

Abstract
Plasmacytoid dendritic cells (pDCs) play a key role in the innate immune response to viral infection, due largely to their ability to produce large quantities of type I IFNs. These cells are also notable for their ability to differentiate into conventional dendritic cells after appropriate stimulation. Here, we show that a splenic population of murine CD11c+ cells expressing pDC markers Gr-1, B220, and PDCA-1 is preferentially parasitized after infection with the virulent RH strain of Toxoplasma gondii. Although these markers are closely associated with pDCs, the population we identified was unusual because the cells express CD11b and higher than expected levels of CD11c. By adoptive transfer of CD45.1-positive cells into CD45.2 congenic mice, we show that CD11c+Gr-1+ cells migrate from the peritoneal cavity to the spleen. During infection, these cells accumulate in the marginal zone region. Recruitment of infected CD11c+Gr-1+ cells to the spleen is partially dependent upon signaling through chemokine receptor CCR2. Intracellular cytokine staining demonstrates that infected, but not noninfected, splenic CD11c+Gr-1+ dendritic cells are suppressed in their ability to respond to ex vivo TLR stimulation. We hypothesize that Toxoplasma exploits pDCs as Trojan horses, targeting them for early infection, suppressing their cytokine effector function, and using them for dissemination within the host.
Toxoplasma gondii is an intracellular protozoan known for its ability to induce strong Th1-type cytokine responses (1). IL-12 and IFN-γ are required to survive acute and chronic infection with this opportunistic pathogen, but overproduction of these cytokines can precipitate proinflammatory pathology that results in host death (2-7). Therefore, there is a need to tightly regulate Th1 cytokine production to allow parasite control without causing host death. Early events in initiation of immunity to Toxoplasma are likely to play a prominent role in determining the strength and pattern of cytokine production during infection.
Dendritic cells (DCs)3 are now recognized as central in immune response initiation, due to their ability to acquire Ag, migrate from peripheral tissues to lymphoid organs, activate naive T cells, and secrete immune-polarizing cytokines such as IL-12. DC biology is complex insofar as there are many distinct subpopulations with discrete phenotypic characteristics and functional activities (8, 9). Broadly, DCs can be subdivided into conventional DCs and type I IFN-producing plasmacytoid DCs (pDCs). Conventional DCs can be further subdivided based upon tissue localization and surface marker expression. Furthermore, other types of DCs arise in inflammatory or infection conditions, such as the TNF- and inducible NO synthase-expressing DCs that emerge during mouse infection with Listeria monocytogenes and monocyte-derived DCs that appear at the infection site during Leishmania major infection (10-12).
DCs are required to survive infection with T. gondii as shown in a recent study where diptheria toxin was used to deplete cells expressing the diptheria toxin receptor under the control of the CD11c promoter (13). Older studies showed that CD8α+ DCs in the spleen are an important IL-12 source after i.v. injection of parasite lysate Ag (14). The identification of a Toxoplasma profilin molecule that induces high level IL-12 production from splenic DCs through activation of TLR11 provides a molecular explanation for this effect (15). Unexpectedly, it was recently reported that pDCs, which are usually associated with responses to viruses, acquire T. gondii-derived Ag and produce IL-12 during infection with this parasite (16). At the same time, other studies provide evidence that DC infection with Toxoplasma down-modulates their capacity to produce IL-12 (17). Discrepancies in how these cells respond to infection may result from using different DC subsets, or they may reflect differences in DC responses to extracellular parasite molecules vs their inherent ability to produce cytokines after parasite infection.
In addition to their role in initiation of immunity to Toxoplasma, there is evidence that DCs serve as vehicles for dissemination during infection. During early oral infection, infected CD11c+ cells were identified in the lamina propria, Peyer's patches, and mesenteric lymph nodes, although CD11c−CD11b+ cells were also found to be a reservoir of infection at these locations (18). Interestingly, another study reported that infection of human monocyte-derived DCs as well as mouse bone marrow-derived DCs induce a state of hyper-motility that, at least in mice, potentiates dissemination in the host (19). Therefore, it is seemingly paradoxical that Toxoplasma may target for infection cells that themselves play a predominant role in triggering anti-parasite immunity.
To gain insight into in vivo interactions between DCs and Toxoplasma, and to determine the functional consequences of intracellular infection in an in vivo situation, we examined recruitment and invasion of DCs during the early immune response. We chose to use an i.p. infection model so that we could precisely control delivery of parasites and examine early cell recruitment and invasion at the site of infection. Most of the infected cells in the peritoneal cavity expressed monocyte/macrophage and neutrophil markers. In striking contrast, in the spleen the major population of infected cells was largely restricted to a subset that expressed surface markers most closely associated with pDCs. These cells were present as a minor population in the peritoneal cavity, but they were markedly more susceptible to infection compared with other cell types. We examined cytokine production by these cells and found that infected DCs’ ability to produce IL-12 was suppressed, whereas the corresponding noninfected population was capable of producing this cytokine. We hypothesize that T. gondii targets pDCs for early infection and dissemination, in the process inhibiting their ability to produce IL-12.
Materials and Methods
Mice
Female C57BL/6 mice 6−8 wk of age were purchased from Charles River Laboratories and Taconic Farms. CCR2−/−, CCR5−/−, and control B6.129 mice were purchased from The Jackson Laboratory. CD45.1 C57BL/6 background congenic mice (strain B6.SJL-Ptprca Pepcb/BoyJ) were maintained as a breeding colony at the Cornell College of Veterinary Medicine Animal Facility and were provided by Dr. J. Appleton (James A. Baker Institute for Animal Health, Ithaca, NY). The animals were housed in the Cornell University College of Veterinary Medicine transgenic mouse core facility, which is accredited by the American Association for Accreditation of Laboratory Animal Care.
Parasites and infections
Tachyzoites of the RH strain were originally purchased from the American Type Tissue Collection, and a transgenic RH strain expressing tandem copies of yellow fluorescence protein (RH-YFP) was a gift from D. Roos (University of Pennsylvania, Philadelphia, PA) and B. Striepen (University of Georgia, Athens, GA). Parasites were maintained by passage in human foreskin fibroblasts in DMEM (Mediatech) supplemented with 1% bovine growth serum (HyClone), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen). Infections were accomplished by i.p. injection of 106 tachyzoites in 100 μl of PBS.
Flow cytometric analysis
Splenocytes were prepared by mechanical disruption of whole spleens, and red cells were lysed using red cell lysis buffer (8.3 g/L ammonium chloride in 0.01 M Tris-HCl buffer; Sigma-Aldrich). Splenocytes and peritoneal exudate cells were filtered through 70 μm filters, and incubated on ice for 15 min with 10% normal mouse serum in FACS buffer (1% BSA and 0.05% sodium azide in PBS). Surface staining was performed for 30 min on ice, and intracellular cytokine staining was performed using the BD Cytofix/Cytoperm fixation/permeabilization solution kit (BD Pharmingen). Cells were stained with the following Ab obtained from BD Pharmingen: anti-CD11c allophycocyanin (clone HL3), anti-Gr-1/Ly6G PerCP (clone RB6−8C5), anti-CD4 PE (clone RM4−5), anti-B220 PE (clone RA3−6B2), and anti-IL-12p40 PE (C15.6). The following Abs were obtained from eBioscience: anti-CD40 PE (clone 1C10), anti-B7.1 PE (clone 16−10A1), anti-B7.2 PE (GL1), anti-CD3 PE (145−2C11), anti-CD11b PE (clone M1/70), anti-MHC class II PE (clone M5/114.15.2), anti-CD8 PE (clone 53−6.7) and anti-CD45.1 PE (clone A20). Anti-mPDCA-1 PE (clone JF05−1C2.4.1) was purchased from Miltenyi Biotec, and anti-T. gondii p30 FITC (clone G-II9) from Argene. Cells were analyzed using CellQuest software and a FACSCalibur cytometer (BD Biosciences).
Adoptive transfer of peritoneal exudate cells
Peritoneal exudate cells were harvested from CD45.1 congenic mice on day 3 after i.p. inoculation with 106 RH-YFP tachyzoites, and 1.5 × 107 cells were injected i.p. into C57BL/6 mice that had been infected on the same day with the same dose of RH-YFP parasites as the donor mice. After 48 h, peritoneal cells and splenocytes were harvested, and presence and phenotype of CD45.1 cells in the spleen were analyzed by flow cytometry.
In vitro culture and analysis of splenic DCs
Splenocytes were harvested from mice 4 days after infection with RH-YFP tachyzoites. DCs were enriched using pan-DC microbeads according to the manufacturer's instructions (Miltenyi Biotec). The cells were subsequently incubated for 24 h with or without Escherichia coli LPS (Sigma-Aldrich) and CpG oligodinucleotide 1826 (Coley Pharmaceutical Group), including Golgi-Stop (BD Pharmingen) for the last 4 h of incubation. The DC-enriched population was washed and stained for surface markers and intracellular IL-12p40, then analyzed by flow cytometry.
Immunofluorescence labeling
To visualize morphology of infected cells, splenocytes were harvested from mice infected i.p. 4 days earlier with RH-YFP tachyzoites and stained (as described above for FACS analysis) with anti-Gr-1 PE (BD Pharmingen). Cells were transferred to a glass slide via cytospin (Shandon), and nuclei were labeled with 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen, Molecular Probes). Staining of spleen sections from infected mice was performed as described previously (20). Briefly, fragments of spleen were embedded in Tissue-Tek OCT (Miles Laboratories), snap-frozen in pre-chilled methyl-butane, and stored at −80°C. Cryostat sections (8 μm) of spleen were fixed in 4% paraformaldehyde/PBS and blocked with 10% normal goat serum and the avidin-biotin blocking kit (Vector Laboratories). Tissue sections were incubated with the following reagents (Ab were obtained from BD Pharmingen, unless specified): 1) biotin-anti-MOMA-1 mAb (Bachem) followed by Cy3-streptavidin (Jackson ImmunoResearch Laboratories) or 2) anti-CD11c mAb plus Cy3-anti-hamster IgG in combination with either biotin-CD3 mAb or biotin-Gr-1 mAb followed by Cy5-streptavidin (Jackson ImmunoResearch Laboratories). Nuclei were stained with DAPI (Molecular Probes).
Statistical analyses
Statistical analysis was performed using Prism 4 software (GraphPad software) and significance was calculated using an unpaired, two-tailed Student's t test.
Results
A rare population of CD11c+ Gr-1+ cells in the peritoneal cavity is preferentially targeted following i.p. infection with tachyzoites
To elucidate cell types parasitized by Toxoplasma during early infection, C57BL/6 mice were infected i.p. with RH-YFP, a genetically engineered parasite strain expressing tandem copies of the gene encoding yellow fluorescence protein, and the peritoneal exudate cells were analyzed by flow cytometry 3 days later. As expected for Toxoplasma, a parasite known for its ability to promiscuously infect a diverse range of cell types, most cells in the peritoneal cavity were parasitized at this time point, including macrophages (F4/80+), neutrophils (Gr-1+), DCs (CD11c+), and B cells (B220+) (Fig. 1A). Nevertheless, while ∼50% of macrophages, neutrophils, and DCs harbored parasites, B lymphocytes and T lymphocytes were considerably more resistant to infection. In each case only a minority of cells were infected (20% and 5% for B220+ and CD3+ lymphocytes, respectively). There are several subsets of DCs, including some that coexpress CD11c and Gr-1. A small population of the CD11c+ Gr-1+ cells (<1%) was detected in the peritoneal cavity 72 h after infection (Fig. 1B). These cells were remarkable inasmuch as over 80% were positive for parasites (Fig. 1C).
FIGURE 1.
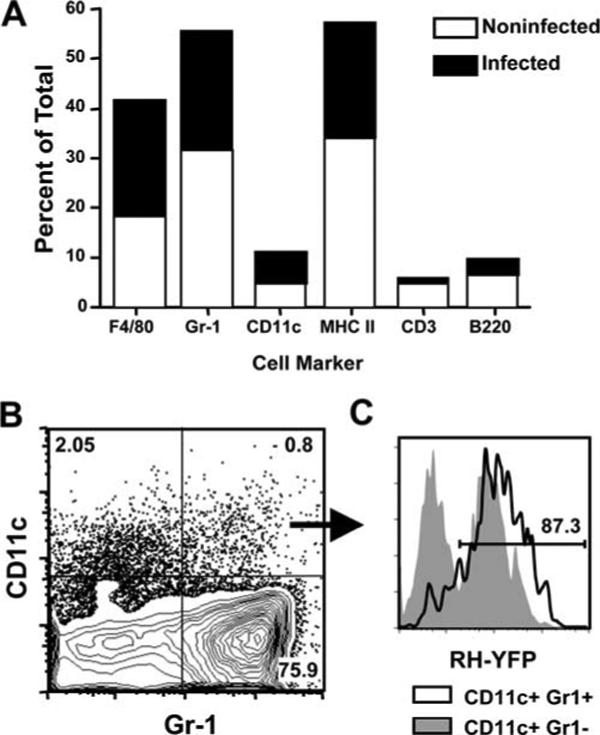
CD11c+Gr-1+ cells in the peritoneal cavity are highly susceptible to infection with T. gondii. A, Mice were infected i.p. with RH-YFP tachyzoites, and peritoneal exudate cells were harvested 3 days postinfection for phenotypic analysis and assessment of infection levels by flow cytometry. B, Costaining for CD11c and Gr-1 reveals a small population of CD11c+Gr-1+ cells in the peritoneal cavity 3 days postinfection. C, Examination of parasite levels in the CD11c+Gr-1+ population reveals that these cells are highly susceptible to infection (black line histogram) compared with CD11c+Gr-1− cells (shaded histogram). The numbers indicate the percent of cells in each quadrant (B) or gate (C). This experiment was repeated three times with similar results.
The major population of infected cells in the spleen predominantly expresses CD11c and Gr-1
We next examined infection in the spleen, an organ targeted by the parasite during acute infection (2, 6). Splenocytes were obtained from mice infected 4 days before by i.p. administration of RH-YFP tachyzoites. At this time point, 5−6% of total splenocytes were positive for T. gondii (Fig. 2A). Gating on the infected population, we found that the majority (∼70%) coexpressed CD11c and Gr-1 (Fig. 2B). We also found that ∼20% of infected cells possessed a CD11c−Gr-1+ phenotype. This population is likely to be neutrophils, but it is also possible that some are Gr-1+ inflammatory monocytes described by Sibley and colleagues (21, 22). We also determined the proportion of total splenic CD11c+Gr-1+ cells that were infected with T. gondii. As shown in Fig. 2C, ∼60% of this population harbored intracellular parasites.
FIGURE 2.
CD11c+Gr-1+ cells are the predominant population of infected cells in the spleen, and exhibit phenotypic characteristics of both plasmacytoid and myeloid dendritic cells. A, Splenocytes were harvested from mice 4 days postinfection and analyzed by flow cytometry for RH-YFP tachyzoites. B, CD11c and Gr-1 expression on YFP-positive cells. C, YFP expression in the total CD11c+Gr-1+ population in the spleen. The numbers indicate the percent of cells falling within the indicated gates (A and C) or quadrants (B). D, Phenotypic analysis of infected CD11c+Gr-1+ (upper panels) and CD11c+Gr-1− cells (lower panels) shown in B. The solid lines represent each respective marker and the shaded histograms indicate isotype controls. One representative of two independent experiments is shown.
We wanted to investigate the phenotype of the CD11c+Gr-1+ population in more detail (Fig. 2D, upper panels). The cells expressed MHC class II, B220, CD11b, CD80/86, and PDCA-1. This profile, in particular expression of PDCA-1, Gr-1, and B220, was suggestive of a pDC-related population, although they are atypical in that they express CD11b and higher than expected levels of CD11c (23-26). We also examined the phenotype of the CD11c−Gr-1+ population (Fig. 2D, lower panels). These cells expressed high levels of CD11b, a molecule expressed by neutrophils. Interestingly, the cells were also positive for MHC class II and CD80.
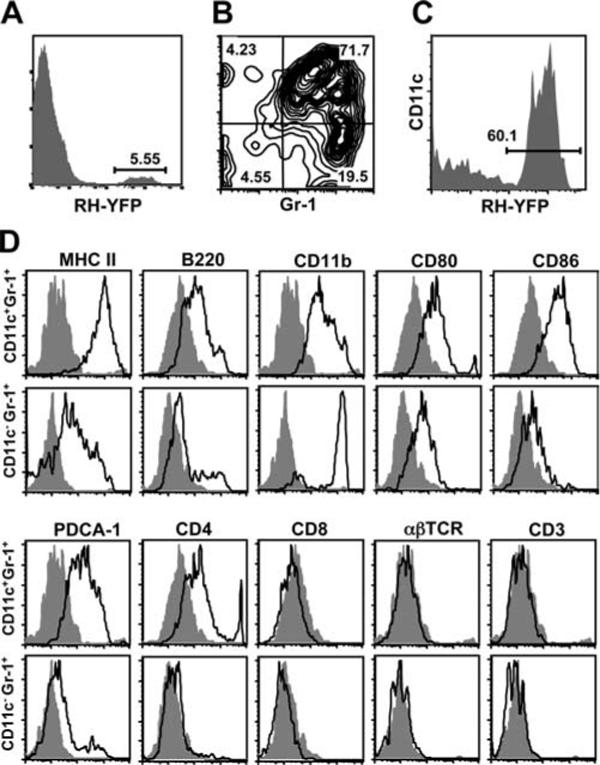
We examined the appearance of Gr-1+ parasite-infected cells in the spleen by fluorescence microscopy (Fig. 3). The vast majority of infected Gr-1+ cells were clearly distinct from the neutrophil population, because they possessed abundant cytoplasm and non-polymorphic nuclei. However, we were also able to detect some infected neutrophils, consistent with our data showing a population of infected CD11c−Gr-1+ cells (Figs. 2B and 3C).
FIGURE 3.
Morphology of Toxoplasma infected cells in the spleen. Mice were infected with RH-YFP tachyzoites (green), then 4 days later splenocytes were isolated and stained for Gr-1 (red) and DNA (DAPI, blue). Most infected cells exhibited a round or bean-shaped nucleus consistent with monocyte/macrophage/DC (A–C) and abundant cytoplasm, in which multiple parasites were found. Lesser numbers of infected polymorphonuclear leukocytes were also observed (C, arrow). The experiment was repeated twice with similar results.
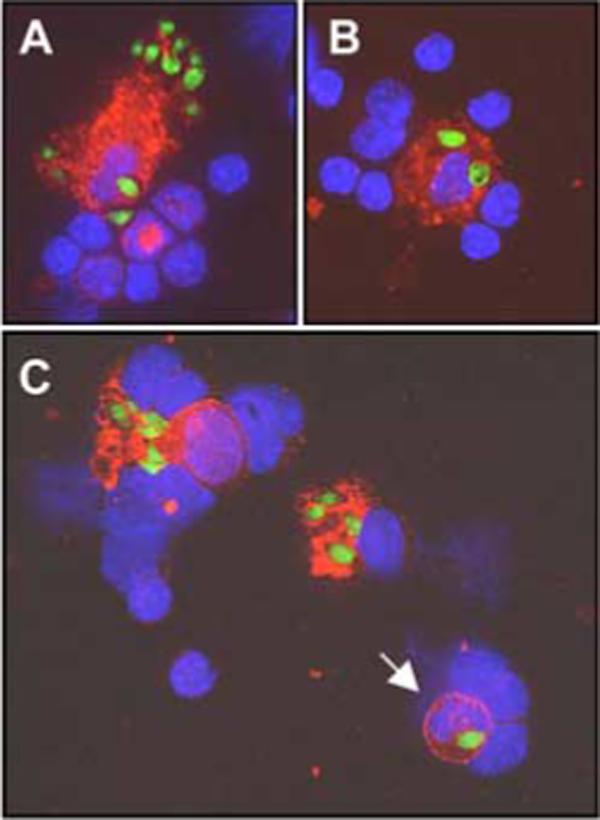
Next, we used immunofluorescence staining of frozen tissue sections to localize infected cells in situ. In spleens of day 4-infected mice, parasites were mainly present in phagocytes of the marginal zone, red pulp, and to a lesser extent in cells of the lymphoid follicles (Fig. 4). Within the splenic follicles, T cell areas also contained CD11c+ DCs infected with T. gondii (Fig. 5, A and B). In Fig. 5, C and D, we used 4-color fluorescence imaging to confirm that infected CD11c+ cells (C) also expressed Gr-1 (D).
FIGURE 4.
Infected cells are located primarily in the marginal zone of the spleen. Spleens were obtained from mice 4 days postinfection and immunofluorescence on frozen sections was performed. Staining for metallophilic macrophages (MOMA-1, red) delineates the edge of the marginal zone (MZ), revealing concentration of infected cells (yellow) in this area (arrowheads), with a small number of infected cells (arrows) in the lymphoid follicle (F). The inset shows at high magnification a MOMA-1+ cell with intracellular parasites. Blue, DAPI stained nuclei. Original magnification ×200. One representative of two independent experiments is depicted.
FIGURE 5.
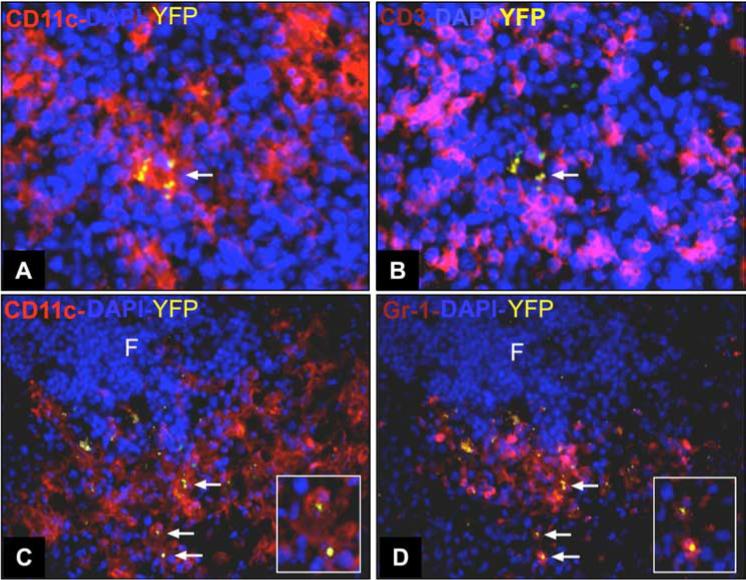
Infected cells in the T cell areas of the spleen coexpress CD11c and Gr-1. Mice were infected with RH-YFP tachyzoites and 4 days later spleens were collected for immunohistochemical staining. A, Four-color immunofluorescence staining of frozen sections shows CD11c+ cells (red) infected with RH-YFP (yellow). B, In the same section, staining for CD3 (pink) reveals T cells surrounding infected CD11c+ cells. Staining for CD11c (C) and Gr-1 (D) shows that most infected cells (arrows) coexpress CD11c (red) and Gr-1 (pink). Insets in C and D shows an expanded view of infected CD11c+Gr-1+ cells. Original magnification ×400 (A and B) and ×200 (C and D). This experiment was repeated twice with similar results.
CD11c+ Gr-1+ cells migrate from the peritoneal cavity to the spleen
Because we found that CD11c+Gr-1+ cells were highly susceptible to infection in the peritoneal cavity, we hypothesized that these cells may be migrating from the inoculation site to the spleen. To determine whether this was the case, we infected congenic B6.SJL mice (CD45.1) with i.p. injected RH-YFP tachyzoites. Two days later we harvested the peritoneal exudate cells from the B6.SJL mice and transferred them by i.p. injection into CD45.2 congenic C57BL/6 mice infected i.p. 2 days before with RH-YFP tachyzoites. Spleen and peritoneal exudate cells were harvested 48 h after transfer. We found a small number of transferred cells, which were primarily noninfected, remaining in the peritoneal cavity at this time (data not shown). As expected, in the spleen there was background staining for CD45.1+ cells in nontransferred animals (Fig. 6A). However, we found adoptively transferred parasite-infected CD45.1+ cells mobilized to the spleen in the CD45.2 congenic hosts (Fig. 6B), and as predicted, these migrating cells coexpressed Gr-1 and CD11c (Fig. 6C). We transferred cells from CD45.1+ mice infected with YFP-RH into CD45.2 animals infected with nonfluorescent RH tachyzoites (Fig. 6D). As shown in the figure, we detected the appearance of YFP-RH infected, CD45.1+ cells in the spleen. As expected, these cells also expressed Gr-1 and CD11c (data not shown). Also evident in Fig. 6D, most of the YFP+ cells were derived from recipient mice, because they do not express CD45.1. The majority of these cells are positive for Gr-1 and CD11c (data not shown), and it seems likely that they originate from host pDC infected in the peritoneal cavity by RH-YFP. We cannot be sure what accounts for the low percentage of CD45.1+ cells that traffic to the spleen in these situations. However, we suspect that the cells are negatively affected by the recovery and transfer protocol, and that many subsequently die after transfer. Regardless, from these data we conclude that infected CD11c+Gr-1+ cells are capable of migrating from the peritoneal cavity to the spleen over the course of infection.
FIGURE 6.
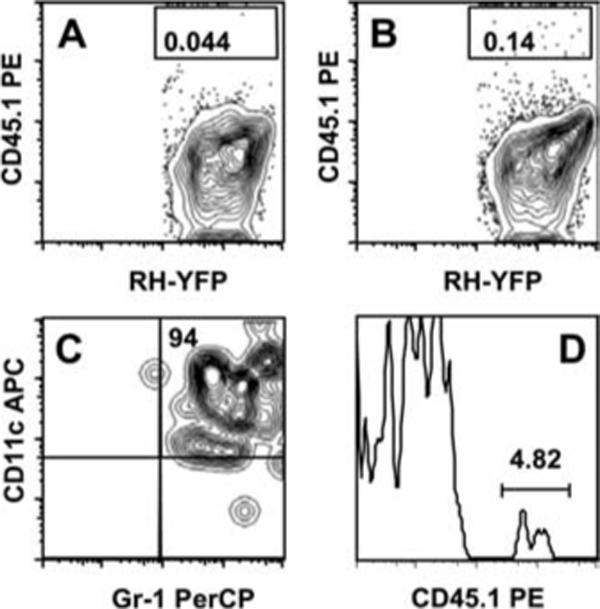
Infected CD11c+Gr-1+ cells migrate from the peritoneal cavity to the spleen. Peritoneal exudate cells from B6.SJL CD45.1 congenic mice were isolated 2 days after i.p. inoculation with RH-YFP tachyzoites. The exudate cells were then adoptively transferred i.p. into congenic C57BL/6 CD45.2 host mice infected 2 days earlier with RH-YFP tachyzoites. Two days later splenocytes were harvested from the host mice and three-color staining for CD45.1, CD11c, and Gr-1 was conducted. A, splenocytes from a control day 4-infected C57BL/6 mouse that received no congenic peritoneal exudate cells, showing background staining for CD45.2. B, splenocytes from a day 4-infected C57BL/6 mouse adoptively transferred with CD45.2-positive peritoneal exudate cells. C, CD11c and Gr-1 expression of infected CD45.2+ cells in the spleen. The numbers indicate the percentage of cells falling within the indicated rectangles (A and B) and quadrants (C). The graphs show results from one representative mouse. In a group of four mice, the percentage of CD45.1+ cells after transfer was 0.14 ± 0.03. In D, CD45.1+ peritoneal exudate cells from mice infected with RH-YFP parasites were transferred into CD45.2+ mice infected with nonfluorescent parasites. Spleen cells were recovered as above and CD45.1 expression was examined on YFP+ cells. The numbers in each graph indicate the percent of cells falling within the indicated quadrants or gates.
CCR2 is partially involved in recruitment of CD11c+ Gr-1+ to the spleen
The chemokine receptor CCR2 is involved in recruitment of inflammatory myeloid cells and DCs during several infections, including Toxoplasma and Listeria monocytogenes (10, 22). Therefore, we sought to determine whether CCR2 might play a role in recruitment of CD11c+Gr-1+ cells during infection. Accordingly, CCR2 knockout mice were infected in parallel with wild-type controls and CD11c+Gr-1+ splenocytes were analyzed 4 days postinfection. We found a major reduction in the number of CD11c+Gr-1+ cells in the spleens of infected mice in the absence of CCR2 signaling (Fig. 7A). Nevertheless, recruitment of CD11c+Gr-1+ DCs to the spleen was not completely dependent upon CCR2 because there was a low, but significant increase in the number of these cells comparing noninfected with infected CCR2−/− mice. We also examined the influence of CCR5, another chemokine receptor implicated in resistance to T. gondii (27-29), on mobilization of CD11c+Gr-1+ cells to the spleen during infection. In this case, absence of CCR5 had no impact on the migration of CD11c+Gr-1+ cells to the spleen following T. gondii infection (Fig. 7B).
FIGURE 7.
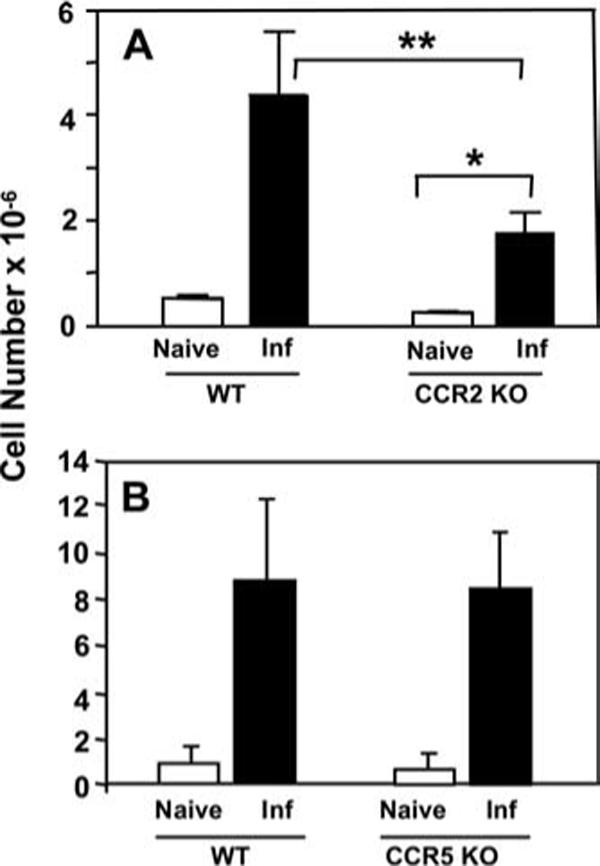
CCR2 is partially involved in recruitment of CD11c+Gr-1+ cells to the spleen during infection. Wild-type and chemokine receptor knockout mice were infected with RH-YFP tachyzoites, and recruitment of CD11c+Gr-1+ cells to the spleen was assessed by flow cytometry. Recruitment of CD11c+Gr-1+ cells was defective in CCR2 knockout mice relative to wild-type controls (A), but unaffected in CCR5 knockout mice (B). Data shown are pooled from three experiments, with two mice per condition. *, p < 0.05, **, p < 0.005.
Toxoplasma-infected CD11c+ Gr-1+ DCs are refractory to TLR-induced IL-12p40 production
Splenic CD8α+ DCs are a well-characterized source of IL-12 after i.v. injection of Toxoplasma lysate Ag (14). This activity is largely due to parasite profilin-TLR11 interactions (15). However, our previous data has shown that Toxoplasma-infected macrophages are suppressed in their ability to produce proinflammatory cytokines such as IL-12 when subjected to TLR stimulation (30). Similar findings have been reported by others using mouse bone marrow-derived DCs (17). Therefore, we sought to determine whether the CD11c+Gr-1+ DCs targeted for infection by T. gondii were stimulated to produce IL-12, or whether the parasite actively suppressed the cytokine.
To discriminate between induction vs suppression of IL-12, splenic DCs from day 4-infected mice were enriched using pan-DC immunomagnetic beads. In this population, ∼14% of the cells coexpressed CD11c and Gr-1 (Fig. 8A). The enriched DC population was cultured for 24 h in the presence of medium alone or with TLR ligands. Because the phenotype of the CD11c+Gr-1+ cells displayed characteristics of both pDC and conventional DC, and because these DC populations express distinct TLR ligands, we used a combination of TLR9 and TLR4 ligands (CpG and LPS, respectively) for stimulation. We then examined IL-12 p40 expression in infected and noninfected CD11c+Gr-1+ DCs. As shown in Fig. 8B, ∼9% of noninfected CD11c+Gr-1+ cells produced IL-12 without further in vitro stimulation. After stimulation with TLR ligands, almost 40% of noninfected CD11c+Gr-1+ DCs expressed IL-12 (Fig. 8C). In marked contrast to these results, only 2% of infected CD11c+Gr-1+ stained positive for IL-12 during culture in medium (Fig. 8D), and with the addition of LPS and CpG, the percentage increased to only ∼14% (Fig. 8E). We conclude from these data that Toxoplasma targets CD11c+Gr-1+ cells for infection, and the parasite suppresses IL-12 production once inside the cells.
FIGURE 8.
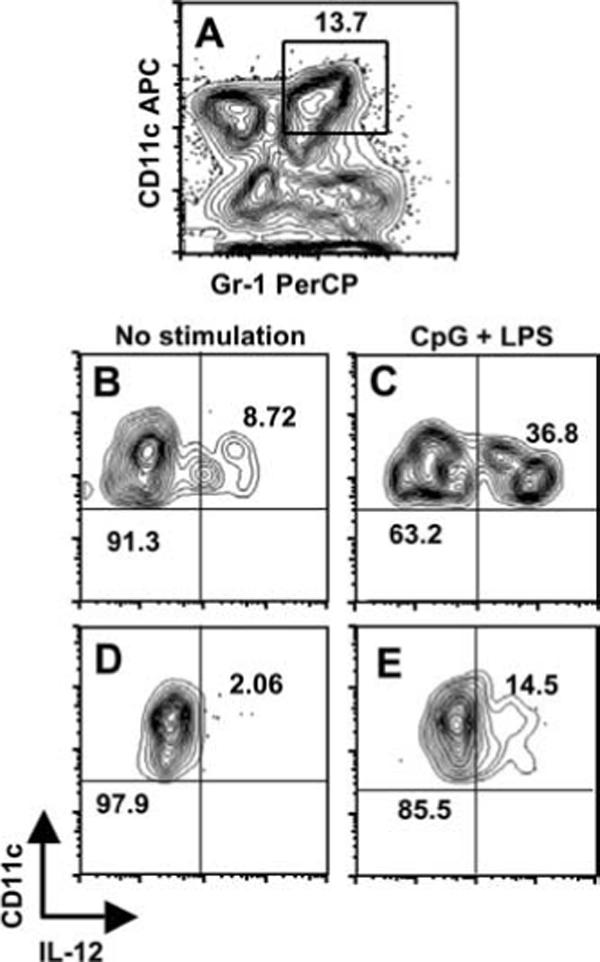
IL-12 production is defective in CD11c+Gr-1+ cells infected with T. gondii. Splenocytes were harvested 4 days postinfection with RH-YFP tachyzoites and enriched for CD11c+ cells using magnetic beads. A, CD11c and Gr-1 expression levels in the enriched DC population. The number indicates the percent of cells in the indicated rectangle. Enriched cells were cultured 24 h in medium alone, or medium with CpG oligodinucleotide (1 μg/ml) and LPS (100 ng/ml). The cells were stained for intracellular IL-12p40 and surface Gr-1 and CD11c. B, IL-12 expression by noninfected CD11c+Gr-1+ cells cultured in medium alone. C, IL-12 expression by parasite-negative CD11c+Gr-1+ cells after in vitro stimulation with LPS + CpG. D, IL-12p40 expression by YFP+ CD11c+Gr-1+ cells with no in vitro stimulation. E, Expression of IL-12p40 in infected CD11c+Gr-1+ cells after TLR ligand-stimulation. In B–D, the numbers indicate the percent of cells falling within the indicated quadrants. One representative of two independent experiments is shown.
Discussion
The results of this study show that inoculation of mice with the tachyzoite stage of T. gondii leads to preferential infection of Gr-1-expressing DCs. CCR2 is involved in the migration of these cells from the peritoneal cavity to the marginal zone of the spleen, where they express markers associated with pDC. Infection with T. gondii suppresses the capacity of these cells to produce the proinflammatory cytokine IL-12 upon ex vivo stimulation with ligands for TLR4 and TLR9, while noninfected cells retain their ability to synthesize IL-12. Recently, inflammatory macrophages have been implicated in the early anti-microbial response to Toxoplasma. Those cells resemble the pDC-like population identified here in that they express Gr-1 and they display CCR2-dependent recruitment (22). However, based upon expression of PDCA-1, CD11c, and B220, it is most likely that the two populations are distinct, and that the cells reported here are related to cells of the pDC lineage.
pDCs are differentiated effector cells specialized for production of anti-viral type I IFN (25). However, they possess the unusual property of being able to differentiate into conventional DCs that activate naive T cells and instruct adaptive immunity through production of mediators such as IL-12 (31). Differentiation can be induced by cytokines such as IL-3, and also microbial stimuli including bacteria and viruses (26, 32). Accompanying pDC differentiation into DCs, the cells acquire dendritic morphology and up-regulate CD11c, MHC class II, and costimulatory molecules. However, there are likely to be functional differences between pDC-derived DCs and conventional DCs insofar as activation of naive T cells by pDC-derived DCs leads to generation of regulatory T cells in several cases (33, 34). pDCs may also differ from conventional DCs in terms of Ag uptake and processing (35, 36).
Although the cells targeted for infection express markers associated with pDCs, namely PDCA-1, Gr-1 and B220, they are nevertheless atypical in that they also express the monocyte lineage marker CD11b. A potential explanation for this unique phenotype may relate to differentiation of pDC into DC under inflammatory conditions. During infection with lymphocytic choriomeningitis virus, bone marrow-derived pDCs down-regulate B220 and Gr-1 and up-regulate CD11b, dependent upon type I IFN (37). Interestingly, this was found to be a two-step process, in which CD11b was first up-regulated, followed by down-regulation of B220. After this transformation, the cells were also newly capable of producing IL-12 in response to TLR4 activation, indicating functional as well as phenotypic conversion. In this regard, it is of interest that Toxoplasma was recently shown to stimulate type I IFN secretion by pDCs (16). We hypothesize that T. gondii infection stimulates an intermediate stage of pDC to conventional DC conversion, associated with coexpression of pDC and myeloid DC markers. Whether this is a stable phenotype associated with infection, or is a transitory state on the way to a conventional DC phenotype remains to be determined.
In addition to type I IFN secretion, pDCs also produce IL-12 upon stimulation with T. gondii or soluble tachyzoite Ag, and they are also capable of presenting Ag during infection (16). This observation suggests a role for pDCs in the induction of the adaptive immune response to Toxoplasma. However, although we did not examine Ag presenting capability, our work argues that T. gondii infection suppresses pDC IL-12 production. In this regard, pDCs cultured in vitro from the bone marrow were used in the experiments showing IL-12 production in response to T. gondii, whereas the pDCs identified in the present study were enriched from the spleens of infected animals. Therefore, it is possible that the cells identified here are functionally distinct from pDCs generated in vitro. Additionally, we used flow cytometry to separate splenic pDCs into infected and noninfected populations. Although the infected pDC population was suppressed in IL-12 production, we found substantial IL-12 production by the noninfected pDC population. Thus, we also found that pDCs present during T. gondii infection are a source of IL-12. However, our study demonstrates that when pDCs are directly parasitized, their ability to produce IL-12 is suppressed.
We, and others, previously reported that T. gondii inhibits TLR-induced IL-12 production by bone marrow-derived macrophages and DCs (17, 38). The present study is significant because it is the first to demonstrate the suppression phenomenon in cells parasitized during in vivo infection. Inhibition of TLR signaling may be a consequence of the need to avoid activation by the parasite's own TLR ligands (15, 39). The mechanisms of suppression are not clear, although the parasite has blocking effects on both NFκB and MAPK signaling pathways (30, 40-42). More recently, IL-10-independent STAT3 activation driven by injection of Toxoplasma rhoptry protein kinases into the host cell cytosol has been implicated in IL-12 inhibition (43, 44).
A key question is why Toxoplasma would target pDCs for preferential infection. As immune effectors, pDCs have two major functions, which are to produce large quantities of anti-viral type I IFN and to present Ag (25). pDCs are not known to possess strong microbicidal activity, unlike macrophages and other myeloid DC populations. Therefore, pDCs may offer a more hospitable environment for the invading parasite relative to other cell types. Furthermore, activated DCs are highly motile, and are sensitive to chemokine-mediated recruitment to lymphoid organs draining the site of infection or inflammation. In this regard, it has long been known that Toxoplasma infection initially spreads through the lymphatics, and lymphoid organs become rapidly parasitized during infection (45). Therefore, pDCs may provide the parasite with a safe vehicle for dissemination during early infection.
It is an unexpected finding that pDCs play a role in the immune response to Toxoplasma. Although we used a virulent parasite strain that leads to 100% lethality in this study, preliminary data (not shown) suggests that low virulence strains capable of establishing long-term infection also preferentially parasitize pDCs. Other protozoa, including Plasmodium and Leishmania spp. are known to display complex interactions with DCs (46, 47). Whether these pathogens also target the pDC population remains to be determined.
Acknowledgments
We thank B. Butcher and C. Egan for useful discussion and critical review of the manuscript.
Footnotes
This work was supported by Public Health Service Grants AI50617 (E.Y.D.), T32 AI07643 (A.L.B.) and HL075512 and HL077545 (A.E.M.).
Abbreviations used in this paper: DC, dendritic cell; pDC, plasmacytoid DCs; DAPI, 4′,6′-diamidino-2-phenylindole.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Denkers EY, Gazzinelli RT. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gavrilescu LC, Denkers EY. IFN-γ overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J. Immunol. 2001;167:902–909. doi: 10.4049/jimmunol.167.2.902. [DOI] [PubMed] [Google Scholar]
- 3.Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. Parasite-induced IL-12 stimulates early IFN-γ synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- 4.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent upon CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ, and TNF-α. J. Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 5.Liesenfeld O, Kosek J, Remington JS, Suzuki Y. Association of CD4+ T cell-dependent, IFN-γ-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J. Exp. Med. 1996;184:597–607. doi: 10.1084/jem.184.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mordue DG, Monroy F, La Regina M, Dinarello CA, Sibley LD. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 2001;167:4574–4584. doi: 10.4049/jimmunol.167.8.4574. [DOI] [PubMed] [Google Scholar]
- 7.Scharton-Kersten TM, Wynn TA, Denkers EY, Bala S, Showe L, Grunvald E, Hieny S, Gazzinelli RT, Sher A. In the absence of endogenous IFN-γ mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 1996;157:4045–4054. [PubMed] [Google Scholar]
- 8.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741–750. doi: 10.1016/j.immuni.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Geissmann F, Auffray C, Palframan R, Wirrig C, Ciocca A, Campisi L, Narni-Mancinelli E, Lauvau G. Blood monocytes: distinct subsets, how they relate to dendritic cells, and their possible roles in the regulation of T-cell responses. Immunol. Cell Biol. 2008;86:398–408. doi: 10.1038/icb.2008.19. [DOI] [PubMed] [Google Scholar]
- 10.Serbina NV, Salazar-Mathar TP, Biron CA, Kuziel WA, Pamer EA. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 11.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity. 2007;26:519–531. doi: 10.1016/j.immuni.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, Machado FS, Aliberti J. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J. Immunol. 2006;177:31–35. doi: 10.4049/jimmunol.177.1.31. [DOI] [PubMed] [Google Scholar]
- 14.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. In vivo microbial stimulation induces rapid CD40L-independent production of IL-12 by dendritic cells and their re-distribution to T cell areas. J. Exp. Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yarovinsky F, Zhang D, Anderson JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science. 2005;308:1626–1629. doi: 10.1126/science.1109893. [DOI] [PubMed] [Google Scholar]
- 16.Pepper M, Dzierszinski F, Wilson E, Tait E, Fang Q, Yarovinsky F, Laufer TM, Roos D, Hunter CA. Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J. Immunol. 2008;180:6229–6236. doi: 10.4049/jimmunol.180.9.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKee AS, Dzierszinski F, Boes M, Roos DS, Pearce EJ. Functional inactivation of immature dendritic cells by the intracellular protozoan Toxoplasma gondii. J. Immunol. 2004;173:2632–2640. doi: 10.4049/jimmunol.173.4.2632. [DOI] [PubMed] [Google Scholar]
- 18.Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. CD11c and CD11b expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. 2006;107:309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell. Microbiol. 2006;8:1611–1623. doi: 10.1111/j.1462-5822.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 20.Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, Wang Z, Watkins SC, Falo LD, Jr., Thomson AW. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 2003;101:611–620. doi: 10.1182/blood-2002-06-1769. [DOI] [PubMed] [Google Scholar]
- 21.Mordue DG, Sibley LD. A novel population of Gr-1+-activated macrophages induced during acute toxoplasmosis. J. Leukocyte Biol. 2003;74:1–11. doi: 10.1189/jlb.0403164. [DOI] [PubMed] [Google Scholar]
- 22.Robben PM, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O'Garra A, Vicari A, Trinchieri G. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J. Exp. Med. 2005;201:1157–1167. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J. Immunol. 2003;171:6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- 25.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 26.O'Keeffe M, Hochrein H, Vremec D, Caminschi I, Miller JL, Anders EM, Wu L, Lahoud MH, Henri S, Scott B, Hertzog P, Tatarczuch L, Shortman K. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8+ dendritic cells only after microbial stimulus. J. Exp. Med. 2002;196:1307–1319. doi: 10.1084/jem.20021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aliberti J, Reis e Sousa C, Schito M, Hieny S, Wells T, Huffnage GB, Sher A. CCR5 provides a signal for microbial induced production of IL-12 by CD8α+ dendritic cells. Nat. Immunol. 2000;1:83–87. doi: 10.1038/76957. [DOI] [PubMed] [Google Scholar]
- 28.Luangsay S, Kasper LH, Rachinel N, Minns LA, Mennechet FJD, Vandewalle A, Buzoni-Gatel D. CCR5 mediates specific migration of Toxoplasma gondii-primed CD8+ lymphocytes to inflammatory intestinal epithelial cells. Gastroenterology. 2003;125:491–500. doi: 10.1016/s0016-5085(03)00903-x. [DOI] [PubMed] [Google Scholar]
- 29.Khan IA, Thomas SY, Moretto MM, Lee FS, Islam SA, Combe C, Schwartzman JD, Luster AD. CCR5 is essential for NK cell trafficking and host survival following Toxoplasma gondii infection. PLoS Pathog. 2006;2:e49. doi: 10.1371/journal.ppat.0020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butcher BA, Kim L, Johnson PF, Denkers EY. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-κB. J. Immunol. 2001;167:2193–2201. doi: 10.4049/jimmunol.167.4.2193. [DOI] [PubMed] [Google Scholar]
- 31.Soumelis V, Liu YJ. From plasmacytoid to dendritic cell: morphological and functional switches during plasmacytoid pre-dendritic cell differentiation. Eur. J. Immunol. 2006;36:2286–2292. doi: 10.1002/eji.200636026. [DOI] [PubMed] [Google Scholar]
- 32.Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J. Exp. Med. 1997;185:1101–1111. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J. Exp. Med. 2002;195:695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, Blazar BR, Chen W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J. Immunol. 2004;173:4433–4442. doi: 10.4049/jimmunol.173.7.4433. [DOI] [PubMed] [Google Scholar]
- 35.Krug A, Veeraswamy R, Pekosz A, Kanagawa O, Unanue ER, Colonna M, Cella M. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J. Exp. Med. 2003;197:899–906. doi: 10.1084/jem.20021091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salio M, Palmowski MJ, Atzberger A, Hermans IF, Cerundolo V. CpG-matured murine plasmacytoid dendritic cells are capable of in vivo priming of functional CD8 T cell responses to endogenous but not exogenous antigens. J. Exp. Med. 2004;199:567–579. doi: 10.1084/jem.20031059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat. Immunol. 2004;5:1227–1234. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butcher BA, Denkers EY. Mechanism of entry determines ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect. Immun. 2002;70:5216–5224. doi: 10.1128/IAI.70.9.5216-5224.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debierre-Grockiego F, Campos MA, Azzouz N, Schmidt J, Bieker U, Resende MG, Mansur DS, Weingart R, Schmidt RR, Golenbock DT, Gazzinelli RT, Schwarz RT. Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J. Immunol. 2007;179:1129–1137. doi: 10.4049/jimmunol.179.2.1129. [DOI] [PubMed] [Google Scholar]
- 40.Bach EA, Aguet M, Schreiber RD. The IFN-γ receptor: a paradigm for cytokine receptor signaling. Annu. Rev. Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 41.Kim L, Butcher BA, Denkers EY. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J. Immunol. 2004;172:3003–3010. doi: 10.4049/jimmunol.172.5.3003. [DOI] [PubMed] [Google Scholar]
- 42.Shapira SS, Speirs K, Gerstein A, Caamano J, Hunter CA. Suppression of NF-κB activation by infection with Toxoplasma gondii. J. Infect. Dis. 2002;185(Suppl):66–72. doi: 10.1086/338000. [DOI] [PubMed] [Google Scholar]
- 43.Butcher BA, Kim L, Panopoulos A, Watowich SS, Murray PJ, Denkers EY. Cutting edge: IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-α in host macrophages. J. Immunol. 2005;174:3148–3152. doi: 10.4049/jimmunol.174.6.3148. [DOI] [PubMed] [Google Scholar]
- 44.Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sumyuen MH, Garin YJ, Derouin F. Early kinetics of Toxoplasma gondii infection in mice infected orally with cysts of an avirulent strain. J. Parasitol. 1995;81:327–329. [PubMed] [Google Scholar]
- 46.Soong L. Modulation of dendritic cell function by Leishmania parasites. J. Immunol. 2008;180:4355–4360. doi: 10.4049/jimmunol.180.7.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson MM, Urban BC. Antigen presentation and dendritic cell biology in malaria. Parasite Immunol. 2006;28:5–14. doi: 10.1111/j.1365-3024.2006.00772.x. [DOI] [PubMed] [Google Scholar]