expression in WT and mutant GISTs as analyzed by IHC. Primary antibodies used include IGF-2 (1:500 dilution, Abcam), IGF-1R (1:50 dilution, Cell Signaling) and KIT (1:2000 dilution, Dako).
We were also interested in evaluating whether the expression of genes in the IGF pathway are altered in response to treatment with imatinib. Trent and colleagues have demonstrated that expression of the IGF-BP3 is regulated differentially in GISTs in response to short-term IM treatment.20 They compared the level of IGF-BP3 mRNA in biopsy specimens and surgically resected GISTs, before and after 3-7 days of IM therapy, and correlated these results to early response to IM therapy as indicated by FDG-PET scan of the tumors. In a small sample set of paired samples, they found an ∼ two-fold increase in IGF-BP3 mRNA in imatinib-responsive tumors (mainly c-KIT exon 11 mutants) following IM treatment, while the gene was down-regulated in 3 of 3 non-responsive tumors that lacked exon 11 mutations.
We evaluated whether expression of IGF-1R, IGF-2R, IGF-1 and IGF-2 change upon treatment with IM. To do this, we utilized fresh-frozen biopsies from 4 GIST patients obtained prior to and after up to 8 weeks of IM therapy. qPCR analysis was performed and gene expression was compared in the pre-IM versus the post-IM samples. Table 2 shows that IGF-1R expression increases in all post-IM samples, ranging from ∼2- to ∼47-fold. IGF-1 expression also increased upon IM treatment in 3 out of 4 samples, ranging from ∼2-fold to 69-fold. When IGF-2R and IGF-2 transcript expression were examined the opposite effect was observed. In all 4 samples, both IGF-2R and IGF-2 mRNA levels decreased upon IM treatment. This general trend, an increase in IGF-1R and IGF-1 gene expression and decreased expression of IGF-2R and IGF-2 upon IM therapy, observed in a small sample set, is of early interest and will require further validation in a larger set of samples.
Table 2.
Effect of IM on expression of IGF Pathway Genesa
| Sample | KIT/PDGFRα genotype | IGF1R | IGF2R | IGF1 | IGF2 |
|---|---|---|---|---|---|
| 1 | wild type | 17.3 | 0.86 | 69 | 0.15 |
| 2 | not available | 1.60 | 0.50 | 0.66 | 0.60 |
| 3 | KIT exon 11 mutant | 29.1 | 0.11 | 2.18 | 0.04 |
| 4 | KIT exon 11 mutant | 46.5 | 0.43 | 63.9 | 0.55 |
Values given are ratios of RNA expression levels in post/pre samples. RNA, extracted from flash-frozen biopsy specimens and from resected tumors following up to 8 weeks of IM treatment was subjected to quantitative RT-PCR analysis using TaqMan gene expression assays. RNA levels were first normalized to HPRT.
Targeting the IGF Pathway in GISTs
IGF pathway genes are differentially expressed in WT and c-KIT- and PDGFRα-mutated GISTs, and there is evidence that the level of expression of some of these genes may be predictive of risk potential, immediate response to IM therapy, and long-term outcome in patients. With respect to IGF-1R expression and its contribution to downstream signaling pathways, we showed that the small molecule tyrosine kinase inhibitor NVP-AEW541 (Novartis), which has activity against IGF-1R47, decreases ligand-stimulated IGF-1R auto-phosphorylation in the GIST cell lines T1 and 882 without affecting phospho-KIT levels, and furthermore decreases the phosphorylation of downstream signaling molecules AKT, MAPK1/2 and GSKβ.21 In addition, we found using standard cytotoxicity assays that these two cell lines, previously shown to exhibit differential sensitivity to IM treatment48, were in fact equally sensitive to the IGF-1R inhibitor. There was an additive effect when the cell lines were treated with the two drugs in combination.21 These results suggest that the use of IGF-1R inhibitors should be investigated not only as a single agent targeting those GISTs that do not respond well to imatinib and that have been shown to overexpress IGF-1R, but also as a combination agent to enhance therapeutic activity in IM-responsive tumors. It is possible that such an approach of inhibiting both KIT/PDGFR and IGF-1R pathways could extend the length of time that imatinib-based therapy is effective, which is typically 2 years.14, 49, 50 This may benefit patients, as second line therapies, such as sunitinib (Sutent; SU112488; Pfizer), have been shown to control disease for approximately 6 months.51
Although NVP-AEW541 may never reach human clinical trials, a number of other compounds targeting IGF-1R are currently in development to treat a wide variety of tumors. OSI-906 (OSI), another small molecule inhibitor of IGF-1R is currently in phase I trials to treat solid tumors. In addition, several monoclonal antibodies to IGF-1R are also in clinical development to treat many tumor types such as lymphoma, bladder, breast, gastric, head/neck, melanoma, soft tissue sarcomas, among others.24 A recent phase I dose escalation study of CP-751,871 in refractory solid tumors demonstrated the compound to be well tolerated and inhibited growth of tumors in the majority of patients in the study.52 Phase II and phase III trials for CP-751,871 are currently underway to treat NSCLC, HRPC and breast malignancies.24 IMC-A12 has shown potential in targeting a wide range of cancers in xenograft models as a single agent, however, much greater effects have been achieved when used in combination with other cytotoxic agents.53 IMC-A12 is currently in phase II trial to treat colorectal cancer.24 There is also evidence demonstrating the potential value of combining IGF-1R inhibitors with additional signal transduction inhibitors. Bertrand and colleagues showed that IMC-A12 could be use to induce apoptosis in hematopoietic cells as a single agent, but in combination with small molecule inhibitors targeting either the AKT or MAPK pathways this effect was augmented.54 These findings suggest a potential for combined approaches in GIST treatment using IGF-1R targeting compounds.
In addition to previously published reports showing IGF-1R inhibition successfully inhibits rhabdomyosarcoma cell growth55-57, two recent reports have suggested that, based on preclinical studies, targeting IGF-1R in other sarcomas show promise in achieving anti-tumor effects.58, 59 A number of IGF-1R inhibitors are currently in clinical trials for treatment of Ewing's sarcoma, osteosarcoma, as well as rhabdomyosarcoma, offering even further evidence of the need to get these compounds into clinical trials in GISTs. Early trials of R1507 as well as other IGF-1R antibodies demonstrated objective responses in Ewing's sarcoma; a phase II study is further defining the efficacy of R1507 in advanced Ewing's Sarcoma, synovial sarcoma, rhabdomyosarcoma, osteosarcoma as well as some other chemo-insensitive sarcomas. In addition, based on our recent findings and others, it may also be interesting to study the effects of targeting the ligands of IGF-1R in GISTs.
Final Thoughts
Although imatinib mesylate has greatly improved the oncologist's abilities to treat GIST, the clinical reality is that many GISTs are non-responsive to treatment and nearly half of metastatic GISTs do not shrink significantly using current drug regimens. Furthermore, primary and secondary drug-resistance has become a major clinical concern.60 As such, second line therapies are necessary to overcome primary and secondary resistance to imatinib mesylate. Sunitinib, an orally administered, multi-targeted tyrosine kinase inhibitor with activity against KIT, PDGFRα, vascular endothelial growth factor receptor (VEGFR) and FLT-1 is available for treatment following progression on IM. Objective response rates to sunitinib are low, 10%, with an additional 50% of patients achieving disease stabilization. The benefits of sunitinib; however, are limited with median progression free survival of 6 months. Additional agents are in clinical trials, primarily targeting KIT and PDGFR. It is not clear how we will identify patients for therapy with each of these tyrosine kinase inhibitors. Alternate targets may prove to have a greater impact on the management of advanced GIST (see review in ref. 61).
It is becoming apparent that the current treatment paradigm for the clinical management of GIST must change (Figure 5A). Personalizing patient care will be dependent on the molecular characterization of individual tumors prior to treatment, e.g., pre-selecting patients for treatment with imatinib based on c-KIT and PDGFRα mutational status. Recent studies have suggested that patients with exon 9 mutations may benefit from higher doses of IM.62 Furthermore, studies of gene expression profiles may one day help to identify gene signatures that could designate patients' GISTs that are likely to be more responsive to IM63 (Rink et al., unpublished data) or other targeted agents, thus paving the way to potentially more effective treatment models in the future. Finally, our recent studies have highlighted the need to apply genomic and proteomic approaches to identifying additional factors driving tumorigenesis that may ultimately help identify new markers of tumor behavior, prognosis, and drug response (Figure 5B). Demonstrating that IGF-1R is amplified and overexpressed in a portion of GISTs, especially those lacking c-KIT or PDGFRα mutations is an additional step to better understanding the pathogenesis of this disease. More importantly, we demonstrated that imatinib sensitive and resistant GIST cells may be responsive to IGF-1R-targeted therapies, suggesting an alternative and/or complementary therapeutic regimen in the clinical management of GIST, especially in tumors that respond less favorably to imatinib-based therapy, including pediatric cases. These findings are particularly exciting given the number of agents targeting IGF-1R that are currently being tested in clinical trials. Clinical trials using IGF-1R-targeted therapies for imatinib-refractory GIST patients, initially focusing on adult and pediatric GIST patients lacking c-KIT or PDGFRα mutations are in development.
Figure 5. Current and future paradigms for treatment of GISTs.

A. First line therapy for treatment of GISTs is IM, however, primary and secondary resistance has become a major clinical obstacle. Currently the second line therapy is sunitinib, which shows limited benefits and has led to the movement of additional agents into clinical trials. B. The future paradigm for GIST treatment will most likely include a molecular characterization (i.e. genotypic status, expression profiles) of individual tumors before treatment decision. This type of characterization has the potential to lead to more effective treatment models in the future.
Grant supports
This work was supported in part by NIH grants (CA106588 and a supplement to U10 CA21661) to A.K.G., an award by the FCCC Translational Research Committee to M.v.M. and A.K.G as part of the FCCC core grant (P30 CA006927), an NIH Training Grant (Institutional NRSA) Appointment (CA009035-31) to L.R, and by Tania Stutman and the GIST Cancer Research Fund.
Abbreviations
- GIST
gastrointestinal stromal tumor
- RTK
receptor tyrosine kinase
- PDGFR
platelet-derived growth factor receptor
- IGF
insulin-like growth factor
Footnotes
Conflict of interest statement: There are no conflicts of interest.
References
- 1.Corless CL, Heinrich MC. Molecular Pathobiology of Gastrointestinal Stromal Sarcomas. Annu Rev Pathol. 2007 doi: 10.1146/annurev.pathmechdis.3.121806.151538. [DOI] [PubMed] [Google Scholar]
- 2.Tarn C, Godwin AK. The molecular pathogenesis of gastrointestinal stromal tumors. Clin Colorectal Cancer. 2006;6(Suppl 1):S7–17. doi: 10.3816/ccc.2006.s.002. [DOI] [PubMed] [Google Scholar]
- 3.Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, Hibbard MK, Chen CJ, Xiao S, Tuveson DA, Demetri GD, Fletcher CD, Fletcher JA. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–21. [PubMed] [Google Scholar]
- 4.Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160:1567–72. doi: 10.1016/S0002-9440(10)61103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 6.Tarn C, Merkel E, Canutescu AA, Shen W, Skorobogatko Y, Heslin MJ, Eisenberg B, Birbe R, Patchefsky A, Dunbrack R, Arnoletti JP, von Mehren M, Godwin AK. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res. 2005;11:3668–77. doi: 10.1158/1078-0432.CCR-04-2515. [DOI] [PubMed] [Google Scholar]
- 7.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 8.Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008 doi: 10.1111/j.1365-2559.2008.02977.x. [DOI] [PubMed] [Google Scholar]
- 9.Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs) Oncogene. 2004;23:3999–4006. doi: 10.1038/sj.onc.1207525. [DOI] [PubMed] [Google Scholar]
- 10.Atfi A, Prunier C, Mazars A, Defachelles AS, Cayre Y, Gespach C, Bourgeade MF. The oncogenic TEL/PDGFR beta fusion protein induces cell death through JNK/SAPK pathway. Oncogene. 1999;18:3878–85. doi: 10.1038/sj.onc.1202734. [DOI] [PubMed] [Google Scholar]
- 11.Shen Y, Devgan G, Darnell JE, Jr., Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A. 2001;98:1543–8. doi: 10.1073/pnas.041588198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Ilasaca M, Li W, Uren A, Yu JC, Kazlauskas A, Gutkind JS, Heidaran MA. Requirement of phosphatidylinositol-3 kinase for activation of JNK/SAPKs by PDGF. Biochem Biophys Res Commun. 1997;232:273–7. doi: 10.1006/bbrc.1997.6289. [DOI] [PubMed] [Google Scholar]
- 13.Kabarowski JH, Allen PB, Wiedemann LM. A temperature sensitive p210 BCR-ABL mutant defines the primary consequences of BCR-ABL tyrosine kinase expression in growth factor dependent cells. Embo J. 1994;13:5887–95. doi: 10.1002/j.1460-2075.1994.tb06934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–34. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 15.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 16.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 17.Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, Dimitrijevic S, Sciot R, Stul M, Vranck H, Scurr M, Hagemeijer A, van Glabbeke M, van Oosterom AT. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–95. doi: 10.1016/j.ejca.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 18.van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, Martens M, Webb A, Sciot R, Van Glabbeke M, Silberman S, Nielsen OS. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358:1421–3. doi: 10.1016/s0140-6736(01)06535-7. [DOI] [PubMed] [Google Scholar]
- 19.Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC, Fletcher JA. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67:9084–8. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- 20.Trent JC, Ramdas L, Dupart J, Hunt K, Macapinlac H, Taylor E, Hu L, Salvado A, Abbruzzese JL, Pollock R, Benjamin RS, Zhang W. Early effects of imatinib mesylate on the expression of insulin-like growth factor binding protein-3 and positron emission tomography in patients with gastrointestinal stromal tumor. Cancer. 2006;107:1898–908. doi: 10.1002/cncr.22214. [DOI] [PubMed] [Google Scholar]
- 21.Tarn C, Rink L, Merkel E, Flieder D, Pathak H, Koumbi D, Testa JR, Eisenberg B, von Mehren M, Godwin AK. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc Natl Acad Sci U S A. 2008;105:8387–92. doi: 10.1073/pnas.0803383105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, Maki RG, DeMatteo RP, Besmer P, Antonescu CR. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204–15. doi: 10.1158/1078-0432.CCR-07-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braconi C, Bracci R, Bearzi I, Bianchi F, Sabato S, Mandolesi A, Belvederesi L, Cascinu S, Valeri N, Cellerino R. Insulin-like growth factor (IGF) 1 and 2 help to predict disease outcome in GIST patients. Ann Oncol. 2008;19:1293–8. doi: 10.1093/annonc/mdn040. [DOI] [PubMed] [Google Scholar]
- 24.Ryan PD, Goss PE. The emerging role of the insulin-like growth factor pathway as a therapeutic target in cancer. Oncologist. 2008;13:16–24. doi: 10.1634/theoncologist.2007-0199. [DOI] [PubMed] [Google Scholar]
- 25.Sachdev D, Yee D. The IGF system and breast cancer. Endocr Relat Cancer. 2001;8:197–209. doi: 10.1677/erc.0.0080197. [DOI] [PubMed] [Google Scholar]
- 26.Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 27.Miller BS, Yee D. Type I insulin-like growth factor receptor as a therapeutic target in cancer. Cancer Res. 2005;65:10123–7. doi: 10.1158/0008-5472.CAN-05-2752. [DOI] [PubMed] [Google Scholar]
- 28.Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun K, Maw MA, Smith PJ, Reeve AE. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms' tumour. Nature. 1993;362:749–51. doi: 10.1038/362749a0. [DOI] [PubMed] [Google Scholar]
- 29.Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest. 1994;94:445–8. doi: 10.1172/JCI117344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ueda S, Tsuda H, Sato K, Takeuchi H, Shigekawa T, Matsubara O, Hiraide H, Mochizuki H. Alternative tyrosine phosphorylation of signaling kinases according to hormone receptor status in breast cancer overexpressing the insulin-like growth factor receptor type 1. Cancer Sci. 2006;97:597–604. doi: 10.1111/j.1349-7006.2006.00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Railo MJ, von Smitten K, Pekonen F. The prognostic value of insulin-like growth factor-I in breast cancer patients. Results of a follow-up study on 126 patients. Eur J Cancer. 1994;30A:307–11. doi: 10.1016/0959-8049(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 32.Peyrat JP, Bonneterre J, Laurent JC, Louchez MM, Amrani S, Leroy-Martin B, Vilain MO, Delobelle A, Demaille A. Presence and characterization of insulin-like growth factor 1 receptors in human benign breast disease. Eur J Cancer Clin Oncol. 1988;24:1425–31. doi: 10.1016/0277-5379(88)90332-x. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu C, Hasegawa T, Tani Y, Takahashi F, Takeuchi M, Watanabe T, Ando M, Katsumata N, Fujiwara Y. Expression of insulin-like growth factor 1 receptor in primary breast cancer: immunohistochemical analysis. Hum Pathol. 2004;35:1537–42. doi: 10.1016/j.humpath.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 34.Bauer TW, Liu W, Fan F, Camp ER, Yang A, Somcio RJ, Bucana CD, Callahan J, Parry GC, Evans DB, Boyd DD, Mazar AP, Ellis LM. Targeting of urokinase plasminogen activator receptor in human pancreatic carcinoma cells inhibits c-Met- and insulin-like growth factor-I receptor-mediated migration and invasion and orthotopic tumor growth in mice. Cancer Res. 2005;65:7775–81. doi: 10.1158/0008-5472.CAN-05-0946. [DOI] [PubMed] [Google Scholar]
- 35.Dunn SE, Torres JV, Nihei N, Barrett JC. The insulin-like growth factor-1 elevates urokinase-type plasminogen activator-1 in human breast cancer cells: a new avenue for breast cancer therapy. Mol Carcinog. 2000;27:10–7. doi: 10.1002/(sici)1098-2744(200001)27:1<10::aid-mc3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 36.Feldser D, Agani F, Iyer NV, Pak B, Ferreira G, Semenza GL. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999;59:3915–8. [PubMed] [Google Scholar]
- 37.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–11. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 38.Mira E, Manes S, Lacalle RA, Marquez G, Martinez AC. Insulin-like growth factor I-triggered cell migration and invasion are mediated by matrix metalloproteinase-9. Endocrinology. 1999;140:1657–64. doi: 10.1210/endo.140.4.6623. [DOI] [PubMed] [Google Scholar]
- 39.Zhang D, Brodt P. Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene. 2003;22:974–82. doi: 10.1038/sj.onc.1206197. [DOI] [PubMed] [Google Scholar]
- 40.DiGiovanni J, Kiguchi K, Frijhoff A, Wilker E, Bol DK, Beltran L, Moats S, Ramirez A, Jorcano J, Conti C. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc Natl Acad Sci U S A. 2000;97:3455–60. doi: 10.1073/pnas.97.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang YZ, Wong YC. Sex hormone-induced prostatic carcinogenesis in the noble rat: the role of insulin-like growth factor-I (IGF-I) and vascular endothelial growth factor (VEGF) in the development of prostate cancer. Prostate. 1998;35:165–77. doi: 10.1002/(sici)1097-0045(19980515)35:3<165::aid-pros2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 42.Yang XF, Beamer WG, Huynh H, Pollak M. Reduced growth of human breast cancer xenografts in hosts homozygous for the lit mutation. Cancer Res. 1996;56:1509–11. [PubMed] [Google Scholar]
- 43.Wu Y, Cui K, Miyoshi K, Hennighausen L, Green JE, Setser J, LeRoith D, Yakar S. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res. 2003;63:4384–8. [PubMed] [Google Scholar]
- 44.Prakash S, Sarran L, Socci N, DeMatteo RP, Eisenstat J, Greco AM, Maki RG, Wexler LH, LaQuaglia MP, Besmer P, Antonescu CR. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol. 2005;27:179–87. doi: 10.1097/01.mph.0000157790.81329.47. [DOI] [PubMed] [Google Scholar]
- 45.Baird K, Davis S, Antonescu CR, Harper UL, Walker RL, Chen Y, Glatfelter AA, Duray PH, Meltzer PS. Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer Res. 2005;65:9226–35. doi: 10.1158/0008-5472.CAN-05-1699. [DOI] [PubMed] [Google Scholar]
- 46.Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006;30:477–89. doi: 10.1097/00000478-200604000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Hopfner M, Huether A, Sutter AP, Baradari V, Schuppan D, Scherubl H. Blockade of IGF-1 receptor tyrosine kinase has antineoplastic effects in hepatocellular carcinoma cells. Biochem Pharmacol. 2006;71:1435–48. doi: 10.1016/j.bcp.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Tarn C, Skorobogatko YV, Taguchi T, Eisenberg B, von Mehren M, Godwin AK. Therapeutic effect of imatinib in gastrointestinal stromal tumors: AKT signaling dependent and independent mechanisms. Cancer Res. 2006;66:5477–86. doi: 10.1158/0008-5472.CAN-05-3906. [DOI] [PubMed] [Google Scholar]
- 49.Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 50.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–32. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 51.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 52.Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, Yap TA, Roberts ML, Sharma A, Gualberto A, Adjei AA, de Bono JS. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res. 2007;13:5834–40. doi: 10.1158/1078-0432.CCR-07-1118. [DOI] [PubMed] [Google Scholar]
- 53.Rowinsky EK, Youssoufian H, Tonra JR, Solomon P, Burtrum D, Ludwig DL. IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res. 2007;13:5549s–55s. doi: 10.1158/1078-0432.CCR-07-1109. [DOI] [PubMed] [Google Scholar]
- 54.Bertrand FE, Steelman LS, Chappell WH, Abrams SL, Shelton JG, White ER, Ludwig DL, McCubrey JA. Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia. 2006;20:1254–60. doi: 10.1038/sj.leu.2404217. [DOI] [PubMed] [Google Scholar]
- 55.Kalebic T, Tsokos M, Helman LJ. In vivo treatment with antibody against IGF-1 receptor suppresses growth of human rhabdomyosarcoma and down-regulates p34cdc2. Cancer Res. 1994;54:5531–4. [PubMed] [Google Scholar]
- 56.Kalebic T, Blakesley V, Slade C, Plasschaert S, Leroith D, Helman LJ. Expression of a kinase-deficient IGF-I-R suppresses tumorigenicity of rhabdomyosarcoma cells constitutively expressing a wild type IGF-I-R. International journal of cancer. 1998;76:223–7. doi: 10.1002/(sici)1097-0215(19980413)76:2<223::aid-ijc9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 57.Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou XM, Blattler WA, Chittenden T, Singh R. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63:5073–83. [PubMed] [Google Scholar]
- 58.Scotlandi K, Picci P. Targeting insulin-like growth factor 1 receptor in sarcomas. Current opinion in oncology. 2008;20:419–27. doi: 10.1097/CCO.0b013e328302edab. [DOI] [PubMed] [Google Scholar]
- 59.Magenau JM, Schuetze SM. New targets for therapy of sarcoma. Current opinion in oncology. 2008;20:400–6. doi: 10.1097/CCO.0b013e328303671d. [DOI] [PubMed] [Google Scholar]
- 60.Tarn C, Godwin AK. Molecular research directions in the management of gastrointestinal stromal tumors. Curr Treat Options Oncol. 2005;6:473–86. doi: 10.1007/s11864-005-0026-x. [DOI] [PubMed] [Google Scholar]
- 61.von Mehren M. Imatinib-refractory gastrointestinal stromal tumors: the clinical problem and therapeutic strategies. Curr Oncol Rep. 2006;8:192–7. doi: 10.1007/s11912-006-0019-3. [DOI] [PubMed] [Google Scholar]
- 62.Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Van Glabbeke M, Hagemeijer A, Judson I. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 63.Frolov A, Chahwan S, Ochs M, Arnoletti JP, Pan ZZ, Favorova O, Fletcher J, von Mehren M, Eisenberg B, Godwin AK. Response markers and the molecular mechanisms of action of Gleevec in gastrointestinal stromal tumors. Mol Cancer Ther. 2003;2:699–709. [PubMed] [Google Scholar]