

Abstract
Despite the significant successes in the area of anti-HBV agents, resistance and cross resistance against available therapeutics are the major hurdles in drug discovery. The present investigation is to understand the molecular basis of drug resistance conferred by the B and C domain mutations of HBV polymerase on the binding affinity of five anti-HBV agents [lamivudine (3TC, 1), adefovir (ADV, 2), entecavir (ETV, 3), telbivudine (LdT, 4) and clevudine (L-FMAU, 5)]. In this regard, homology modeled structure of HBV polymerase were used for minimization, conformational search and induced fit docking followed by binding energy calculation on wild type as well as on mutant-HBV polymerases (L180M, M204V, M204I, L180M+M204V, L180M-M204I). Our studies suggest a significant correlation between the fold resistances and the binding affinity of anti-HBV nucleosides. The binding mode studies reveals that the domain C residue M204 is closely associated with sugar/pseudosugar ring positioning in the active site. The positioning of oxathiolane ring of 3TC (1) is plausible due the induced fit orientation of the M204 residue in wild type, and further mutation of M204 to V204 or I204 reduces the final binding affinity which leads to the drug resistance. The domain B residue L180 is not directly close (~6Å) to the nucleoside/nucleoside analogs, but indirectly associated with other active-site hydrophobic residues such as A87, F88, P177 and M204. These five hydrophobic residues can directly affect on the incoming nucleoside analogs in terms of its association and interaction that can alter the final binding affinity. There was no sugar ring shifting observed in the case of adefovir (2) and entecavir (3), and the position of sugar ring of 2 and 3 is found similar to the sugar postion of natural substrate dATP and dGTP respectively. The exocyclic double bond of entecavir (3) occupied in the backside hydrophobic pocket (made by residues A87, F88, P177, L180 and M204), which enhances the overall binding affinity. The active site binding of LdT (4) and L-FMAU (5) showed backward shifting along with upward movement without enforcing M204 residue and this significant different binding mode makes these molecules as polymerase inhibitors, without being incorporated into the growing HBV-DNA chain. Structural results conferred by these L- and D-nucleosides, explored the molecular basis of drug resistance which can be utilized for future anti-HBV drug discovery.
1. Introduction
More than 350 million people are chronically-infected with hepatitis B virus (HBV), resulting about 1 million death per year (Lai et al. 2003b). HBV, a member of the hepadnavirus family, is an enveloped virus that contains a partially double stranded DNA genome (3 kbp). In addition to regular transcription and translation processes, it has a reverse transcription process similar to HIV. This is mediated by a single enzyme, catalyzing RNA- and DNA-dependent DNA polymerase, RNase H and protein priming activities (Seeger and Mason 2000). Analogous to HIV, the HBV polymerase is also a good target for inhibiting the viral replication. Several nucleoside-analogs such as lamivudine (1, 3TC, a cytosine L-nucleoside analog) (Dienstag et al. 1995), adefovir (2, ADV, a adenosine analog), entecavir (3, ETV, a carbocyclic guanosine analog) (de Man et al. 2001) and telbivudine (4, LdT, a thymidine L-nucleoside analog) (Lai et al. 2004) have been approved by the US-FDA for the treatment of chronic HBV infection (Figure 1).
Figure 1.
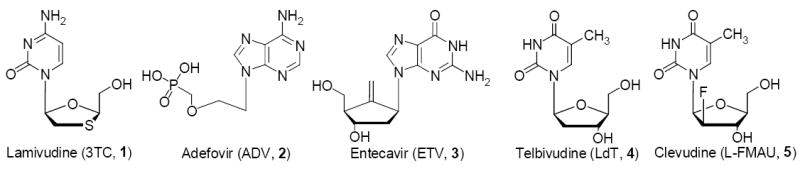
The chemical structure of potential HBV-polymerase inhibitors.
Previously, we have synthesized a number of L-nucleosides. Among which clevudine (5, L-FMAU, a thymidine L-nucleoside analog) has been discovered as a potent anti-HBV agent (Chu et al. 1995; Marcellin et al. 2004; Yoo et al. 2007a; Yoo et al. 2007b) and more recently it was approved for the treatment of chronic hepatitis B virus infection in South Korea in November 13, 2006. Currently, it is undergoing Phase III clinical trials in US and Europe (Marcellin, Mommeja-Marin et al. 2004).
Despite the above significant successes in the discovery of an anti-HBV agent (Ferir et al. 2008; Palumbo 2008), the critical issue is the development of drug resistance and cross resistance against available therapeutics. Recent studies reveal that adefovir resistance increases to 29% after 5 years of use (Locarnini et al. 2005). Therefore, increasing use of adefovir against lamivudine-resistant HBV infection can increase a risk of multidrug-resistant HBV. The development of drug resistance is not unexpected if viral replication continues during the current monotherapy. The prevention of resistance requires the adoption of strategies that can more effectively control the virus replication, including the combination therapy (Sasadeusz et al. 2007; Hui et al. 2008). For the past several years, our group has been involved in understanding the HBV drug resistance issue at molecular level by molecular modeling (Chong and Chu 2002; Yadav and Chu 2004).
Recently, several publications related to HBV drug resistance issue appeared on different classes of anti-HBV agents (Ayres et al. 2004; Bartholomeusz et al. 2004; Chong and Chu 2004; Tenney et al. 2004; Jacquard et al. 2006; Locarnini and Mason 2006). HBV-polymerase has several domains that are homologous to other RNA-dependent DNA polymerases (Poch et al. 1989; Lesburg et al. 1999). A revised numbering system for HBV polymerase in comparison to HIV RT proposed by Stuyver et al (Stuyver et al. 2001), has been generally accepted and it is being used in this article.
A current anti-HBV agent (1-5) causes a specific primary mutation during the treatment and the published biological data (Table 1) (Chin et al. 2001; Delaney et al. 2001a; Delaney et al. 2001b; Levine et al. 2002; Ono-Nita et al. 2002; Angus et al. 2003; Locarnini 2003; Angus and Locarnini 2004; Locarnini and Mason 2006) provides an opportunity to understand the structural and functional insight of the active-site of HBV-polymerase.
Table 1.
Antiviral activity (IC50 in μM) and fold resistance (FR)a of anti-HBV agents (1-5) against wild-type and 3TC-resistant HBV polymerases.(Chin, Shaw et al. 2001; Locarnini 2003; Brunelle et al. 2005)
| HBV-strain | 3TC (1) | ADV (2) | ETV (3) | LdT (4) | L-FMAU (5) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | FR | IC50 | FR | IC50 | FR | IC50 | FR | IC50 | FR | |
| Wild-type | 0.6 | 1.0 | 3.9 | 1 | 0.8 | 1 | 0.17 | 1 | 0.1 | 1 |
| L180M | 0.80 | 1.7 | 2.0 | 0.5 | na | 1b | 1.96 | >10 | >100 | >100 |
| M204V | 8.5 | 18 | 2.8 | 0.7 | na | 10b | na | na | 1.5 | 15 |
| M204I | >50 | >100 | 2.7 | 0.7 | na | na | 39.5 | >100 | >100 | >100 |
| L180M-M204Vc | >50 | >100 | 0.6 | 0.2 | 5.0 | 6 | 22.2 | >100 | >100 | >100 |
Fold resistance = (mutant IC50)/(wt IC50)
Separate study on ETV, shows that M204V mutation had 10 fold reduction on the potency of ETV, while L180M had no significant effect.(Levine, Hernandez et al. 2002)
Dual mutant 3TC resistant strain (L180M-M204V and L180M-M204I) having similar pattern of resistance profile against most of the potential compounds (1-5).(Das et al. 2001; Langley et al. 2007)
Several groups have developed the structural models of HBV RT based on HIV RT (Allen et al. 1998; Bartholomeusz et al. 1998; Das, Xiong et al. 2001), although homology models may not be accurate due to low sequence homology between HIV and HBV. Fortunately, the active site residues of HBV polymerase are highly conserved in with respect to HIV RT (Bartholomeusz, Tehan et al. 2004). Therefore HBV polymerase modeled structure can be utilized to draw some useful conclusions. A similar right hand structure (fingers, palm, and thumb subdomains) were assigned for the modeled structure of HBV polymerase with respect to HIV RT (Figure 2a). The catalytic site residues are highly conserved (Bartholomeusz, Tehan et al. 2004) and is responsible for polymerase residues-recognition with the template, primer or an incoming nucleotide analog.
Figure 2.
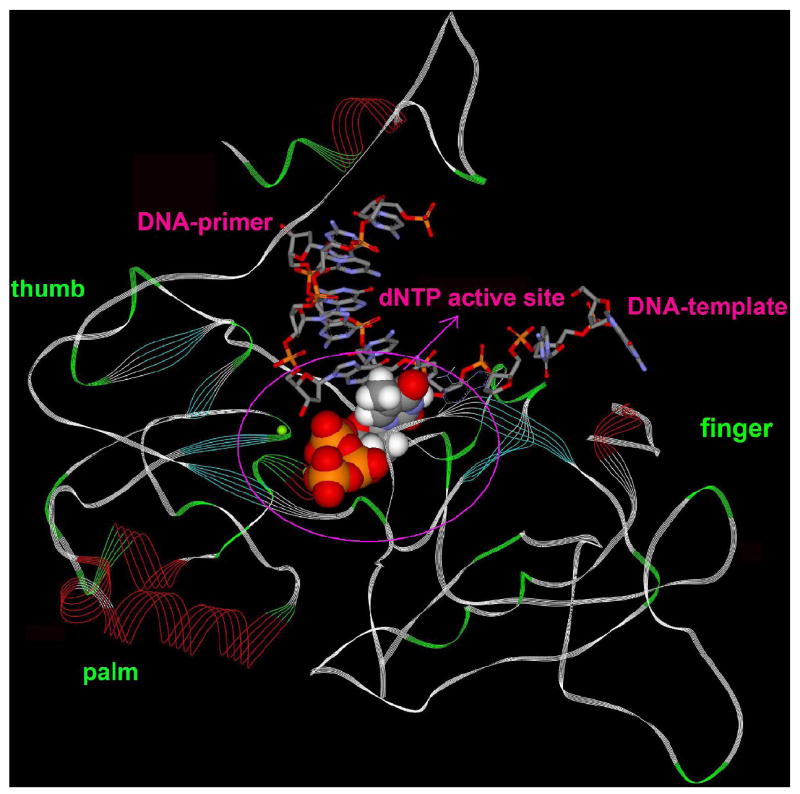
a. BV-polymerase (model structure) with DNA double strand (primer and template) and natural nucleoside, highlighting the position of catalytic site.
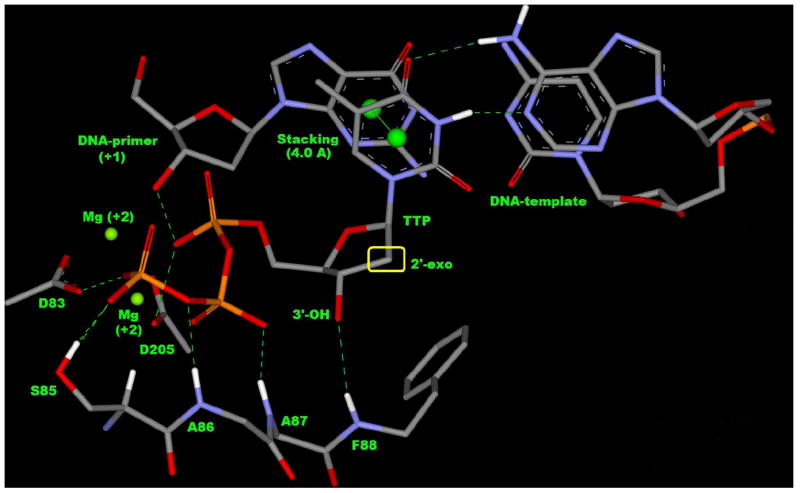
b. Binding mode of TTP (thymidine triphosphate) in the active site showing 2’-exo conformation of ribose, H-bonding (green dashed line), stacking (centroid distance = 4 Å) with DNA-primer (+1), base pairing with DNA-template by H-bond (green dashed line) and catalytic residues D83, D205 with two Mg+2 cation.
In our molecular modeling studies, we focused our attention on highly conserved regions surrounding the binding site of natural substrate thymidine triphosphate (TTP). The initial modeling of TTP highlighted its binding mode in the active site of wild type HBV-polymerase (Figure 2b). Some of the important non-covalent interactions (Table 2) that stabilize the complex (HBV-poly-DNA-TTP) and required for any molecule to fit into the active-site to behave as a perfect mimic of incoming natural nucleoside (TTP) are shown in Figure 2b. The strong hydrogen bonding network between the triphosphate moiety with HBV-polymerase residues (S85, A86, A87) is conserved for all nucleoside reverse transcriptase inhibitor-triphosphates (NRTI-TPs) and natural nucleosides.
Table 2.
Molecular recognition element (non-covalent interaction) required for NRTIs to mimic dNTP in HBV-polymerase based on Figure 2b (dTTP-binding).
| NRTIs | Interaction detail with HBV-polymerase |
|---|---|
| Base Pairing | Thymine base is forming two strong H-bond with Adenine of DNA-template. |
| Base Stacking | Thymine base is showing stacking interaction (Centroid Distance ~ 4Å) with the guanine base of DNA-primer (+1) nucleotide. |
| Two-metal-ion mechanism | Conserved catalytic residue D83 and D205 with two Mg+2 is responsible for dNTP addition to the DNA-primer (+1) for chain elongation. |
| Triphosphate positioning | Triphosphate positioning is mediated by H-bonding with residues S85, A86 and A87. |
Figure 2b also focuses on the two-metal-ion (Mg+2) mechanism (Steitz 1998) for facilitating the 3’O- attack on the α-phosphate with help of the conserved catalytic residues D83 and D205 for the addition of TTP to the primer (+1) for DNA chain elongation. Table 2 clearly describes the strong interaction (recognition elements) that may be the minimum criteria for any nucleoside analogs to become NRTIs or a potential drug candidate. A recent modeling study reveals the role of 3’-OH group in entecavir (3) to form the H-bond with the backbone of F88 (NH) (Langley, Walsh et al. 2007). In addition, entecavir (3) showed other conserved interactions as mentioned in Table 2, and these structural features make entecavir a perfect mimic of natural substrate.
The NRTIs for HIV or HBV include those compounds, which can mimic endogenous pyrimidine or purine nucleosides, and all of them are required for an intracellular phosphorylation to their triphosphate form. The first phosphorylation step is often considered as rate limiting, which is dependent on deoxynucleoside kinases, such as thymidine kinase, deoxycytidine kinase (dCK), and deoxyguanosine kinase (dGK). Deoxycytidine kinase does not discriminate between the enantiomeric (D & L) forms of substrates and is able to phosphorylate L-nucleosides (Krishnan et al. 2003).
In view of this, we conducted our molecular modeling calculations based on the triphosphate-form of NRTIs. The aim of present molecular modeling study is to elucidate the structural features of HBV-polymerase in terms of qualitative and quantitative changes conferred by B and C domain mutations (3TC; Lamivudine associated resistance: rtL180M and rtM204V/I) on the binding of different class of anti-HBV nucleosides (1-5).
2. Methods
HBV polymerase model has been constructed and refined according to the previously reported method (Das, Xiong et al. 2001; Chong and Chu 2002; Yadav and Chu 2004) using the crystal structure of HIV RT (PDB code: 1RTD) (Huang et al. 1998). The Composer Module of Sybyl 7.1 (Tripos Inc. 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA) and Prime Module of Schrödinger Suite 2006 (Schrödinger Inc. LLC, New York, NY) have been used to obtain the HBV polymerase model structure. The natural substrate thymidine triphosphate (TTP) and short chain of template-primer duplex (7/4) along with two Mg+2 cation were placed on the conserved active site of HBV RT polymerase according to HIV RT crystal structure (1RTD). The HBV polymerase model was subjected for 500 ps molecular dynamics using OPLS_2005 force field (Macromodel 9.1, Schrödinger Inc., LLC) to confirm the stability of the HBV polymerase model. The above model was then manually inspected for accuracy in the χ2 dihedral angle of Asn and His residues and the χ3 angle of Gln, and rotated by 180° when needed to maximize hydrogen bonding. All Asp, Glu, Arg, and Lys residues were checked for their charged state. The corrected model of HBV polymerase in terms of proper bond order, hydrogen atoms and charges was further subjected for a single run of minimization in protein preparation module of Maestro (Schrödinger, LLC) to relieve any clashes and also to optimize the overall structure ensuring the chemical correctness of the starting structure. The minimization was terminated when the RMSD reached a maximum value of 0.3 Å.
The starting ligand conformation of all the nucleoside triphosphates were obtained after performing a 5000 steps of Monte Carlo conformational search using Macromodel 9.1 (Schrödinger, LLC). The MMFFs force field has been used for the conformational search with GB/SA continuum water solvation model. This was followed by the Polak-Ribiere Conjugate Gradient (PRCG) energy minimization of 5000 steps or until the energy difference between subsequent structures was 0.05 kJ/mol.
The heterocyclic moiety of the fifth nucleotide in the template was modified to the appropriate base complementary (A-T, G-C) to the incoming nucleotide required for DNA-primer elongation. The HBV polymerase model contains TTP in the active site, therefore, the induced fit docking protocol (Sherman et al. 2006) has been used to obtained the model structure of natural nucleoside triphosphates (CTP, ATP, GTP and TTP) and anti-HBV compounds (1-5) for a comparative study.
The induced fit docking (IFD) was run from the Maestro 8.0 (Schrödinger, LLC) graphical user interface. In the first stage of induced fit docking protocol, Glide 4.0 (Schrödinger, LLC) was used to generate 20 initial poses through softened-potential docking step, which enabling tolerance of more steric clashes than in a normal docking run. The SP scoring function was used in all docking stages. Further in the second stage, a full cycle of protein refinement was performed through the Prime 1.5 modules (Schrödinger, LLC) to judge the active site conformational change around 5 Ǻ of the initial poses. The side-chains, as well as the backbone and ligand, underwent subsequent energy minimization using OPLS_2001 force field and Prime’s implicit solvent model. After successful active-site modification, refinement by prime and re-docking by Glide, (Halgren et al. 2004) the model structure has been generated.
Finally, the eMBrAcE module (MacroModel, v9.1, Schrödinger, LLC) has been used to determine the binding energy differences through minimization using MMFFs force field. All the calculations were performed with GB/SA continuum water solvation model. During eMBrAcE minimization, residues further away by more than 16 Å from the nucleoside binding site were frozen and residues from 6 Å to 16 Å were constrained by harmonic constraints. Only residues inside a 6 Å sphere from the nucleoside triphosphate were allowed to move freely. Minimization calculations were performed for 5000 steps or until the energy difference between subsequent structures was 0.05 Kj/mol. The above calculations (eMBrAcE) provided total energy difference (ΔETotal = Ecomplex − Eligand − Eprotein), which has been used for the comparative binding energy study as shown in tables.
Various mutants (L180M, M204V, M204I, L180M+M204V and L180M+M204I) of HBV polymerase were generated using builder module of Maestro (Schrödinger, LLC) and were subsequently subjected for all the modeling calculations as described above.
3 Results and Discussion
3.1 Lamivudine (3TC) in wild type HBV-polymerase
The extensive use of Lamivudine (or 3TC) [(-)-β-L-2’-3’ dideoxythiacytidine)] (1) for the treatment of chronic HBV infection resulted in the emergence of resistant mutants (24% after a one year therapy, while increased to 70% after four years of monotherapy) (Leung 2001; Lai et al. 2003a; Bottecchia et al. 2008). 3TC is a cytosine analog; therefore we performed molecular modeling studies of 3TC with respect to the natural substrate deoxycytosine triphosphate (dCTP) for their binding mode in order to understand the detail molecular mechanism of the resistance.
Figure 3a shows the overall binding mode of dCTP in HBV polymerase and it clearly indicates the similar pattern of binding mode, which is previously explained in the Figure 2b and Table 2 (TTP-binding mode). The cytosine base of dCTP has stacking interaction with the DNA-primer (+1) (gray) and the base-pairing with DNA-template (gray) as shown in Figure 3a. The two-metal ion mechanism (pink) is required for the DNA elongation and the triphosphate positioning is mediated by the interaction with S85, A86 and A87 residues (gold). The residues A87, F88, P177, L180 and M204 (Figure 3) are not directly involved with dCTP binding. However these residues are playing a crucial role in the recognition (Wilchek et al. 2006) of NRTIs for their binding affinity by altering hydrophobic and other non-covalent interactions (Williams et al. 2004). Further, L180 and M204 are important for detailed structural investigation as they are associated with the mutation during lamivudine (3TC) therapy.
Figure 3.
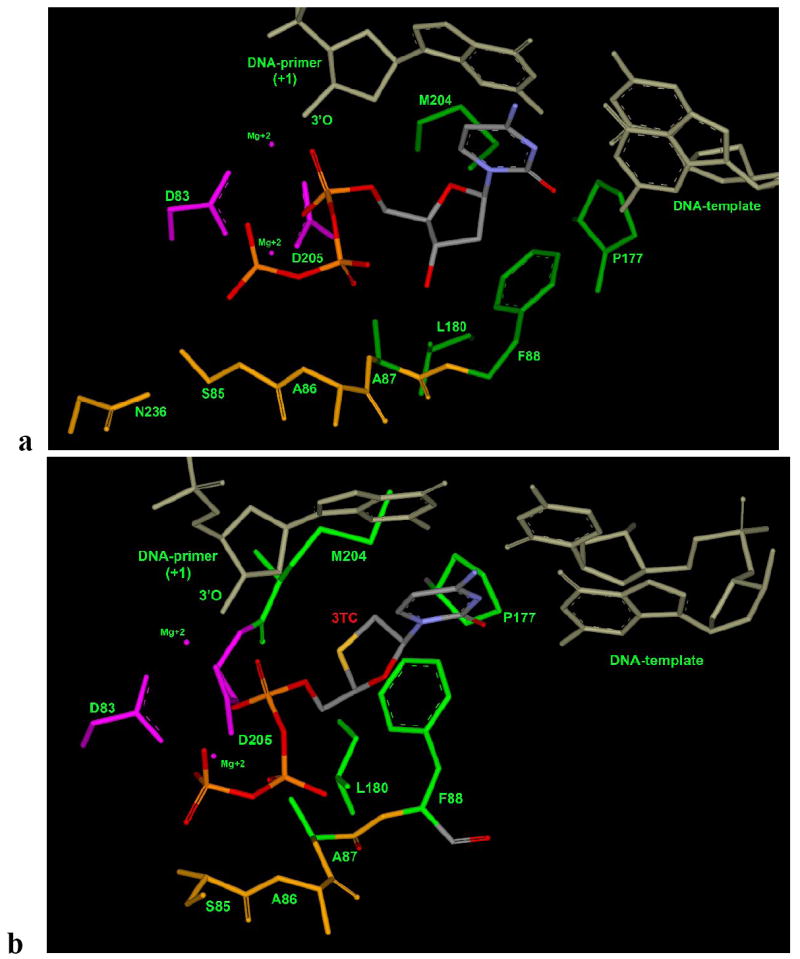
Natural substrate (Figure 3a: dCTP) and 3TC (Figure 3b: 3TC-TP binding mode in wild type HBV polymerase (wt-hbv): two conservered aspartic-acid (D83 and D205) with two-metal-ion (Mg+2) in pink color; residues N236, S85, A86, and A87 responsible for phosphate interaction are shown in gold color. Residues A87, F88, P177, L180 and M204 responsible for hydrophobic interaction and also for proper positioning the sugar ring are shown in green color.
3TC (1) is a cytosine analog with a L-oxathiolane ring, while dCTP is a cytosine analog with D-ribose. Therefore, these compounds are structurally different, and 3TC should not be expected to fit in the HBV-polymerase in exactly the same way as that of dCTP. In view of these differences, we used a recently developed induced fit methodology in place of rigid docking (McGovern 2007) by Schrödinger that has wide applications (Sherman, Day et al. 2006) to handle the receptor flexibility for a conformationally different ligand. The binding mode of 3TC, obtained through successful induced fit docking followed by minimization, shows that it occupies the active site properly and shows all the necessary interactions as mentioned in Table 2. The position of oxathiolane ring has been slightly shifted towards the DNA-primer nucleotide in comparison to ribose moiety of dCTP. Due to the absence of a 3’-OH group, 3TC looses one hydrogen-bond with the backbone (NH) of F88 causing shifting of F88 towards the oxathiolane ring of 3TC. This shifting of F88 enhances arene interaction (H…π interaction) with the oxathiolane ring. The structural studies clearly reveals that L-configuration, presence of a sulphur atom and absence of 3’-OH group in 3TC enforces M204 and F88 to adopt different conformation in comparison to the dCTP binding mode as shown in Figure 4 and Figure 5.
Figure 4.
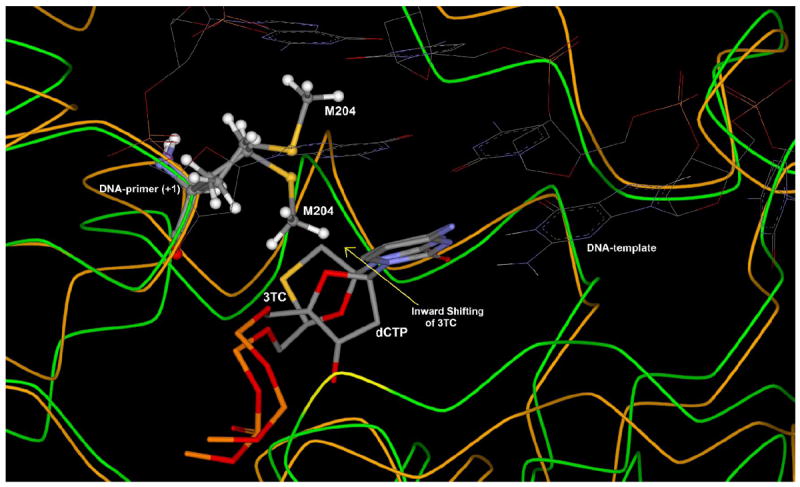
Superposition study of 3TC-HBV and dCTP-HBV complex after flexible docking, showing the different orientation of M204 to accommodate shifted oxathiolane ring of lamivudine triphosphate (3TC-TP) in the active site in comparison to dCTP. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
Figure 5.
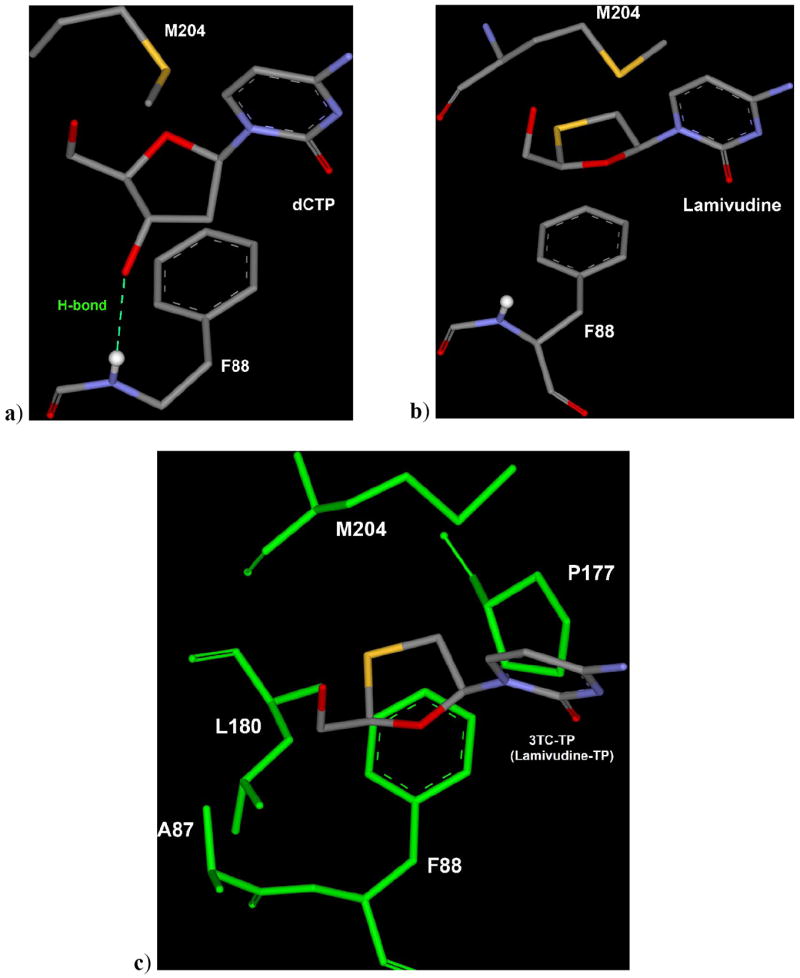
a) M204 residue of HBV-polymerase oriented towards sugar of dCTP and 3’-OH of dCTP forming H-bond with the backbone (NH) of F88. b) Lamivudine triphosphate (3TC-TP) binding mode showing different orientation of M204 and slight shifting of F88 in comparison to dCTP binding mode to allow the proper accommodation of oxathiolane ring. c) Sulphur-atom of oxathiolane ring of 3TC-TP utilizes the small backside hydrophobic pocket formed by A87, F88, P177, L180 and M204 residues (green). [triphosphate moiety not shown for the sake of clarity]
Binding mode of dCTP and 3TC in wild type HBV-polymerase suggests that 3TC can accommodate perfectly in the active site due to induced fit movement of M204, which allows inward shifting of oxathiolane ring of 3TC (Figure 4). Structural studies also suggest the important role of F88 residues to provide stability by stacking with the ribose/pseudo-ribose ring of incoming nucleotide in the active site of HBV-polymerase.
Thus, it is clear that 3TC has structural features to accommodate in the HBV-active site, which leads to significant activity against wild type HBV polymerase. Energy calculations are shown in Table 3 for 3TC in comparison to dCTP for wild type HBV polymerase.
Table 3.
Multi-ligand bimolecular association with energetics (eMBrAcE) calculation of dCTP and 3TC after induced fit docking and minimization in wild type HBV-RT.
| Compound | Energy Difference Results (ΔE: kj/mol) |
|||
|---|---|---|---|---|
| Electrostatic | VdWa | Total | Δ Eb | |
| DCTP | -5567.0 | 229.0 | -351.9 | -8.1 |
| Lamivudine triphosphate (3TC-TP) | -5018.9 | 126.3 | -360.0 | |
van der Waals interaction.
ΔE = Energy difference in comparison to dCTP.
The result of the energy correlations justifies the significant activity of 3TC in comparison to dCTP. Favorable vander Waals energy for 3TC in wild type HBV polymerase arises by the occupation of the sulphur atom and shifting of the oxathiolane ring into the hydrophobic pocket as shown in Figure 5c. The loss of electrostatic interaction in 3TC can be explained by missing one H-bond (Figure 5a and Figure 5b) due to absence of the 3’-OH group. Overall, the total energy difference is consistent with the in vitro anti-HBV activity of 3TC against wild type HBV (Table 1). It is concluded that the significant activity of 3TC against wild type HBV polymerase is a direct outcome from the flexibility of M204 (induced fit effect) that allow 3TC to fit into the active site. Consequently, the long-term clinical use of 3TC to treat HBV-infected patients enforces virus to mutate M204 to develop 3TC-resistant HBV.
3.2 Lamivudine (3TC) in mutant HBV-polymerase
Four major mutational patterns (M204V, M204I, L180M-M204V and L180M-M204I) of 3TC-resistant HBV with significant reduced antiviral activity are observed in patients (Table 1), while individuals with L180M mutation show only 1.7 fold resistance. The present molecular modeling study shows the significant binding energy differences between dCTP and 3TC in mutant HBV polymerases. Consequently, 3TC looses the binding affinity with all five mutants in comparison to natural substrate dCTP as shown in Table 4.
Table 4.
Multi-ligand bimolecular association with energetics (eMBrAcE) calculation of Lamivudine triphosphate (3TC-TP) in comparision to dCTP after induced fit docking and minimization in mutant HBV RT.
| Mutant Strain HBV polymerase | Energy Difference Results (ΔE: Kj/mol) | ||||
|---|---|---|---|---|---|
| Electrostatic | VdWa | Total | Δ Eb | ||
| L180M | dCTP | -5118.0 | 189.2 | -412.3 | 50.7 |
| 3TC-TP | -5009.8 | 126.9 | -361.6 | ||
| M204V | dCTP | -5094.6 | 194.2 | -411.3 | 36.0 |
| 3TC-TP | -5026.4 | 127.9 | -375.3 | ||
| M204I | dCTP | -5229.5 | 186.1 | -426.5 | 79.8 |
| 3TC-TP | -5149.7 | 135.6 | -346.7 | ||
| L180M-M204V | dCTP | -5282.9 | 193.8 | -424.6 | 55.6 |
| 3TC-TP | -5223.2 | 133.0 | -369.0 | ||
| L180M-M204I | dCTP | -5261.9 | 188.6 | -435.5 | 94.0 |
| 3TC-TP | -5166.5 | 136.5 | -341.5 | ||
van der Waals interaction.
ΔE = Energy difference (-: favorable, +: worse) in comparison to dCTP.
L180 residue is not interacting directly with 3TC, but one of its methyl group is oriented towards the small hydrophobic pocket formed by the residues A87, F88, P177, L180 and M204 (Figure 5c). In case of L180M HBV, the resulting M180 is oriented away from the hydrophobic pocket and looses the participation of one methyl group (Figure 6a). This may be the reason for the modest resistance profile of 3TC (1.7 fold, Table 1) by the L180M mutation.
Figure 6.
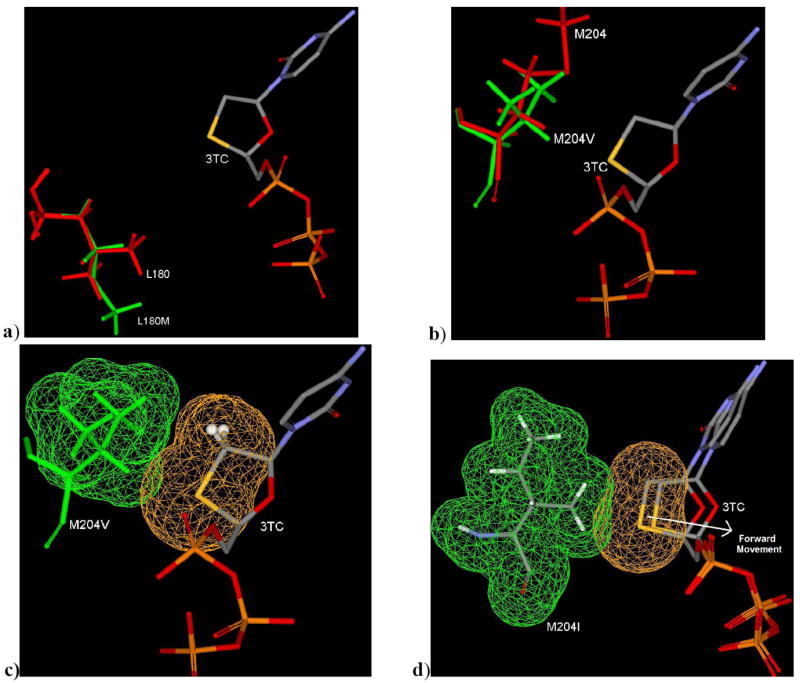
a) Binding mode (3TC-TP in L180M HBV) showing the loosing of one methyl contribution in small hydrophobic pocket of L180M HBV (green) in compare to wild type HBV (red; L180). b) Binding mode (3TC-TP in M204V HBV) showing possible steric clash due to bulky V204 (green) in comparision to wild type HBV (M204; red). c) Binding Mode of 3TC-TP in M204V showing partial steric clash by vander waal’s surface area. d) Binding Mode of 3TC-TP in M204I showing extensive steric clash by vander waal surface area resulting further unfavorable forward movement of 3TC in comparision to wild type HBV.
The shifting of oxathiolane ring of 3TC utilizes the small hydrophobic pocket due to induced fit orientation of M204 (Figure 4). In M204V HBV, shifting of oxathiolane ring is disfavored due to bulkiness of branched residue V204 (Figure 6b & 6c), which ultimately leads to an 18 fold resistance (Table 1). It’s obvious that substitution of M204 to I204 (Isoleucine: more bulky group) will reduce the binding affinity in several folds by enforcing the oxathiolane ring forward to a non-binding mode of 3TC in M204I HBV (Figure 6d).
Energetic results in Table 4 clearly indicates a significant low binding affinity of 3TC-TP (ΔE = -346.7 KJ/mol) in comparison to dCTP (ΔE = -426.5 KJ/mol) in M204I HBV. From above structural studies, it is obvious that the dual mutations L180M+M204V or L180M+M204I won’t allow the movement of sugar or the oxathiolane ring backwards and restrict the utilization of hydrophobic pocket for favorable binding of 3TC. In this regard the binding mode of 3TC in dual mutant HBV (L180M+M204I) shown in Figure 7, shows the poor stacking of oxathiolane ring with F88 and steric clash between I204 and 3TC.
Figure 7.
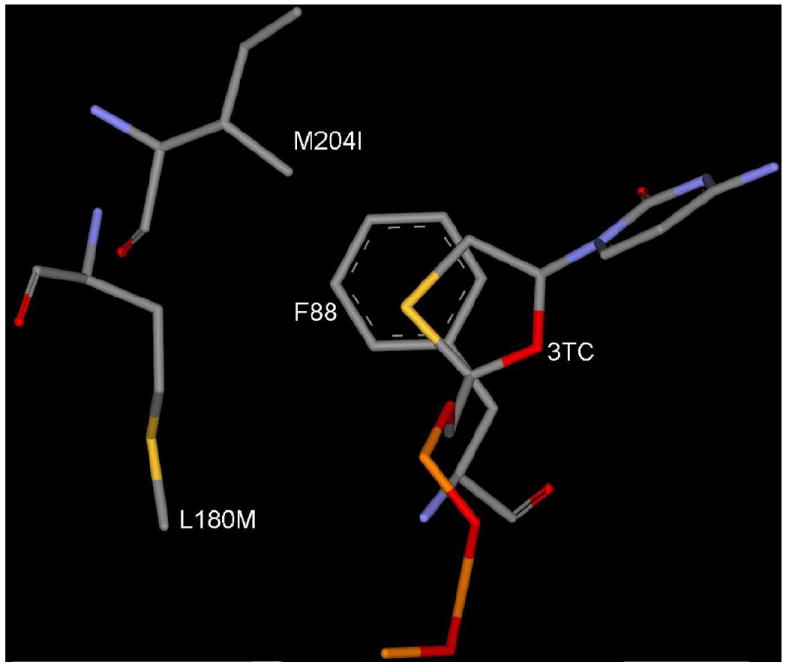
Binding mode of 3TC-TP in L180M+M204I mutant HBV. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
3.3 Adefovir (ADV, 2) in wild type and mutant HBV polymerase
Adefovir (2, Figure 1) is an acyclic adenosine analog, and in this study its binding mode has been investigated in comparison to the natural substrate, dATP. The molecular modeling studies of adefovir in wild type shows that adefovir is occupying the active site in a similar way as natural substrate dATP (Figure 8), while its open chain is oriented away from the small hydrophobic backside pocket (residues A87, F88, P177, L180 and M204).
Figure 8.
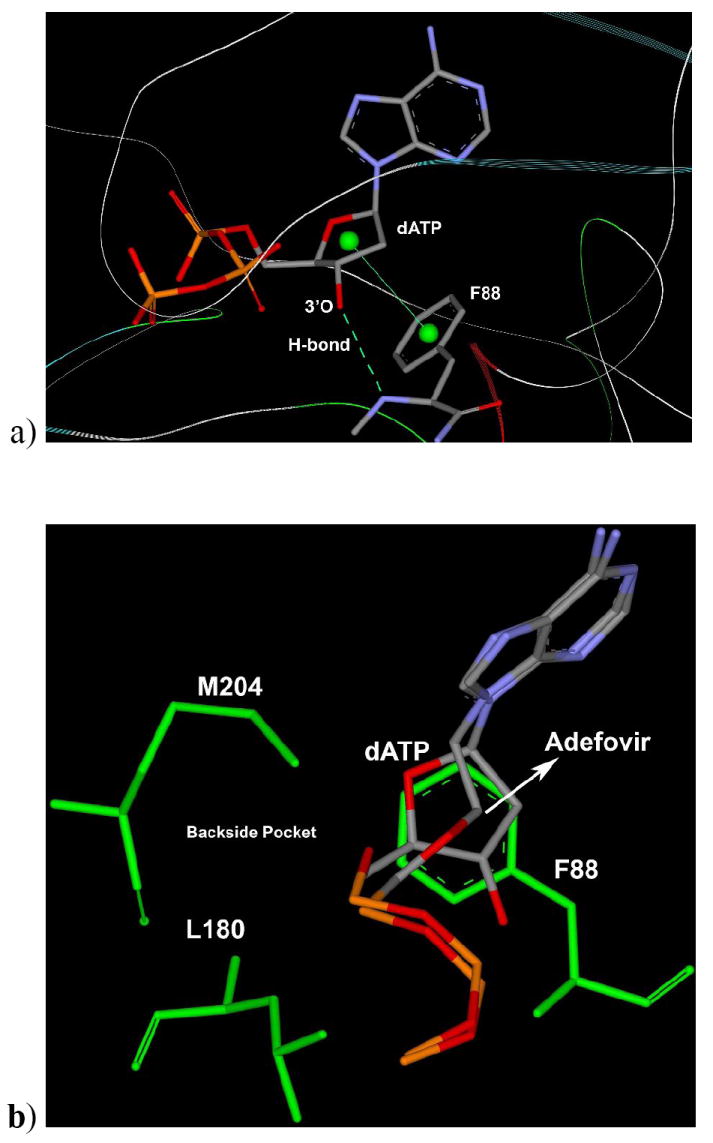
a) Binding mode of dATP in wild type HBV shows H-bond with 3’-OH with NH of F88 residues and stacking support by phenyl (F88) to the sugar ring of dATP. b) Binding mode of Adefovir-TP with superposition of dATP showing the same position for adenosine base and phosphates, while Adefovir-TP open chain is oriented to forward side and it is not affecting or interacting with backside residues such as L180, M204 etc. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
The binding mode study clearly indicates that adefovir may not be affected by the 3TC-associated mutation, which supports the experimental findings regarding the resistance profile of adefovir. Further, the empirical energy calculations (Table 5) reveal that adefovir has better binding affinity in comparison to dATP with the wild type as well as the 3TC-associated mutant HBV (L180 and M204V). However, it did not provide a quantitative correlation with in-vitro activity of adefovir against M204I and the dual mutant. These discussions indicate the pitfalls of the modeling to predict an ideal structural model, which can provide quantitative correlation with biological results. It is well known that the empirical calculations may have uncertainties due to a slight conformational difference (Tirado-Rives and Jorgensen 2006) in the complex-enzyme model. Taken together, these results support the better profile of adefovir resistance and its potency as an anti-HBV agent.
Table 5.
Multi-ligand bimolecular association with energetics (eMBrAcE) calculation of Adefovir triphosphate (ADV-TP) in comparision to dATP after induced fit docking and minimization in HBV polymerase.
| Energy Difference Results (ΔE: kj/mol) | |||||
|---|---|---|---|---|---|
| HBV polymerase | Electrostatic | VdWa | Total | Δ Eb | |
| Wild Type (WT) | dATP | -5133.1 | 217.9 | -386.9 | -33.7 |
| ADV-TP | -5235.5 | 187.3 | -420.6 | ||
| L180M | dATP | -5005.3 | 168.8 | -410.8 | -12.2 |
| ADV-TP | -5193.9 | 179.3 | -423.0 | ||
| M204V | dATP | -4983.9 | 174.1 | -407.9 | -20.7 |
| ADV-TP | -5062.9 | 176.9 | -428.6 | ||
| M204I | dATP | -5247.0 | 167.2 | -431.9 | 11.0 |
| ADV-TP | -5194.8 | 172.8 | -420.9 | ||
| L180M-M204V | dATP | -5300.7 | 175.7 | -429.5 | 14.1 |
| ADV-TP | -5257.1 | 180.6 | -415.4 | ||
| L180M-M204I | dATP | -5278.2 | 169.5 | -431.7 | 8.4 |
| ADV-TP | -5217.8 | 174.7 | -423.3 | ||
van der Waals interaction.
ΔE = Energy difference (-ive: favorable) in comparison to dATP.
3.4 Entecavir (ETV, 3) in wild type and mutant HBV polymerase
The result from entecavir modeling explains its higher potency in comparison to other NRTIs (Table 6). Entecavir forms all the network of H-bonds and interactions with the active site residues, which was observed in the case of natural substrate dGTP (Table 2). Entecavir occupies the active site in a similar way as dGTP with no shifting of carbocyclic ring in comparison to the sugar ring of dGTP (Figure 9a). The exocyclic alkene of ETV occupies the backside hydrophobic pocket near the M204 residue and above the para position of F88 (Figure 9a).
Table 6.
Multi-ligand bimolecular association with energetic (eMBrAcE) calculation of Entecavir triphosphate (ETV-TP) in comparison to dGTP after induced fit docking and minimization in HBV polymerase.
| Energy Difference Results (ΔE: kj/mol) | |||||
|---|---|---|---|---|---|
| HBV polymerase | Electrostatic | VdWa | Total | Δ Eb | |
| Wild Type (WT) | dGTP | -5161.7 | 139.5 | -366.8 | -37.9 |
| ETV-TP | -5465.8 | 196.6 | -404.7 | ||
| L180M | dGTP | -5010.2 | 173.7 | -422.7 | 2.6 |
| ETV-TP | -5009.4 | 163.1 | -420.1 | ||
| M204V | dGTP | -4977.5 | 179.7 | -417.4 | -10.3 |
| ETV-TP | -4972.7 | 163.3 | -427.7 | ||
| M204I | dGTP | -5233.9 | 169.0 | -429.9 | -8.6 |
| ETV-TP | -5277.4 | 180.3 | -438.5 | ||
| L180M-M204V | dGTP | -5293.3 | 179.5 | -449.1 | -3.0 |
| ETV-TP | -5321.3 | 170.7 | -452.1 | ||
| L180M-M204I | dGTP | -5267.1 | 172.2 | -438.5 | 1.0 |
| ETV-TP | -5297.6 | 185.0 | -437.5 | ||
van der Waals interaction.
ΔE = Energy difference (-ive: favorable) in comparison to the dGTP.
Figure 9.
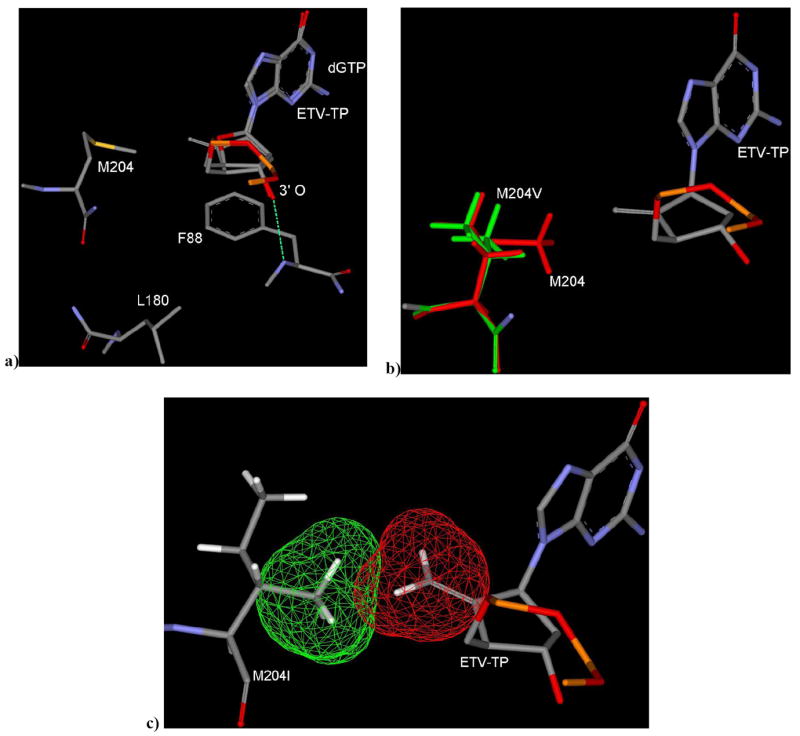
a) ETV binding mode in WT HBV, superimposed with dGTP showing no shifting of sugar ring and there is no induced fit movement of M204. b) ETV binding mode in M204V HBV: exocyclic alkene of ETV oriented towards mutated residue V204 (green) showing no significant steric clash. The exocyclic alkene moiety may loose favorable hydrophobic interaction in the backside pocket due to mutation of M204 to V204. c) M204I mutant shows partial steric clash between I204 and exocyclic alkene as shown by vander Waal’s surface area. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
This result indicates that there is no induced fit movement of the M204 residue as was observed in the case of 3TC (Figure 4). Presence of the 3’-OH group and above described natural substrate dGTP-mimic properties support the previous experimental and modeling studies (Langley, Walsh et al. 2007), which showed ETV is a chain terminator through the incorporation into DNA, followed by the abortive mechanism of ETV-containing DNA.
Further molecular modeling studies have been carried with 3TC resistant HBV to investigate the active site interactions to understand the resistant profile of ETV. The 3TC-associated L180 mutant disfavors the shifting of oxathiolane ring, while there is no significant movement of the carbocyclic ring of ETV in comparison to dGTP (Figure 9a). Thus, it is obvious that the L180M mutation won’t affect on its binding to the active site. In addition, the mutant M204V doesn’t affect much on the binding mode of ETV as well (Figure 9b).
The mutation of M204 to V204 can result in only partial filling of the small hydrophobic pocket which may be the reason for the reduction of potency of ETV in M204V HBV RT (Table 1). Figure 9c shows a possible partial steric clash between the exocyclic alkene and the I204 residue, which supports the reduction of potency of ETV in comparison to M204V HBV. Despite of the partial steric clash, there is no forward movement observed for the exocyclic sugar ring of ETV as it was clearly observed in the case of 3TC (Figure 6c). These results support the low level of cross-resistance to ETV, which is consistent with the experimental results and ETV-TP can still bind to the 3TC-associated mutant HBV. Empirical energy correlation (Table 6) did not provide us a quantitative correlation, but it reveals that there is no significant binding energy difference between natural substrate (dGTP) and ETV in all of the five mutant HBV. Empirical energy supports the experimental evidence (Dienstag et al. 2007) that entecavir would be more potent than that of 3TC.
3.5 Telbivudine (LdT, 4) in wild type and mutant HBV polymerase
Telbivudine triphosphate (LdT-TP) inhibits HBV polymerase by competing with the natural substrate, TTP (Bryant et al. 2001). LdT is not supposed to behave as a perfect mimic of the natural substrate, as it looses some of the well known interaction patterns: H-bonding with F88 and stacking with primer base (Table 2). A molecular modeling study in wild type HBV polymerase indicates the significant difference in the binding mode of LdT in comparison to the natural substrate TTP (Figure 10). But still it occupies the active site and has a small binding energy difference (-346.2 Kj/mol) in comparison to TTP (-337.5 Kj/mol) as shown in Table 7.
Figure 10.
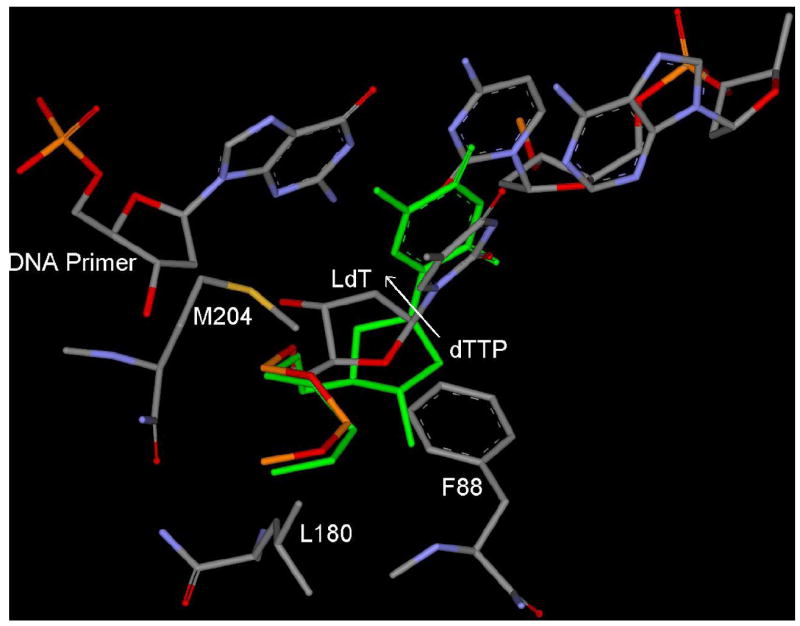
Binding mode of LdT in wild type HBV shows the shifting of sugar ring towards backside small hydrophobic pocket in comparision to TTP (green). 3’-OH of LdT, oriented towards base of DNA primer, while 3’-OH of TTP (green) forms H-bond (not shown) with NH of F88. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
Table 7.
Multi-ligand bimolecular association with energetics (eMBrAcE) calculation of Telbivudine triphosphate (LdT-TP) in comparision to TTP after induced fit docking and minimization in HBV polymerase.
| Energy Difference Results (ΔE: kj/mol) | |||||
|---|---|---|---|---|---|
| HBV polymerase | Electrostatic | VdWa | Total | Δ Eb | |
| Wild Type (WT) | TTP | -4477.4 | 138.8 | -337.5 | -8.7 |
| LdT-TP | -4930.8 | 151.9 | -346.2 | ||
| L180M | TTP | -4810.2 | 171.1 | -400.9 | 59.7 |
| LdT-TP | -4967.8 | 150.6 | -341.2 | ||
| M204V | TTP | -4799.7 | 177.4 | -398.6 | 44.7 |
| LdT-TP | -4926.6 | 160.4 | -353.9 | ||
| M204I | TTP | -4552.0 | 177.6 | -371.0 | 28.5 |
| LdT-TP | -4911.5 | 166.5 | -342.5 | ||
| L180M-M204V | TTP | -4585.5 | 185.5 | -383.6 | 26.2 |
| LdT-TP | -4928.3 | 160.7 | -357.4 | ||
| L180M-M204I | TTP | -4567.0 | 179.2 | -374.5 | 32.5 |
| LdT-TP | -4942.8 | 167.0 | -342.0 | ||
van der Waals interaction.
ΔE = Energy difference (-ive: favorable) in comparison to TTP.
In addition to the backward shifting, there is also an upward shifting of the sugar ring of LdT in comparision to TTP (Figure 10). This upward shifting is due to the presence of 3’-OH, which is enforcing the formation of a H-bond with the nitrogen of the guanine base of DNA primer (+1). Due to upward shifting of the sugar ring, the M204 residue is oriented below the sugar ring. This result indicates that LdT has a different binding mode in comparision to other L-nucleoside such as 3TC and it does not enforce M204 residue to adopt different conformation as previously observed in the case of 3TC (Figure 4).
LdT binding mode in the M204V, M204I and L180M are shown in Figure 11. The M204V mutation clearly reflects the loss of contribution by one methyl group in the back-side hydrophobic pocket to interact with the sugar ring (Figure 11a). Although LdT does not have any steric clash with 3TC associated mutation M204V, but this mutation alters the backside hydrophobic pocket. This alteration reduces the favorable interaction of the ribose ring of LdT in comparison to TTP and indirectly decreases the binding affinity. This result supports the reduced potency of LdT in M204V HBV.
Figure 11.
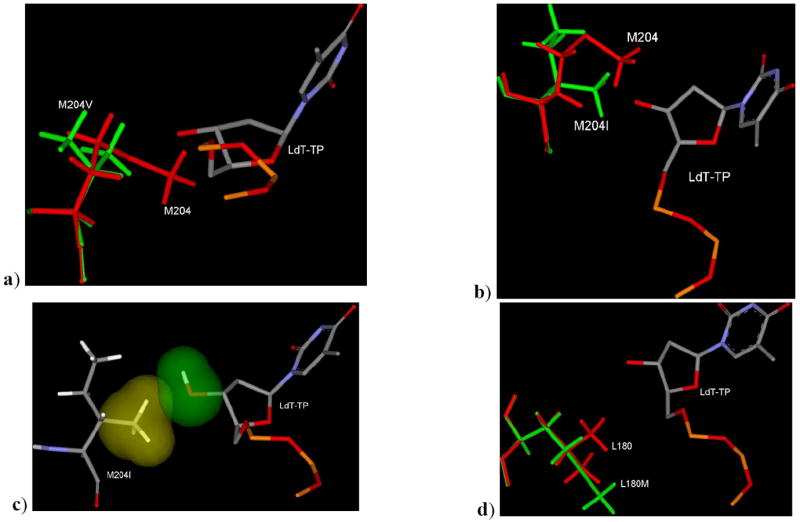
a) Binding mode of LdT in mutant M204V HBV superimposed with M204 residue. b) Binding mode of LdT in mutant M204I HBV superimposed with M204 residue showing the different position and orientation of methyl group in wild type and M204I HBV. c) VdW surface showing the hydrophobic methyl group of I204 is oriented towards hydrophilic group (3’-OH) of LdT and also showed the steric clash. d) Binding mode of LdT in L180M HBV showing the absence of one methyl group in the backside binding pocket of LdT due to mutation of L180 (red) to Met180 (green). [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
Modeling studies of LdT in M204I also clearly shows the significant differences in the binding pocket due to the M204 mutation, in terms of position of the methyl group (Figure 11b) and orientation of the methyl group of I204 residue (Figure 11c). Positional and orientation differences do not support the binding of LdT in the active site of M204I HBV, and this result justifies the several-folds reduced susceptibility of LdT in cell based assays. The binding mode of LdT in L180M HBV (Figure 11d) and the modeling studies indicate the absence of one methyl group contribution in the back-side hydrophobic pocket causing reduced binding affinity. Therefore, the dual mutation, L180M+M204V, can cause removal of the two methyl group (Figure 11a and Figure 11d) from the backside hydrophobic pocket. Consequently, LdT is no longer active in the dual mutant. Above modeling discussion and energetic correlation (Table 7) support the fact that, LdT having significant activity in wild type HBV (EC50: 0.2 μM), while reduced potency in 3TC associated mutants.
In summary, modeling studies also conclude that LdT (L-nucleoside analogs) has a different binding mode in comparison to 3TC (L-nucleoside analog) as well as natural nucleoside (TTP). This study supports a slight difference in the resistance profile of LdT in comparison to 3TC.
3.6 Clevudine (L-FMAU, 5) in wild type and mutant HBV polymerase
Clevudine is an orally active antiviral agent for the treatment of chronic HBV that has been shown to be well tolerated and demonstrated potent antiviral activity in clinical studies (Marcellin, Mommeja-Marin et al. 2004; Lee et al. 2006; Yoo, Kim et al. 2007b). Clevudine has been approved for the treatment of chronic hepatitis B virus infection in South Korea in 2006 and is currently undergoing Phase III clinical trials in US and Europe. Recent phase III clinical trials for 24 weeks (Yoo, Kim et al. 2007a) at 30 mg clearly demonstrated the potent and sustained antiviral effects associated with sustained normalization of alanine aminotransferase (ALT) levels. ALT normalization was found well maintained during the post-treatment follow-up period. Viral suppression was monitored for additional 24 weeks after withdrawal of the treatment and found the sustained antiviral effect, with 3.11 log(10) reduction at week 48 (Yoo, Kim et al. 2007b). This sustained antiviral effect after discontinuation of the drug makes this molecule as a unique antiviral agent in comparison to other approved anti-HBV agents. In addition, there was no evidence found for viral resistance during the 48 weeks treatment period in HBeAg-positive chronic hepatitis B. The plausible mechanism of sustained viral suppression may attribute to the reduction of intrahepatic cccDNA (Zhu et al. 2001). In addition, the immunological factors might be another reason for a sustained anti-HBV activity. These combined mechanisms along with the favorable pharmacokinetic properties may be the reason for the unique antiviral properties by clevudine. This hypothesis still remains to be investigated to prove or disprove the unique antiviral efficacy of clevudine.
Clevudine is a L-nucleoside analog, having additional 2’-fluoro (beta) in comparison to LdT. Modeling studies has been carried out to understand the molecular basis for similarities and differences with respect to other potential anti-HBV agents. Energetic calculations (Table 8) showed that clevudine have a significant binding energy difference in wild type HBV (-349.9 kj/mol) in comparison to the natural substrate TTP (-337.5 kj/mol). However, this energetic qualitative correlation does not fully support the outcome of the clinical potency of clevudine.
Table 8.
Multi-ligand bimolecular association with energetics (eMBrAcE) calculation of L-FMAU-TP in comparision to TTP after induced fit docking and minimization in HBV polymerase.
| Energy Difference Results (ΔE: kj/mol) | |||||
|---|---|---|---|---|---|
| HBV polymerase | Electrostatic | VdWa | Total | Δ Eb | |
| Wild Type (WT) | TTP | -4477.4 | 138.8 | -337.5 | -12.4 |
| L-FMAU-TP | -4653.8 | 153.7 | -349.9 | ||
| L180M | TTP | -4810.2 | 171.1 | -400.9 | 54.0 |
| L-FMAU-TP | -4668.1 | 151.3 | -346.9 | ||
| M204V | TTP | -4799.7 | 177.4 | -398.6 | 46.2 |
| L-FMAU-TP | -4632.2 | 159.7 | -354.7 | ||
| M204I | TTP | -4552.0 | 177.6 | -371.0 | 15.8 |
| L-FMAU-TP | -4916.6 | 171.8 | -355.2 | ||
| L180M-M204V | TTP | -4585.5 | 185.5 | -383.6 | 20.6 |
| L-FMAU-TP | -4922.5 | 153.3 | -363.0 | ||
| L180M-M204I | TTP | -4567.0 | 179.2 | -374.5 | 20.0 |
| L-FMAU-TP | -4936.4 | 170.5 | -354.5 | ||
van der Waals interaction.
ΔE = Energy difference (-ive: favorable) in comparison to TTP.
The calculations (Table 8) in mutant HBV in comparison with natural substrate (TTP) also support the lower potency of clevudine in 3TC-associated mutants. The binding mode of clevudine (Figure 12a) describes the backward and upward shifting of L-sugar ring in the active site without affecting the neighboring amino-acid residue, which makes it suitable for wild type HBV-polymerase. Careful examination of binding mode shows the unfavorable stacking interaction of the thymine base of clevudine with the base of DNA-primer, resulting further a distorted base pairing interaction with DNA-template. These studies supports the fact that clevudine is not a perfect mimic of natural substrate. This could be the reason for non-competitive inhibition, without incorporation into the growing HBV-DNA chain (Balakrishna Pai et al. 1996). This also might be the reason for the high genetic barrier, which lower the chance for the emergence of viral resistance during clevudine therapy.
Figure 12.
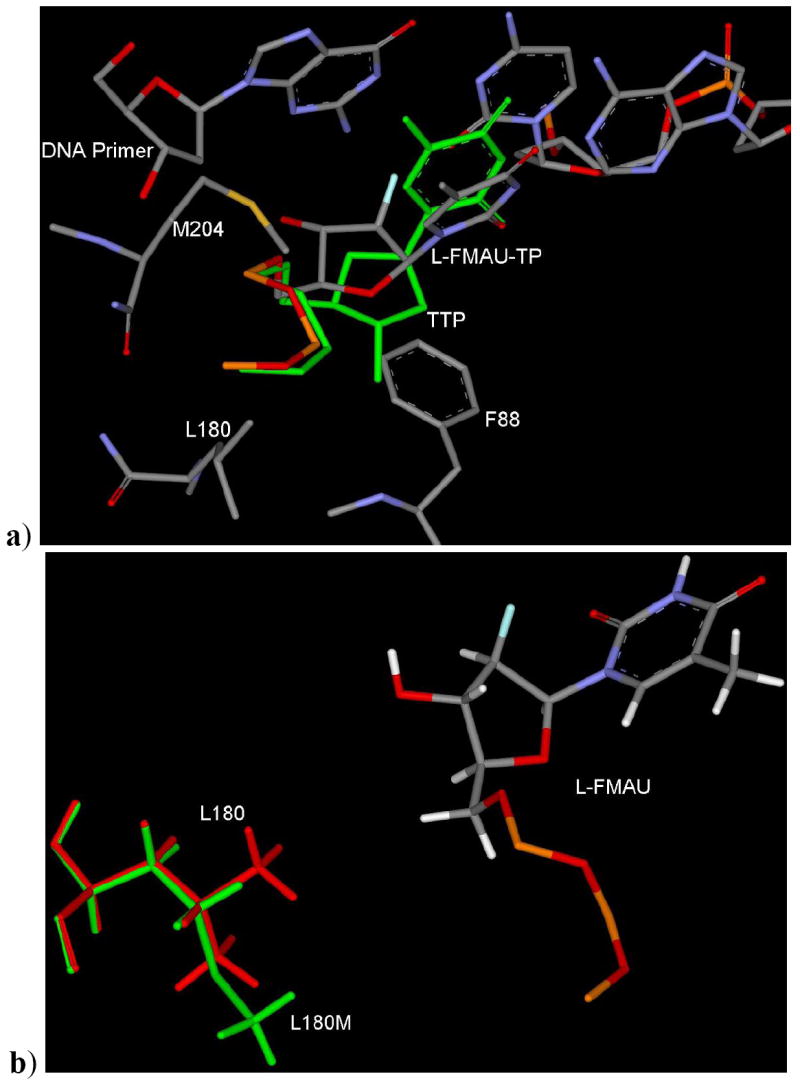
a) Binding mode of L-FMAU-TP in wild type HBV showing the similar binding mode and backward-upward shifting of sugar ring as previously described in the case of LdT (Figure 10). b) Binding mode of L-FMAU-TP in L180M HBV. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
As in the case of L180M, there is absence of a methyl group in the backside hydrophobic pocket (Figure 12b). This mutation decreases the binding affinity of clevudine. The M204 residue orients towards the lower hydrophobic pocket as clevudine binds to the active site with the upward shifting of ribose ring. Due to this fact, M204V mutation may not affect heavily in terms of steric clash (Figure 13a) as it was observed the case of 3TC. These modeling results support the better activity profile of clevudine in comparison to 3TC in M204V.
Figure 13.
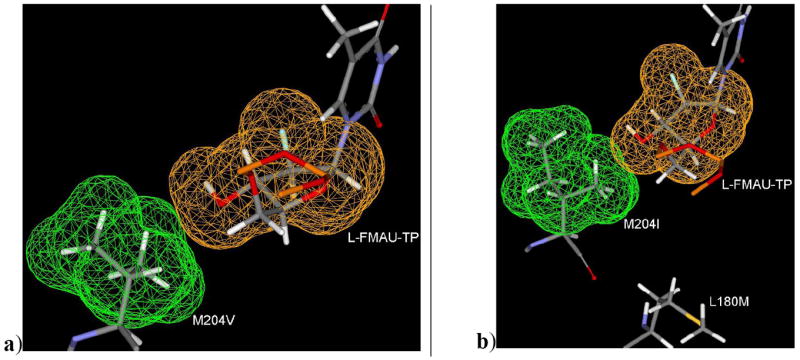
a) Binding mode of L-FMAU-TP along with mutant M204V residue showing neglible steric clash. b) Binding mode of L-FMAU-TP along with M204I and L180M residues (dual mutant L180M+M204I HBV) showing the hydrophobic methyl group of M204I is oriented towards hydrophilic group (3’-OH) of L-FMAU-TP. In addition, the absence of one methyl group in the hydrophobic pocket due to L180M mutation. [Only mainframe of the triphosphate (Pα-O-Pβ-O-Pγ) showed for the sake of clarity]
A further binding mode study reveals a steric clash in case of M204I HBV due to the unfavorable position and orientation of a methyl group (Figure 13b) of I204 residue. This could be the reason for the several folds reduced susceptibility of clevudine in M204I as well as in dual mutant L180M/M204I. Accordingly, the dual mutation L180M+M204V can cause the removal of two methyl group from the backside hydrophobic pocket, and consequently, clevudine is no longer active in this dual mutant.
Summary
The molecular modeling studies revealed significant information about the positioning of different class of molecules in the active site of wild type and 3TC-associated mutant HBV. The oxathiolane ring of 3TC showed backward shifting into the small hydrophobic pocket causing M204 induced fit movement, which is the reason for a significant potency of 3TC in the wild type HBV. This induced fit movement of M204 residue ultimately leads to M204 mutation that is associated with 3TC treatment. There was no acyclic ring shifting observed for adefovir that is the possible reason for significant potency of adefovir against 3TC associated mutants. Entecavir also did not show any backward shifting, but exocyclic double bond occupies the backside small hydrophobic pocket (made by residues A87, F88, P177, L180 and M204). The higher potency of entecavir in wild type as well as in 3TC associated mutant in comparison to 1, 2, 4 and 5 could be due to additional hydrophobic interaction and mimicking of natural substrate. LdT showed backward shifting along with upward movement of sugar ring in comparison to natural substrate. Upward shifting favors the binding of LdT without enforcing M204 for induced fit effect, and also helps in binding to mutant M204V HBV, while M204I does not support LdT binding due to steric clash. Clevudine showed similar binding mode as described in LdT. Backward shifting along with upward shifting of the fluoro-containing sugar in clevudine, makes this molecule as an effective non-competitive inhibitor of HBV, without being incorporated into the growing HBV-DNA chain.
We investigated the molecular basis of drug resistance of clinically effective anti-HBV agents in terms of similarities and differences in the active site of wild type and mutant HBV polymerase. Although the present studies, may not fully explain quantitatively, it highlighted the structural aspect for the mechanism of anti-HBV compounds against wild type and mutant HBV.
Acknowledgments
This research was supported by NIH AI25889.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen MI, Deslauriers M, Andrews CW, Tipples GA, Walters KA, Tyrrell DL, Brown N, Condreay LD. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Hepatology. 1998;27:1670–1677. doi: 10.1002/hep.510270628. [DOI] [PubMed] [Google Scholar]
- Angus P, Locarnini S. Lamivudine-resistant hepatitis B virus and ongoing lamivudine therapy: stop the merry-go-round, it’s time to get off! Antivir Ther. 2004;9:145–8. [PubMed] [Google Scholar]
- Angus P, Vaughan R, Xiong S, Yang H, Delaney W, Gibbs C, Brosgart C, Colledge D, Edwards R, Ayres A. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003;125:292–7. doi: 10.1016/s0016-5085(03)00939-9. [DOI] [PubMed] [Google Scholar]
- Ayres A, Locarnini S, Bartholomeusz A. HBV genotyping and analysis for unique mutations. Methods Mol Med. 2004;95:125–49. doi: 10.1385/1-59259-669-X:125. [DOI] [PubMed] [Google Scholar]
- Balakrishna Pai S, Liu SH, Zhu YL, Chu CK, Cheng YC. Inhibition of hepatitis B virus by a novel L-nucleoside 2’-fluoro-5-methyl-beta-L-arabinofuranosyl uracil. Antimicrob Agents Chemother. 1996;40:380–6. doi: 10.1128/aac.40.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeusz A, Schinazi RF, Locarnini SA. Significance of mutations in the hepatitis B virus polymerase selected by nucleoside analogues and implications for controlling chronic disease. Viral Hepatitis Reviews. 1998;4:167–187. [Google Scholar]
- Bartholomeusz A, Tehan BG, Chalmers DK. Comparisons of the HBV and HIV polymerase, and antiviral resistance mutations. Antivir Ther. 2004;9:149–60. [PubMed] [Google Scholar]
- Bottecchia M, Ikuta N, Niel C, Araujo NM, O KM, Gomes SA. Lamivudine resistance and other mutations in the polymerase and surface antigen genes of hepatitis B virus associated with a fatal hepatic failure case. J Gastroenterol Hepatol. 2008;23:67–72. doi: 10.1111/j.1440-1746.2007.05238.x. [DOI] [PubMed] [Google Scholar]
- Brunelle MN, Jacquard AC, Pichoud C, Durantel D, Carrouee-Durantel S, Villeneuve JP, Trepo C, Zoulim F. Susceptibility to antivirals of a human HBV strain with mutations conferring resistance to both lamivudine and adefovir. Hepatology (Baltimore) 2005;41:1391–1398. doi: 10.1002/hep.20723. [DOI] [PubMed] [Google Scholar]
- Bryant ML, Bridges EG, Placidi L, Faraj A, Loi AG, Pierra C, Dukhan D, Gosselin G, Imbach JL, Hernandez B. Antiviral l-Nucleosides Specific for Hepatitis B Virus Infection. Antimicrobial Agents and Chemotherapy. 2001;45:229–235. doi: 10.1128/AAC.45.1.229-235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin R, Shaw T, Torresi J, Sozzi V, Trautwein C, Bock T, Manns M, Isom H, Furman P, Locarnini S. In vitro susceptibilities of wild-type or drug-resistant hepatitis B virus to (-)-beta-D-2,6-diaminopurine dioxolane and 2’-fluoro-5-methyl-beta-L-arabinofuranosyluracil. Antimicrob Agents Chemother. 2001;45:2495–501. doi: 10.1128/AAC.45.9.2495-2501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Y, Chu CK. Understanding the unique mechanism of L-FMAU (clevudine) against hepatitis B virus: molecular dynamics studies. Bioorg Med Chem Lett. 2002;12:3459–62. doi: 10.1016/s0960-894x(02)00747-3. [DOI] [PubMed] [Google Scholar]
- Chong Y, Chu CK. Molecular mechanism of dioxolane nucleosides against 3TC resistant M184V mutant HIV. Antiviral Res. 2004;63:7–13. doi: 10.1016/j.antiviral.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Chu CK, Ma T, Shanmuganathan K, Wang C, Xiang Y, Pai SB, Yao GQ, Sommadossi JP, Cheng YC. Use of 2’-fluoro-5-methyl-beta-L-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob Agents Chemother. 1995;39:979–81. doi: 10.1128/aac.39.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG, Arnold E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC) J Virol. 2001;75:4771–9. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Man RA, Wolters LM, Nevens F, Chua D, Sherman M, Lai CL, Gadano A, Lee Y, Mazzotta F, Thomas N, DeHertogh D. Safety and efficacy of oral entecavir given for 28 days in patients with chronic hepatitis B virus infection. Hepatology. 2001;34:578–82. doi: 10.1053/jhep.2001.26815. [DOI] [PubMed] [Google Scholar]
- Delaney WE, Edwards R, Colledge D, Shaw T, Torresi J, Miller TG, Isom HC, Bock CT, Manns MP, Trautwein C. Cross-Resistance Testing of Antihepadnaviral Compounds Using Novel Recombinant Baculoviruses Which Encode Drug-Resistant Strains of Hepatitis B Virus. Antimicrobial Agents and Chemotherapy. 2001a;45:1705–1713. doi: 10.1128/AAC.45.6.1705-1713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney WE, Locarnini S, Shaw T. Resistance of hepatitis B virus to antiviral drugs: current aspects and directions for future investigation. Antivir Chem Chemother. 2001b;12:1–35. doi: 10.1177/095632020101200101. [DOI] [PubMed] [Google Scholar]
- Dienstag JL, Perrillo RP, Schiff ER, Bartholomew M, Vicary C, Rubin M. A preliminary trial of lamivudine for chronic hepatitis B infection. N Engl J Med. 1995;333:1657–61. doi: 10.1056/NEJM199512213332501. [DOI] [PubMed] [Google Scholar]
- Dienstag JL, Wei LJ, Xu D, Kreter B. Cross-study analysis of the relative efficacies of oral antiviral therapies for chronic hepatitis B infection in nucleoside-naive patients. Clin Drug Investig. 2007;27:35–49. doi: 10.2165/00044011-200727010-00003. [DOI] [PubMed] [Google Scholar]
- Ferir G, Kaptein S, Neyts J, De Clercq E. Antiviral treatment of chronic hepatitis B virus infections: the past, the present and the future. Rev Med Virol. 2008;18:19–34. doi: 10.1002/rmv.554. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–75. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- Hui CK, Zhang HY, Bowden S, Locarnini S, Luk JM, Leung KW, Yueng YH, Wong A, Rousseau F, Yuen KY, Naoumov NN, Lau GK. 96 weeks combination of adefovir dipivoxil plus emtricitabine vs. adefovir dipivoxil monotherapy in the treatment of chronic hepatitis B. J Hepatol. 2008;48:714–720. doi: 10.1016/j.jhep.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Jacquard AC, Brunelle MN, Pichoud C, Durantel D, Carrouee-Durantel S, Trepo C, Zoulim F. In vitro characterization of the anti-hepatitis B virus activity and cross-resistance profile of 2’,3’-dideoxy-3’-fluoroguanosine. Antimicrob Agents Chemother. 2006;50:955–61. doi: 10.1128/AAC.50.3.955-961.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan P, Gullen EA, Lam W, Dutschman GE, Grill SP, Cheng Y-c. Novel Role of 3-Phosphoglycerate Kinase, a Glycolytic Enzyme, in the Activation of L-Nucleoside Analogs, a New Class of Anticancer and Antiviral Agents. J Biol Chem. 2003;278:36726–36732. doi: 10.1074/jbc.M307052200. [DOI] [PubMed] [Google Scholar]
- Lai CL, Dienstag J, Schiff E, Leung NW, Atkins M, Hunt C, Brown N, Woessner M, Boehme R, Condreay L. Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B. Clin Infect Dis. 2003a;36:687–96. doi: 10.1086/368083. [DOI] [PubMed] [Google Scholar]
- Lai CL, Lim SG, Brown NA, Zhou XJ, Lloyd DM, Lee YM, Yuen MF, Chao GC, Myers MW. A dose-finding study of once-daily oral telbivudine in HBeAg-positive patients with chronic hepatitis B virus infection. Hepatology. 2004;40:719–26. doi: 10.1002/hep.20374. [DOI] [PubMed] [Google Scholar]
- Lai CL, Ratziu V, Yuen MF, Poynard T. Viral hepatitis B. Lancet. 2003b;362:2089–94. doi: 10.1016/S0140-6736(03)15108-2. [DOI] [PubMed] [Google Scholar]
- Langley DR, Walsh AW, Baldick CJ, Eggers BJ, Rose RE, Levine SM, Kapur AJ, Colonno RJ, Tenney DJ. Inhibition of hepatitis B virus polymerase by entecavir. J Virol. 2007;81:3992–4001. doi: 10.1128/JVI.02395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Chung YH, Lee K, Byun KS, Paik SW, Han JY, Yoo K, Yoo HW, Lee JH, Yoo BC. A 12-week clevudine therapy showed potent and durable antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology. 2006;43:982–8. doi: 10.1002/hep.21166. [DOI] [PubMed] [Google Scholar]
- Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–43. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- Leung N. Extended lamivudine treatment in patients with chronic hepatitis B enhances hepatitis B e antigen seroconversion rates: Results after 3 years of therapy. Hepatology. 2001;33:1527–1532. doi: 10.1053/jhep.2001.25084. [DOI] [PubMed] [Google Scholar]
- Levine S, Hernandez D, Yamanaka G, Zhang S, Rose R, Weinheimer S, Colonno RJ. Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob Agents Chemother. 2002;46:2525–32. doi: 10.1128/AAC.46.8.2525-2532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locarnini S. Hepatitis B viral resistance: mechanisms and diagnosis. J Hepatol. 2003;39(Suppl 1):S124–32. doi: 10.1016/s0168-8278(03)00318-0. [DOI] [PubMed] [Google Scholar]
- Locarnini S, Mason WS. Cellular and virological mechanisms of HBV drug resistance. J Hepatol. 2006;44:422–31. doi: 10.1016/j.jhep.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Locarnini S, Qi X, Arterburn S, Snow A, Brosgart CL, Currie G, Wulfsohn M, Miller M, Xiong S. Incidence and predictors of emergence of adefovir resistant HBV during four years of adefovir dipivoxil (ADV) therapy for patients with chronic hepatitis B (CHB) J Hepatol. 2005;42:17. [Google Scholar]
- Marcellin P, Mommeja-Marin H, Sacks SL, Lau GK, Sereni D, Bronowicki JP, Conway B, Trepo C, Blum MR, Yoo BC, Mondou E, Sorbel J, Snow A, Rousseau F, Lee HS. A phase II dose-escalating trial of clevudine in patients with chronic hepatitis B. Hepatology. 2004;40:140–8. doi: 10.1002/hep.20257. [DOI] [PubMed] [Google Scholar]
- McGovern B. Antiretroviral therapy for patients with HIV-hepatitis B virus coinfection. Clin Infect Dis. 2007;44:1012–3. doi: 10.1086/512376. [DOI] [PubMed] [Google Scholar]
- Ono-Nita SK, Kato N, Shiratori Y, Carrilho FJ, Omata M. Novel nucleoside analogue MCC-478 ( LY582563) is effective against wild-type or lamivudine-resistant hepatitis B virus. Antimicrob Agents Chemother. 2002;46:2602–5. doi: 10.1128/AAC.46.8.2602-2605.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo E. New drugs for chronic hepatitis B: a review. Am J Ther. 2008;15:167–72. doi: 10.1097/MJT.0b013e318155a191. [DOI] [PubMed] [Google Scholar]
- Poch O, Sauvaget I, Delarue M, Tordo N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. Embo J. 1989;8:3867–74. doi: 10.1002/j.1460-2075.1989.tb08565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasadeusz JJ, Locarnini SL, Macdonald G. Why do we not yet have combination chemotherapy for chronic hepatitis B? Med J Aust. 2007;186:204–6. doi: 10.5694/j.1326-5377.2007.tb00863.x. [DOI] [PubMed] [Google Scholar]
- Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Steitz TA. A mechanism for all polymerases. Nature. 1998;391:231–2. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- Stuyver LJ, Locarnini SA, Lok A, Richman DD, Carman WF, Dienstag JL, Schinazi RF. Nomenclature for antiviral-resistant human hepatitis B virus mutations in the polymerase region. Hepatology. 2001;33:751–7. doi: 10.1053/jhep.2001.22166. [DOI] [PubMed] [Google Scholar]
- Tenney DJ, Levine SM, Rose RE, Walsh AW, Weinheimer SP, Discotto L, Plym M, Pokornowski K, Yu CF, Angus P, Ayres A, Bartholomeusz A, Sievert W, Thompson G, Warner N, Locarnini S, Colonno RJ. Clinical emergence of entecavir-resistant hepatitis B virus requires additional substitutions in virus already resistant to Lamivudine. Antimicrob Agents Chemother. 2004;48:3498–507. doi: 10.1128/AAC.48.9.3498-3507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado-Rives J, Jorgensen WL. Contribution of conformer focusing to the uncertainty in predicting free energies for protein-ligand binding. J Med Chem. 2006;49:5880–4. doi: 10.1021/jm060763i. [DOI] [PubMed] [Google Scholar]
- Wilchek M, Bayer EA, Livnah O. Essentials of biorecognition: the (strept)avidin-biotin system as a model for protein-protein and protein-ligand interaction. Immunol Lett. 2006;103:27–32. doi: 10.1016/j.imlet.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Williams DH, Stephens E, O’Brien DP, Zhou M. Understanding noncovalent interactions: ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angew Chem Int Ed Engl. 2004;43:6596–616. doi: 10.1002/anie.200300644. [DOI] [PubMed] [Google Scholar]
- Yadav V, Chu CK. Molecular mechanisms of adefovir sensitivity and resistance in HBV polymerase mutants: a molecular dynamics study. Bioorg Med Chem Lett. 2004;14:4313–7. doi: 10.1016/j.bmcl.2004.05.075. [DOI] [PubMed] [Google Scholar]
- Yoo BC, Kim JH, Chung YH, Lee KS, Paik SW, Ryu SH, Han BH, Han JY, Byun KS, Cho M, Lee HJ, Kim TH, Cho SH, Park JW, Um SH, Hwang SG, Kim YS, Lee YJ, Chon CY, Kim BI, Lee YS, Yang JM, Kim HC, Hwang JS, Choi SK, Kweon YO, Jeong SH, Lee MS, Choi JY, Kim DG, Kim YS, Lee HY, Yoo K, Yoo HW, Lee HS. Twenty-four-week clevudine therapy showed potent and sustained antiviral activity in HBeAg-positive chronic hepatitis B. Hepatology. 2007a;45:1172–8. doi: 10.1002/hep.21629. [DOI] [PubMed] [Google Scholar]
- Yoo BC, Kim JH, Kim TH, Koh KC, Um SH, Kim YS, Lee KS, Han BH, Chon CY, Han JY, Ryu SH, Kim HC, Byun KS, Hwang SG, Kim BI, Cho M, Yoo K, Lee HJ, Hwang JS, Kim YS, Lee YS, Choi SK, Lee YJ, Yang JM, Park JW, Lee MS, Kim DG, Chung YH, Cho SH, Choi JY, Kweon YO, Lee HY, Jeong SH, Yoo HW, Lee HS. Clevudine is highly efficacious in hepatitis B e antigen-negative chronic hepatitis B with durable off-therapy viral suppression. Hepatology. 2007b doi: 10.1002/hep.21800. In Press. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yamamoto T, Cullen J, Saputelli J, Aldrich CE, Miller DS, Litwin S, Furman PA, Jilbert AR, Mason WS. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J Virol. 2001;75:311–22. doi: 10.1128/JVI.75.1.311-322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]