

Abstract
The mammalian molecular circadian clock in the suprachiasmatic nuclei (SCN) regulates locomotor activity rhythms as well as clocks in peripheral tissues (Reppert and Weaver, 2002; Ko and Takahashi, 2006). Constant light (LL) can induce behavioral and physiological arrhythmicity by desynchronizing clock cells in the SCN (Ohta et al., 2005). We examined how the disordered clock cells resynchronize by probing the molecular clock and measuring behavior in mice transferred from LL to constant darkness (DD). The circadian locomotor activity rhythms disrupted in LL become robustly rhythmic again from the beginning of DD, and the starting phase of the rhythm in DD is specific, not random, suggesting that the desynchronized clock cells are quickly reset in an unconventional manner by the L/D transition. By measuring mPERIOD protein rhythms, we showed that the SCN and peripheral tissue clocks quickly become rhythmic again in phase with the behavioral rhythms. We propose that this resetting mechanism may be different from conventional phase shifting, which involves light induction of Period genes (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997). Using our functional insights, we could shift the circadian phase of locomotor activity rhythms by 12 h using a 15 h LL treatment: essentially producing phase reversal by a single light pulse, a feat that has not been reported previously in wild-type mice and that has potential clinical utility.
Keywords: circadian rhythms, LL, entrainment, type 0 resetting, PERIOD, suprachiasmatic nuclei, SCN
Introduction
In the absence of external time cues, animals continue to exhibit circadian rhythms: behavioral and physiological oscillations with a period of ∼24 h. Circadian rhythms are driven by endogenous circadian clocks and include activities as diverse as sleep/wake cycles, hormone production, and digestion rhythms (Allada et al., 2001; Panda et al., 2002; Reppert and Weaver, 2002; Hastings et al., 2003; Ko and Takahashi, 2006). In mammals, a circadian clock operates within individual neurons of the suprachiasmatic nuclei (SCN) in the anterior hypothalamus. These neurons form a tissue-level “master clock” that regulates behavior and synchronizes clocks in peripheral tissues through neuronal and humoral outputs (Silver et al., 1996; Ueyama et al., 1999; Terazono et al., 2003). SCN neurons entrain to the external environment through various cues, the most important of which is the light/dark (LD) cycle of day and night (Hattar et al., 2003; Panda et al., 2003; Panda, 2007).
Whereas LD and constant darkness (DD) favor the expression of circadian rhythms, constant light (LL) disrupts those rhythms (Daan and Pittendrigh, 1976). Nocturnal rodents in LL exhibit reduced locomotor activity and an unusually long circadian period, and at high light intensities they become arrhythmic (Aschoff, 1960; Daan and Pittendrigh, 1976). Two main explanations have been proposed for LL-induced arrhythmicity (Winfree, 1980): (1) LL desynchronizes clock cells, disrupting only the tissue-level clock, or (2) LL stops the intracellular clock from working. The latter mechanism explains LL-induced arrhythmicity in Drosophila (Skopik and Pittendrigh, 1967). However, for mammals, LL desynchronizes individual SCN neurons without disrupting the intracellular clock (Ohta et al., 2005).
Because the mechanism for LL-induced arrhythmicity is now established (Ohta et al., 2005), we explored what happens when LL is lifted. We measured locomotor activity and molecular markers of circadian time (CT) (mPERIOD or mPER proteins) from mice experiencing various lighting regimes and inferred how quickly desynchronized clocks resynchronize after transfer from LL to DD. In addition, we performed the unprecedented feat of reversing behavioral rhythms (producing a 12 h phase shift) in wild-type mice using a single light pulse. Clock entrainment in mammals usually requires multiple days to produce large phase shifts, frequently leading to problems in modern human society. When shift workers and long-distance travelers cannot immediately adjust their internal clocks to fit their external environment and behavioral activities, they often experience drowsiness, reduced performance, and even serious health risks; for example, shift workers experience a higher rate of accidents than the general population (Vitaterna et al., 2001; Hastings et al., 2003; Knutsson, 2003; Ko and Takahashi, 2006). If rapid phase shifting by a single light pulse is possible for humans, our findings may have clinical implications in treating shift workers and sufferers of jet lag and other disorders associated with temporal clock malfunction (Vitaterna et al., 2001; Hastings et al., 2003; Knutsson, 2003).
Materials and Methods
Animals and maintenance.
Eight- to twelve-week-old male C57BL/6J mice were purchased from Charles River Laboratories. They were maintained in a climate-controlled room and used according to the Florida State University Animal Care and Use Committee's guidelines. All experiments involving animals were performed according to a Committee-approved protocol.
Behavioral experiments.
Each mouse was housed in a cage with a running wheel. Spontaneous locomotor activity was recorded in 5 min bins using the Stanford Software System. The mice were entrained in 12 h LD cycles for 2 weeks before being released into DD. After 9–12 d in DD, lights (∼500 lux) were turned on for at least 9 d (LL). The light intensity in LL, although within the range of normal indoor illumination, is stronger than those most other groups have used to induce arrhythmicity in mice (Sudo et al., 2003; Muñoz et al., 2005; Ohta et al., 2005). We believe that the majority of mice exhibited severely compromised behavioral rhythms in a short time because of this relatively strong light intensity, as has been shown previously (Daan and Pittendrigh, 1976). On the last day in LL, mice were divided into four groups, and each group was moved into a DD compartment at 6 h intervals except the last group, which was placed in DD by turning off the lights in the same room that was previously used for LL treatment. Period and amplitude were calculated by χ2-periodogram and Fourier analysis, respectively, using the Stanford Software System. Where possible, phase was measured in LL mice; however, the activity of most LL mice was so weakly rhythmic or arrhythmic that accurate phase determination was not possible. A couple of weakly rhythmic mice are shown in Figure 1. The first mouse in Figure 1 had 20% amplitude in LL compared with that in first DD. Sixty-one percent of LL mice showed <20% of amplitude compared with that in DD. The starting phase in the second DD in Figure 1 was calculated as follows: a hypothetical activity onset time on the first day in the second DD was calculated by extrapolating a line through activity onset times backward to the first day in the second DD. This hypothetical time (A) was defined as CT 12 (CT 0 is the time of subjective dawn, whereas CT 12 is the time of subjective dusk; mice begin their nocturnal activities around dusk.), and (A) always preceded the transfer time (B) except in 1 of 46 mice. dT was the time difference between A and B. The starting phase was CT 12 + (dT/tau) × 24, where tau is the period of the second DD free-running rhythm. To determine whether the starting phases are different among the four groups, one-way ANOVA was applied. The phase shift in Figure 4 was determined as follows: a hypothetical activity onset time on the first day in DD was calculated as above. The amount of phase shift was the time (hour) between the previous zeitgeber time (ZT) 12 (activity onset in 12:12 LD; ZT indicates the time in LD with ZT 0 being the beginning of the light cycle) and the hypothetical onset time. For Figure 5, phase shifts were calculated as in Figure 4, except that the onset time on the first day in DD was used instead of the hypothetical one.
Figure 1.
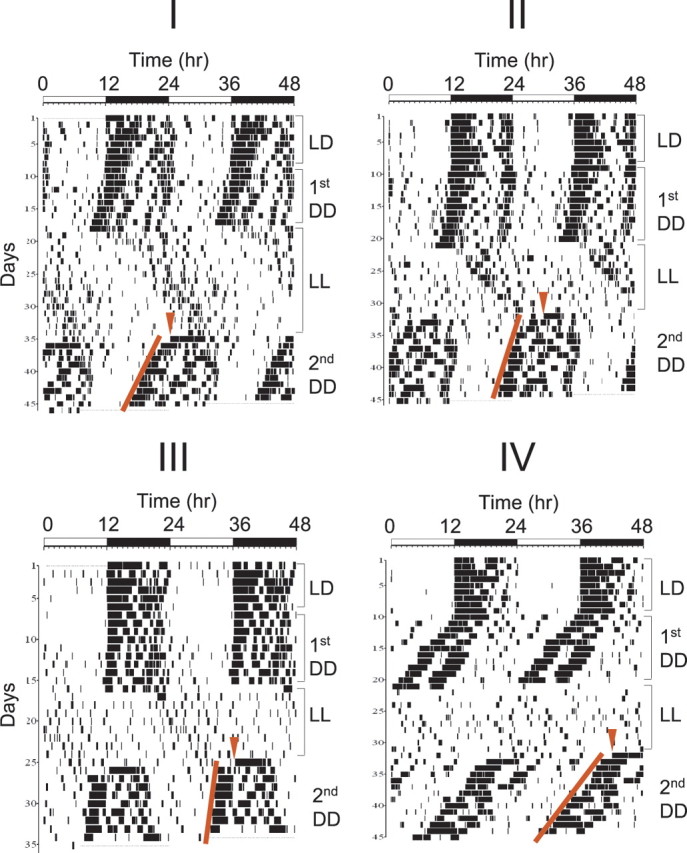
Arrhythmic mice in LL become rhythmic in locomotor activity when they are transferred to DD, and their new starting phase is ∼CT 15. Representative double-plotted actograms from 4 out of 46 mice (used in 4 independent experiments) are shown. Black marks indicate wheel-running activity at each set of time coordinates. Each of the groups I, II, III, and IV were moved from LL to DD in 6 h intervals. The red arrowhead indicates transfer time from LL to DD, and the red line indicates the new phase of activity onset times in DD. Note that the red arrowhead moves 6 h from group to group. The starting phase was calculated relative to the stable phase indicated by the red line after mice were moved into DD.
Figure 4.

The phase of behavioral rhythms can be reversed by constant light in a predictable manner. A, Schematic diagram of 15 h, 3, or 5 d of constant light treatment. Each bar (full panel) represents 24 h. The white and dark areas denote light and dark, respectively. In LD, onset of activity occurs at ZT 12 (lights off; old activity onset). However, if our hypothesis is correct, the onset of activity would be shifted ∼12 h after the constant light treatment. B, Representative actograms of mice before and after the constant light treatments. Mice were entrained in LD at least for 2 weeks, subjected to LL for 5 d (15 + 96 h), 3 d (15 + 48 h), or 15 h at ZT 12, and then placed in DD. The red line represents onset times of activity. If phase shifts were bigger than 10 h, they were considered phase reversals. The phase shifts on the bottom are represented as mean ± SEM.
Figure 5.

A reversed light/dark cycle resets behavioral rhythms more slowly than does constant light. A, Actograms of mice after 1, 3, or 5 d of DL cycle. Mice were entrained in LD for 2 weeks. After the last dark period, lights remained off for another 12 h followed by 12 h of light to switch the mice to DL. Mice were in DL for 1, 3, or 5 d before they were placed in DD. Phase shifts are shown as the mean ± SEM. B, A representative actogram after 1 d of reversed LD cycle in the alternative reversal design (DL→LD). C, Immunoblots showing mPER rhythms in liver in DL mice. The second, fifth, and eighth day samples were collected after 1, 4, and 7 d of DL cycle (LD→DL). These samples were compared with control samples (top) collected during LD.
Tissue collection.
For Figures 2 and 3, mice were entrained in LL for at least 10 d before they were transferred to DD. The mice were killed at indicated times by CO2 asphyxiation. Tissues were rapidly removed and frozen on dry ice. For SCN studies, the SCN region was punched out using a 1.25 mm diameter Harris UNiform Interface to COmputing REsources puncher (Electron Microscopy Sciences) from an ∼1 mm coronal section as described previously (Lee et al., 2001). Five SCN punches were pooled for each time point. A single SCN punch did not provide enough protein for an immunoblot with our antibodies.
Figure 2.

Liver clock is rapidly synchronized to the same circadian time as behavior in LL→DD mice. A, Immunoblotting for mPERs in liver. Representative results are shown from three independent experiments. DD control mice (top panel of each pair) were killed at 4 h intervals on the first day in DD after transfer from LD. CT 0 is subjective dawn and CT 12 is subjective dusk. LL mice were killed at 6 h intervals on the last day in at least 10 d of LL before the LL→DD transition. Two other independent experiments showed similar near steady-state mPER rhythms in LL (data not shown). LL→DD mice were collected at 4 h intervals. The first time point (4 h) in LL→DD mice was collected 4 h after the transfer. B, Quantitative real-time PCR analysis of mPer1 and 2 mRNA levels. Each point represents mean ± SEM from 3 mice. For each transcript, LL→DD, and control mice were aligned at time 0 h and CT 16, respectively, to show that the starting phase (0 h) is consistent with behavior (∼CT 15). The control CT 16 and 20 data were plotted twice to compare with the 24 and 28 h samples of LL→DD mice. The gray graphs (with triangles) and black graphs (with squares) represent LL→DD and LD→DD control mice, respectively. AU, Arbitrary unit.
Figure 3.
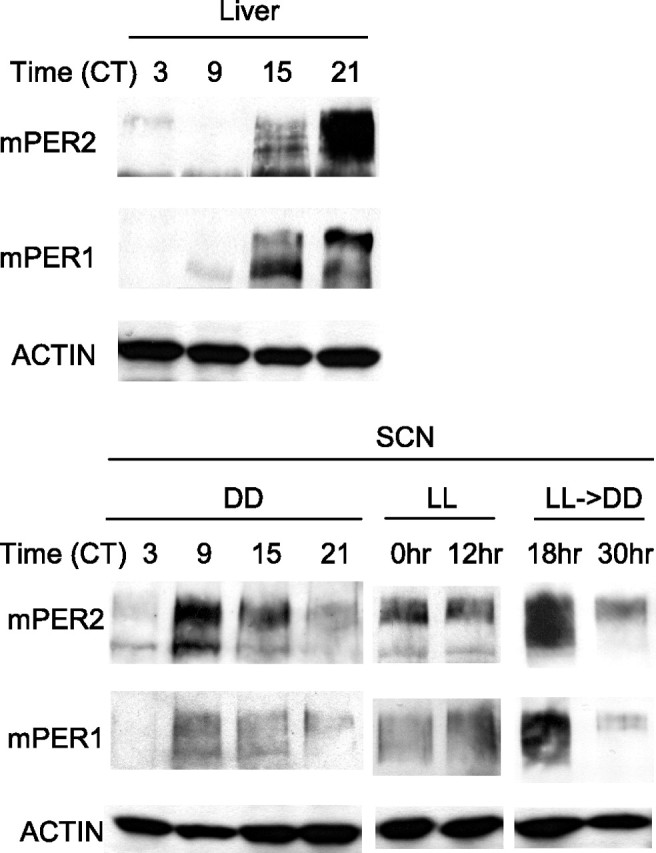
Immunoblots showing mPER protein rhythms in SCN of LL and LL→DD mice. A representative result of two independent experiments. DD mice were collected on the first day in DD. LL mice were collected twice, 12 h apart, after at least 10 d of LL entrainment. LL→DD mice were collected at 18 and 30 h after transfer to DD. Each of the SCN time point samples was extracted from a pool of SCN from 5 mice. Liver samples were prepared from 1 of the 5 DD mice. mPER1, mPER2, and (nonoscillating) actin immunoblots are shown. Note that mPER protein peaks earlier in SCN than in liver.
Quantitative real-time reverse transcriptase-PCR.
Total RNA was extracted from the liver using TRIzol Reagent according to the manufacturer's protocol (Invitrogen), and DNA contamination was eliminated by DNase I treatment. One microgram of total RNA was reverse transcribed using Bio-Rad iScript cDNA kit. Quantitation of mRNA levels was performed by real-time PCR using an iCycler iQ PCR System (Bio-Rad). Analyses were performed using the standard curve method with β-actin as a normalizing endogenous control. The primer sequences were as follows: Per1 sense 5′-TCCCTGTTTCGTCCTCCACT-3′, antisense 5′-CTTGAGCCATTGCTGTTTGC-3′; Per2 sense 5′-ATGAATGGATACGTGGACTTCTCCCCA-3′, antisense 5′-CAGGGTTGCCAGCGTGCTGGCCTT-3′; β-actin sense 5′-ATG GGTCAGAAGGACTCCTATGTGGG-3′, antisense 5′-GGCCACACGCAGCTCATTGTAGAAGG-3′.
Immunoblotting.
Immunoblotting was performed as described previously (Lee et al., 2001). Antibodies to mPER1 (PER1-1-GP) and 2 (PER2-1-GP) were described previously (Lee et al., 2001). Anti-actin antibody was purchased from Sigma-Aldrich.
Results
Behaviorally arrhythmic or weakly rhythmic mice in LL become immediately rhythmic when they are transferred to DD
Because locomotor activity rhythms are believed to be a direct reflection of the current status of the SCN clock, we measured these rhythms to discover how SCN rhythmicity changes in different lighting conditions. Specifically, we measured how behaviorally disrupted mice in LL become rhythmic again, when they are transferred to DD. The data may allow us to deduce two pieces of information. First, the behavioral changes would reveal how quickly the SCN can be resynchronized after its component oscillators have been desynchronized by constant light as shown by Ohta et al. (2005). We expected that mice would remain arrhythmic as long as the desynchrony is maintained, becoming weakly rhythmic and then more robustly rhythmic as the oscillators gradually become synchronized. Second, our experiment would provide clues as to what factors affect the starting phase of the behavioral rhythm in DD. For example, would the starting phase be tied to the LL–DD transition and therefore be about CT 12 in most mice (the time of the light-to-dark transition in a 12:12 LD cycle)? Or would the starting phase be effectively random, because it is strongly influenced by unpredictable factors such as metabolic variations among mice or variations in the underlying desynchrony (i.e., unequal mixtures of oscillators with different phases in different mice)?
We monitored mice in LD and then DD before constant light treatment to exclude any animals that behaved erratically under those conditions. After this initial screening, mice with large amplitudes and unambiguous phases were placed in LL (∼500 lux) for at least 9 d to induce arrhythmicity or severely attenuate the rhythmicity before they were transferred to DD (second DD). The mice were divided into four groups, and each group was moved at 6 h intervals to second DD over one circadian cycle (Fig. 1). Most of the mice became immediately rhythmic after transfer with little transitional time. Interestingly, the starting phases in second DD relative to later stable phases were not randomly distributed over the entire circadian cycle; rather, they were concentrated in a narrow time window around CT 15 (Table 1). The starting phase in second DD was determined by extrapolating the phase of activity onset in second DD back to the day of the LL-second DD transfer; the extrapolated onset phase on the day of the transfer was considered to be CT 12 (because activity onset in mice is a marker for subjective dusk) (Schwartz and Zimmerman, 1990), and the starting phase was the time of the LL–DD transfer relative to the extrapolated CT 12. In most mice, the starting phases were around CT 15 regardless of transfer time, and phases (timing of activity onset) were stable from the second day onwards. It is clear that the starting phases were not affected by previous phases in LD and first DD, because mice that were transferred from LL to second DD 6 h apart exhibited similar starting phases in second DD, not starting phases with a 6 h difference. In a minority of mice, a weak behavioral rhythm persisted in LL, but this rhythm was not consistently related to the starting phase in second DD (Fig. 1; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Table 1.
Summary of behavioral data obtained from the experiment presented in Figure 1.
| Group | Starting phase (mean ± SEM) | Number of mice |
|---|---|---|
| I | 14.17 ± 0.59 | 10 |
| II | 16.09 ± 0.59 | 12 |
| III | 15.86 ± 0.56 | 13 |
| IV | 14.40 ± 0.65 | 11 |
| Total | 15.19 ± 0.32 | 46 |
A total of 46 mice were used in four independent experiments. The extrapolated starting phase of each group in the 2nd DD is presented as mean ± SEM. The starting phases among the groups were not significantly different by ANOVA (F = 2.783; p = 0.53).
mPERs in liver in LL exhibit near steady-state arrhythmicity, but they also resume rhythmicity quickly in DD in phase with behavior
Our above data showed that behaviorally arrhythmic mice in LL become quickly rhythmic again with a specific starting phase (∼CT 15) in subsequent DD. Like behavior, peripheral clocks are regulated by the output of the SCN master clock. Furthermore, peripheral clocks may themselves regulate behavioral rhythms. Previous work showed that when peripheral clocks are uncoupled from the SCN by nonphotic cues such as restricted feeding, behavioral rhythms become disrupted (Damiola et al., 2000; Stokkan et al., 2001). Peripheral clocks that are out of phase with the SCN may therefore affect behavioral rhythms, perhaps by interfering with output signals from the SCN. For all these reasons, we considered how peripheral clocks are affected by LL and LL→DD. Because the behavioral rhythms in the second DD seemed to be as robust as in the first DD from the second day onward in most mice, and because the starting phase of the behavior was consistently ∼CT 15, we hypothesized that peripheral clocks may also be quickly reset by the LL→DD treatment from a steady-state to a synchronized rhythm starting ∼CT 15.
To test this hypothesis, we measured mPER rhythms in liver, assessed the effects of LL→DD treatment, and determined the starting phase of the rhythm in peripheral clocks. In liver, both mPER1 and 2 exhibited a near steady-state arrhythmicity in mice collected every 6 h in LL, suggesting that peripheral tissues also lose rhythmicity in LL, probably through desynchronization of their constituent oscillators (Nagoshi et al., 2004; Ohta et al., 2005) (Fig. 2 A). Regardless of collection time, liver in most LL mice contained mPER across the full range of normal phosphorylation states, from hypophosphorylated mPER (such as ∼CT 12) to hyperphosphorylated mPER (such as ∼CT 0), consistent with a mixture of mPER from clocks at different phases. After mice were transferred to DD, mPER1 and 2 over the course of 28 h were gradually phosphorylated and disappeared, similar to mPERs in control mice. mPERs 24 h after the transfer are similar to mPERs between CT 12 and CT 16 in control mice, based on abundance and phosphorylation status, which implies that the extrapolated starting phase in the liver clock in DD is similar to that of behavioral rhythms (∼CT 15). Thus, liver oscillators seem to quickly resynchronize to the same circadian time as the behavior. Although there was a little more variation in mPER phase among mice collected at the same time during LL→DD than those collected during LD→DD (data not shown), we could see a robust rhythm even with a sample size of only two per time point. In addition, we measured mRNA rhythms of mPer1 and 2 in liver to confirm that mRNA rhythms fit the protein rhythms. When time 0 (LL→DD) was aligned with CT 16 in control (Fig. 2 B), mRNA levels of mPer in LL-treated mice were not as rhythmic as in control mice in the first ∼12 h after DD transfer, but became robustly rhythmic and closely matched those in control mice in the later part of the cycle.
Together, our data suggest that the robust behavioral rhythms arising immediately after the LL→DD transition may result from the rapid resynchronization of desynchronized oscillators to the same circadian time in peripheral tissues as well as in SCN (see below).
mPER proteins in the SCN also resume rhythmicity rapidly after LL to DD transition
Because the behavioral rhythm phase seemed to be stabilized from the second day in second DD in most mice (Fig. 1), and the liver clock is also quickly rhythmic again, we hypothesized that the desynchronized oscillators in the SCN (as a key regulator of both behavioral and peripheral rhythms) must have been rapidly resynchronized by the LL to DD transition within a day or two. To test this prediction, we analyzed molecular rhythms in the SCN from mice subjected to LL→DD treatment. Specifically, we dissected SCN from these mice and measured mPER1 and 2 by immunoblotting to see whether mPER is arrhythmic in LL and becomes rhythmic again in DD. First, we confirmed that mPER1 and 2 oscillate in abundance and phosphorylation in SCN on the first day in DD after LD entrainment as has been shown previously (Lee et al., 2001). The phase of mPER1 and 2 rhythms was compared between SCN and liver from the same mice to verify that the two tissues differ in phase (Lee et al., 2001). As has been demonstrated previously by immunocytochemistry (Reppert and Weaver, 2001), both mPER1 and 2 peaked several hours earlier in our SCN tissue compared with liver (Fig. 3). Additionally, our immunoblotting confirmed that both proteins oscillate robustly in abundance and phosphorylation in the SCN.
Next, we measured mPER proteins in SCN from mice in constant light and mice transferred from LL to DD (Fig. 3). If constant light desynchronized the oscillators in the SCN, we expected to see an LL mPER immunoblot profile resembling a mixture of the four control samples, and the absence of an mPER tissue-level rhythm. Although mPER1 in SCN of LL mice did resemble an even mixture of mPER1 from different phases, hyperphosphorylated isoforms of mPER2 were more pronounced than expected from an even mixture of the four control samples. However, as predicted, mPER1 and mPER2 protein in SCN from mice collected at two times (12 h apart) in LL did not change significantly between the two times. Furthermore, mPER1 and mPER2 in SCN at 18 and 30 h after the transfer from LL to second DD were similar to mPERs at CT 09 and 21, respectively, in control mice (Fig. 3), which suggests that the starting phase (0 h) of mPERs in second DD is close to CT 15, the same starting phase as the behavioral rhythms. However, mPER rhythms could not be detected in SCN at the previous time points of 6 and 12 h (data not shown), which was surprising because rhythms in liver were detectable at those times (see Discussion). mPER levels at these times were constant, similar to those in LL.
Together, our data show that peripheral and SCN clocks become rapidly rhythmic again, consistent with the behavioral data, during LL→DD transfer. In addition, extrapolated starting phases in the second DD are similar (∼CT 15) in locomotor activity rhythms, liver, and SCN.
The phase of behavioral rhythms can be reversed by only a 15 h light pulse at ZT 12
Because our data showed that behavioral rhythms can be disrupted and rapidly reset to a specific phase by LL followed by DD, we set out to measure the minimum duration of constant light that can reset the behavioral rhythm to that phase. To differentiate LL→DD induced resetting of the clock from conventional light-induced phase shifting, we examined LL durations that would generate the maximal phase shift of ∼12 h. To our knowledge, such a large phase shift by a single light pulse has not been reported in wild-type mice. As LL→DD resets the circadian system from arrhythmic to ∼CT 15 at the transition (Fig. 1; Table 1), a 15 h light pulse (LL treatment) at ZT 12 (the end of the light period in a 12:12 LD cycle), followed by release into DD, may induce the maximum phase shift of ∼12 h. The unshifted clock would be at CT 03, but the LL-treated clock would be at ∼CT 15, provided that such a short LL treatment exerts the same effects as the long light treatment. In case this is not enough, we also tested 3 d (15 h + 2 d) and 5 d (15 h + 4 d) of constant light to see how much constant light is required to reverse the behavioral rhythm (Fig. 4 A, diagram). In every mouse (6 of 6), 5 d of constant light induced phase reversal, consistent with our prediction and previous data (Fig. 4 B). Three days of constant light also elicited phase reversal in 7 of 9 mice and induced 7–8 h of phase shift in the other 2 mice (Fig. 4 B). Although not in every mouse, a 15 h light pulse at ZT 12 also caused clear phase reversal in 8 of 12 mice (Fig. 4 B). However, the same light pulse only elicited 2–3 h phase shifts in the other 4 mice (data not shown), suggesting that the 15 h light pulse at ZT 12 is close to a minimum LL treatment capable of resetting the rhythm. We observed that the level of light is critical to induce the phase reversal. When we used lower intensity light (∼250 lux), we could not induce large phase shifts in any of the subjected mice (n = 4) with a 15 h light pulse at ZT 12; we could induce only 3–4 h phase shifts (data not shown). We did not test extensively whether different conditions such as longer light pulses than 15 h (e.g., 18 h) and/or initiating the pulse at times other than ZT 12 would also produce phase reversal. However, because our data showed that LL followed by DD sets the clock to ∼CT 15 regardless of the phase of previous rhythms, we expect that phase reversal depends on very specific combinations of light pulse duration and phase. Thus, an 18 h pulse initiated at ZT 12 or a 15 h pulse initiated at ZT 15 would produce smaller phase shifts. Consistent with this prediction, longer light pulses do not necessarily produce larger phase shifts. Comas et al. (2006) demonstrated that a 9 h light pulse can produce a larger phase shift than an 18 h light pulse in mice.
It takes >5 d to reverse the behavioral phase by a reversed light/dark cycle
To evaluate the power of the LL treatment to reset the clock, we compared it to a different lighting regimen: reversing the light/dark cycle, or shifting the light/dark cycle by 12 h. We compared the ability of this lighting condition to produce the maximal behavioral phase shift in the same environment with the same light intensity as used previously for the LL resetting. We measured behavioral and mPER protein rhythms after the LD cycle is reversed (shifted by 12 h) (Fig. 5). Mice were entrained in a 12 h LD cycle for >2 weeks before they were subjected to 1, 3, or 5 d of a reversed LD cycle (DL) followed by DD (Fig. 5 A). As expected from previous work (Daan and Pittendrigh, 1976), locomotor activity was immediately suppressed by light during DL, and this “masking” effect caused the phase of the locomotor activity rhythm to appear normal (i.e., activity onset occurred around lights off) during DL after 3 d. However, analysis of true circadian phase manifested in DD revealed that 3 d of DL only elicited 7.73 h of phase shift on average and only 5 d of DL induced a nearly complete phase reversal: 11.55 h of phase shift (Fig. 5 A).
It is possible that the modest phase shifts produced by 1 d of the reversed DL cycle may be attributable to the method used for reversing the cycle, which involved repeating the “D” cycle twice (LD→DL). Thus, we also tested whether bigger phase shifts could be induced when mice were exposed to two consecutive “L” cycles during the reversal (DL→LD). The second L was followed by DD. In another words, mice were exposed to 24 h of light before DD. However, this treatment also produced small phase shifts (3.11 ± 0.67 h, n = 4) in behavioral rhythms similar to those produced by the 1 d after LD→DL transfer in Figure 5 A (Fig. 5 B). None of the mice exhibited phase reversal. The DL→LD treatment is similar to the application of a 12 h light pulse at ZT 12, which is only 3 h shorter than the 15 h LL treatment described above. Thus, the failure of this treatment to produce phase reversal adds further support to our conclusion that the 15 h light pulse at ZT 12 is close to the minimum duration of light pulse required (under our laboratory conditions) to induce such an unprecedented magnitude of phase resetting. The small phase shift produced by a 12 h light pulse at ZT 12 is likely a conventional phase resetting response which is mediated by rapid induction of Per gene expression in the SCN (Shearman et al., 1997; Shigeyoshi et al., 1997; Albrecht et al., 2001).
Because it takes ∼5 d to stably reverse the SCN clock using DL cycles, as judged by behavioral rhythms, we predicted that at least the same number of DL cycles is required to phase-reverse peripheral clocks. To determine how fast peripheral clocks are reset in DL, we measured mPER1 and 2 rhythms in liver on the second, fifth, and eighth day in a DL cycle (Fig. 5 C). Consistent with behavioral rhythms, both mPER1 and 2 rhythms were not significantly changed on the second day in DL. However, on the fifth day in DL, mPER1 and 2 rhythms were dramatically changed compared with those in control mice. The mPER profile (protein levels and phosphorylation status) on the fifth day in DL was apparently completely reversed compared with that in control mice (Fig. 5 C). For example, mPER1 and 2 at ZT 12 in DL were similar to those at ZT 12 in LD (before the light/dark cycle was switched). In addition, there was no further change on the eighth day, indicating that 5 d of DL cycle were enough to almost reverse the liver clock as was the case for behavioral rhythms. This suggests that phase resetting in liver may not lag behind that of the SCN significantly, assuming that the behavioral rhythms accurately reflect the status of the SCN clock.
Together, our data show that constant light is a much more efficient way to induce a large phase shift than a shifted light/dark cycle such as the reversed LD cycle. More importantly, the underlying mechanism for the large phase shift response may differ from the mechanism mediating the conventional small phase shift responses.
Discussion
Although arrhythmic behavior in LL was recognized decades ago, it remained unclear how the animals would behave and how the molecular clock would be reset when the LL is removed. Here, we show that the disruption of behavioral rhythms and of the molecular clock by LL is quickly corrected after transfer to DD. Our methods (behavioral and tissue-level protein analyses) do not reveal the activities of individual cells and, therefore, do not directly show desynchronization and resynchronization of those cells. However, Ohta et al. (2005) previously demonstrated using single-cell reporter assays that LL-induced arrhythmicity is caused by SCN neural desynchronization. Reporter genes have proven extremely useful for revealing intrinsic properties of the circadian oscillator and the coupling of cells within the SCN (Yamazaki et al., 2000; Yoo et al., 2004; Ohta et al., 2005), because they allow measurement of rhythms in real time from individual cells as well as from cultured tissue. However, this ex vivo approach may not be appropriate to probe the in vivo clock for circadian properties such as phase and amplitude. Recently, Yoshikawa et al. (2005) explicitly demonstrated that the timing of tissue preparation for ex vivo measurement of reporter activity dramatically affects these properties, as can simple media changes (Yoo et al., 2004; Liu et al., 2007). Thus, although our in vivo method does not measure individual oscillators, it has the advantage of avoiding complications associated with ex vivo tissue preparation, and is appropriate and adequate for measuring the rhythmic output of synchronized cells. Because previous work has shown that LL causes desynchronization of neurons, we can conclude from our data that emergence of new rhythmicity after LL results from resynchronization.
Both locomotor behavior and mPER status in liver lost rhythmicity in LL and regained robust rhythmicity rapidly after transfer to DD. However, we were surprised to find that oscillator resynchronization in the SCN seemed a little slower than these peripheral cell types; despite several attempts, we could not detect consistent mPER phosphorylation and abundance rhythms in SCN tissue earlier than 18–24 h after the LL–DD transfer. We do not know whether this apparent slowness is attributable to our SCN sampling method (see Materials and Methods), heterogeneity of SCN tissue (Lee et al., 2003), or a different mechanism of SCN synchrony by LL→DD than is found in peripheral cells. It should be noted that unlike liver cells, the oscillator cells in SCN are coupled to each other through neural connections. Thus, to restore normal rhythms in SCN, the individual cells must be not only synchronized, but also properly coupled to each other to restore network function, which may explain the apparent slowness. In any case, the relative slowness of SCN resetting begs a question regarding the hierarchy of the circadian system: are the peripheral clocks and behavioral rhythms reset independent of the SCN during the early stage of resynchronization? One might assume that the SCN drives peripheral clock resynchronization because the SCN receives light input from the retina, whereas the liver does not. However, certain zeitgebers can affect peripheral clocks without phase shifting the SCN clock (Damiola et al., 2000), and light/darkness can affect the body independent of the SCN [e.g., masking effects described by Redlin (2001)]. We therefore speculate that the LL→DD transition may generate multiple internal time cues. Some of these cues (perhaps hormonal) may have reset the desynchronized liver oscillators before the SCN itself became fully resynchronized. Consistent with this idea, peripheral oscillators can be resynchronized or reset by numerous signals (Balsalobre et al., 2000a,b). If peripheral oscillators were synchronized earlier than the SCN cells, then both behavior and the SCN may have received reinforcing signals from the peripheral clocks. Our study does not address this hierarchy of synchronization between peripheral and SCN clocks, but our results suggest that further study may be warranted.
Worthy of comment is the fact that some mice maintain weak behavioral rhythmicity in LL. We hypothesize that the weak phase was generated by a small minority of still-synchronized clock cells in the SCN. Because behavioral rhythms are driven by the combined output of individual clock cells (Low-Zeddies and Takahashi, 2001), a weak amplitude oscillation from whole SCN could be generated by the small number of synchronous cells over the baseline of the majority of desynchronized cells. After transfer from LL→DD, the phase of the minority of cells producing the LL rhythm would become irrelevant, overwhelmed by the synchronous oscillations of the majority, whose phase is set by the transfer. Consistent with this hypothesis, the phases of weak rhythms in LL do not correlate with the new phase generated when mice are transferred to DD, and the new phase is similar to that seen in arrhythmic mice.
Our LL treatment represents an example of type 0 entrainment (Dunlap et al., 2004), in which the new phase after the stimulus (LL) is the same regardless of the old phase before the stimulus. The old phase would have been widely distributed over the circadian cycle among mice after >9 d in the first DD, but the new phase was the same after the LL treatment. Furthermore, the timing of the DD→LL transition did not affect our results, because three different times of the transition did not produce a significant difference in the starting phase of rhythms in the subsequent DD (data not shown). Type 1 entrainment, more typical of mammals, involves phase shifts of limited magnitude; thus, the final phase depends critically on stimulus timing (Dunlap et al., 2004). To our knowledge, type 0 phase resetting has not been reported in wild-type mice (Comas et al., 2006). There has, however, been reports of large phase shifts in Clock Δ19 mutant mice (Vitaterna et al., 2006) and in tau mutant hamsters (Shimomura and Menaker, 1994). Vitaterna et al. (2006) suggested that the 6 h light pulse can induce a type 0 response in Clock Δ19 heterozygous mice, but not in wild-type mice, because the amplitude of Per gene oscillation is reduced in the SCN of the mutant mice; thus, the same stimulus can produce a bigger effect in the mutant mice based on the limit-cycle model (Winfree, 1980; Johnson et al., 2003). Interestingly, tau mutant hamsters also show reduced amplitude of Per oscillation (Lowrey et al., 2000). It is unlikely that our type 0 entrainment is mediated through the reduction in the amplitude of PER oscillations in individual oscillators, because Ohta et al. (2005) showed that LL did not significantly reduce the amplitude of mPer1 oscillation in individual SCN neurons. Both Clock Δ19 and tau mutations damage the molecular mechanism of the clock, making the rodents bearing those mutations unusually susceptible to large phase shifts. However, our experiments show that even rodents with normal clocks can exhibit type 0 resetting when given the right stimulus.
In our LD→DL or DL→LD experiments, the lighting regimens may be viewed as a series of 12 h phase shifts administered over 1, 3, and 5 d. Circadian phase was gradually shifted after each light pulse and reached an equilibrium after 5 d. However, a single 15 h light pulse at ZT 12 achieved the same phase shift produced by five 12 h light pulses. It is conceivable that a different molecular mechanism may be responsible for the two processes, as stated previously. We propose that type 0 resetting may occur in wild-type mice through rapid desynchronization and resynchronization of oscillators based on our present studies and those by Ohta et al. (2005). However, because we did not measure the circadian oscillators at the single cell level, we cannot rule out that the phase reversal by a single light pulse may be also mediated by a more exaggerated form of the conventional phase shifting mechanism; the LL treatment may have reduced the amplitudes of mPER oscillations, thus increasing the likelihood of type 0 phase resetting as discussed above. We believe that our data [as an extension of Ohta et al. (2005)] fit the desynchronization–resynchronization model better, but future studies will have to explore alternatives such as LL-mediated exaggeration of the conventional phase-shifting response.
Interestingly, type 0 phase resetting with a major shift of 10.8 h has also been reported in humans (Czeisler et al., 1989). However, the large phase shift was induced by a 5 h light stimulus with a very strong intensity (∼10,000 lux) repeated over 3 d. If (as suggested by homology) our results in mice (by whichever mechanism) can be applied to humans, then it may be possible to shift the human clock 12 h in just 1 d using an appropriate constant light stimulus. This would provide substantial benefits to shift workers, jet lag sufferers, and patients with disorders of circadian phase.
Footnotes
This work was supported by National Institutes of Health Grant NS-053616 awarded to Choogon Lee. We thank Ailing Zheng for excellent technical assistance and Kyunghye Kim for data analysis during the project. We thank Dennis Chang for assistance with manuscript revisions.
References
- Albrecht U, Sun ZS, Eichele G, Lee CC. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- Albrecht U, Zheng B, Larkin D, Sun ZS, Lee CC. MPer1 and mper2 are essential for normal resetting of the circadian clock. J Biol Rhythms. 2001;16:100–104. doi: 10.1177/074873001129001791. [DOI] [PubMed] [Google Scholar]
- Allada R, Emery P, Takahashi JS, Rosbash M. Stopping time: the genetics of fly and mouse circadian clocks. Annu Rev Neurosci. 2001;24:1091–1119. doi: 10.1146/annurev.neuro.24.1.1091. [DOI] [PubMed] [Google Scholar]
- Aschoff J. Exogenous and endogenous components in circadian rhythms. Cold Spring Harb Symp Quant Biol. 1960;25:11–28. doi: 10.1101/sqb.1960.025.01.004. [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schütz G, Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000a;289:2344–2347. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Marcacci L, Schibler U. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts. Curr Biol. 2000b;10:1291–1294. doi: 10.1016/s0960-9822(00)00758-2. [DOI] [PubMed] [Google Scholar]
- Comas M, Beersma DG, Spoelstra K, Daan S. Phase and period responses of the circadian system of mice (Mus musculus) to light stimuli of different duration. J Biol Rhythms. 2006;21:362–372. doi: 10.1177/0748730406292446. [DOI] [PubMed] [Google Scholar]
- Czeisler CA, Kronauer RE, Allan JS, Duffy JF, Jewett ME, Brown EN, Ronda JM. Bright light induction of strong (type 0) resetting of the human circadian pacemaker. Science. 1989;244:1328–1333. doi: 10.1126/science.2734611. [DOI] [PubMed] [Google Scholar]
- Daan S, Pittendrigh CS. A functional analysis of circadian pacemakers in nocturnal rodents. J Comp Physiol. 1976;106:267–290. [Google Scholar]
- Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000;14:2950–2961. doi: 10.1101/gad.183500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings MH, Reddy AB, Maywood ES. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci. 2003;4:649–661. doi: 10.1038/nrn1177. [DOI] [PubMed] [Google Scholar]
- Hattar S, Lucas RJ, Mrosovsky N, Thompson S, Douglas RH, Hankins MW, Lem J, Biel M, Hofmann F, Foster RG, Yau KW. Melanopsin and rod-cone photoreceptive systems account for all major accessory visual functions in mice. Nature. 2003;424:76–81. doi: 10.1038/nature01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CH, Elliott JA, Foster R. Entrainment of circadian programs. Chronobiol Int. 2003;20:741–774. doi: 10.1081/cbi-120024211. [DOI] [PubMed] [Google Scholar]
- Johnson CH, Elliott J, Foster R, Honma K, Kronauer R. Fundamental properties of circadian rhythms. In: Dunlap JC, Loros JJ, Decoursey PJ, editors. Chronobiology. Sunderland, MA: Sinauer; 2005. pp. 67–106. [Google Scholar]
- Knutsson A. Health disorders of shift workers. Occup Med (Lond) 2003;53:103–108. doi: 10.1093/occmed/kqg048. [DOI] [PubMed] [Google Scholar]
- Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15:R271–R277. doi: 10.1093/hmg/ddl207. Spec No 2. [DOI] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- Lee HS, Billings HJ, Lehman MN. The suprachiasmatic nucleus: a clock of multiple components. J Biol Rhythms. 2003;18:435–449. doi: 10.1177/0748730403259106. [DOI] [PubMed] [Google Scholar]
- Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, Buhr ED, Singer O, Meeker K, Verma IM, Doyle FJ, 3rd, Takahashi JS, Kay SA. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell. 2007;129:605–616. doi: 10.1016/j.cell.2007.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low-Zeddies SS, Takahashi JS. Chimera analysis of the Clock mutation in mice shows that complex cellular integration determines circadian behavior. Cell. 2001;105:25–42. doi: 10.1016/s0092-8674(01)00294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science. 2000;288:483–492. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M, Peirson SN, Hankins MW, Foster RG. Long-term constant light induces constitutive elevated expression of mPER2 protein in the murine SCN: a molecular basis for Aschoff's rule? J Biol Rhythms. 2005;20:3–14. doi: 10.1177/0748730404272858. [DOI] [PubMed] [Google Scholar]
- Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U. Circadian gene expression in individual fibroblasts: cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell. 2004;119:693–705. doi: 10.1016/j.cell.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Ohta H, Yamazaki S, McMahon DG. Constant light desynchronizes mammalian clock neurons. Nat Neurosci. 2005;8:267–269. doi: 10.1038/nn1395. [DOI] [PubMed] [Google Scholar]
- Panda S. Multiple photopigments entrain the Mammalian circadian oscillator. Neuron. 2007;53:619–621. doi: 10.1016/j.neuron.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Panda S, Hogenesch JB, Kay SA. Circadian rhythms from flies to human. Nature. 2002;417:329–335. doi: 10.1038/417329a. [DOI] [PubMed] [Google Scholar]
- Panda S, Provencio I, Tu DC, Pires SS, Rollag MD, Castrucci AM, Pletcher MT, Sato TK, Wiltshire T, Andahazy M, Kay SA, Van Gelder RN, Hogenesch JB. Melanopsin is required for non-image-forming photic responses in blind mice. Science. 2003;301:525–527. doi: 10.1126/science.1086179. [DOI] [PubMed] [Google Scholar]
- Redlin U. Neural basis and biological function of masking by light in mammals: suppression of melatonin and locomotor activity. Chronobiol Int. 2001;18:737–758. doi: 10.1081/cbi-100107511. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Schwartz WJ, Zimmerman P. Circadian timekeeping in BALB/c and C57BL/6 inbred mouse strains. J Neurosci. 1990;10:3685–3694. doi: 10.1523/JNEUROSCI.10-11-03685.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF, Jr, Reppert SM. Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- Shimomura K, Menaker M. Light-induced phase shifts in tau mutant hamsters. J Biol Rhythms. 1994;9:97–110. doi: 10.1177/074873049400900201. [DOI] [PubMed] [Google Scholar]
- Silver R, LeSauter J, Tresco PA, Lehman MN. A diffusible coupling signal from the transplanted suprachiasmatic nucleus controlling circadian locomotor rhythms. Nature. 1996;382:810–813. doi: 10.1038/382810a0. [DOI] [PubMed] [Google Scholar]
- Skopik SD, Pittendrigh CS. Circadian systems, II. The oscillation in the individual Drosophila pupa; its independence of developmental stage. Proc Natl Acad Sci U S A. 1967;58:1862–1869. doi: 10.1073/pnas.58.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M. Entrainment of the circadian clock in the liver by feeding. Science. 2001;291:490–493. doi: 10.1126/science.291.5503.490. [DOI] [PubMed] [Google Scholar]
- Sudo M, Sasahara K, Moriya T, Akiyama M, Hamada T, Shibata S. Constant light housing attenuates circadian rhythms of mPer2 mRNA and mPER2 protein expression in the suprachiasmatic nucleus of mice. Neuroscience. 2003;121:493–499. doi: 10.1016/s0306-4522(03)00457-3. [DOI] [PubMed] [Google Scholar]
- Terazono H, Mutoh T, Yamaguchi S, Kobayashi M, Akiyama M, Udo R, Ohdo S, Okamura H, Shibata S. Adrenergic regulation of clock gene expression in mouse liver. Proc Natl Acad Sci U S A. 2003;100:6795–6800. doi: 10.1073/pnas.0936797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama T, Krout KE, Nguyen XV, Karpitskiy V, Kollert A, Mettenleiter TC, Loewy AD. Suprachiasmatic nucleus: a central autonomic clock. Nat Neurosci. 1999;2:1051–1053. doi: 10.1038/15973. [DOI] [PubMed] [Google Scholar]
- Vitaterna MH, Takahashi JS, Turek FW. Overview of circadian rhythms. Alcohol Res Health. 2001;25:85–93. [PMC free article] [PubMed] [Google Scholar]
- Vitaterna MH, Ko CH, Chang AM, Buhr ED, Fruechte EM, Schook A, Antoch MP, Turek FW, Takahashi JS. The mouse Clock mutation reduces circadian pacemaker amplitude and enhances efficacy of resetting stimuli and phase-response curve amplitude. Proc Natl Acad Sci U S A. 2006;103:9327–9332. doi: 10.1073/pnas.0603601103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winfree AT. The geometry of biological time. New York: Springer; 1980. [Google Scholar]
- Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, Ueda M, Block GD, Sakaki Y, Menaker M, Tei H. Resetting central and peripheral circadian oscillators in transgenic rats. Science. 2000;288:682–685. doi: 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Yamazaki S, Menaker M. Effects of preparation time on phase of cultured tissues reveal complexity of circadian organization. J Biol Rhythms. 2005;20:500–512. doi: 10.1177/0748730405280775. [DOI] [PMC free article] [PubMed] [Google Scholar]