

Abstract
Purpose
Retinitis Pigmentosa (RP) is a progressive neurodegenerative disease resulting in blindness for which there is no current treatment. While the members of the family of RP diseases differ in etiology, their outcomes are the same: apoptosis of rods followed by cones. Recently, the bile acid, tauroursodeoxycholic acid (TUDCA), has been shown to have anti-apoptotic properties in neurodegenerative diseases, including those of the retina. In this study we examine the efficacy of TUDCA on preserving rod and cone function and morphology at post-natal day 30 (P30) in the rd10 mouse, a model of RP.
Methods
Wild-type C57BL/6J and rd10 mice were systemically injected with TUDCA (500 mg/kg) every three days from P6-P30 and compared to vehicle (0.15M NaHCO3). At P30, retinal function was measured with electroretinography (ERG) and morphological preservation of the rods and cones assessed with immunohistochemistry.
Results
Dark-adapted ERG responses were two-fold greater in rd10 mice treated with TUDCA compared to vehicle, while light-adapted responses were two-fold larger in TUDCA-treated mice compared to controls, at the brightest ERG flash intensities. TUDCA-treated rd10 retinas had five-fold more photoreceptors than vehicle-treated. TUDCA treatments did not alter retinal function or morphology of wild-type mice when administered to age-matched mice.
Conclusions
TUDCA is efficacious and safe in preserving vision in the rd10 mouse model of RP when treated between P6 and P30. At P30, a developmental stage at which nearly all rods are absent in the rd10 mouse model of RP, TUDCA treatment preserved both rod and cone function and greatly preserved overall photoreceptor numbers.
Introduction
Retinitis Pigmentosa (RP) is a family of diseases characterized by night blindness and loss of peripheral vision followed by progressive loss of central vision. RP affects approximately 50,000-100,000 people in the United States and about 1.5 million people worldwide 1. Currently ∼200 genes have been identified that cause RP or related retinal diseases (http://www.sph.uth.tmc.edu/Retnet/), showing the genetic diversity of this group of diseases. However, regardless of the mutation, the final common pathway is programmed photoreceptor cell death or apoptosis 2.
Bear bile has a history of positive effects in ancient Chinese medicine 3, 4, but Western medicine has only recently investigated the anti-apoptotic effects of bile acids including tauroursodeoxycolic acid (TUDCA), the taurine conjugate of ursodeoxycholic acid (UDCA). The bulk of the therapeutic effects of TUDCA and UDCA have been shown in the treatment of a wide range of liver and gall bladder diseases 5-7. More recently, TUDCA has been shown to be neuroprotective in animal models of Huntington's Disease 8, 9, Parkinson's Disease 10, and acute stroke 11, 12. TUDCA and UDCA are anti-apoptotic agents, and while the exact mechanisms of apoptosis prevention are still under investigation, several key signaling pathways have been implicated. TUDCA modulates cell cycle effector genes including cyclinD1 and P53 13-17. Phosphytidal-inositol-3-kinase (PI3K) 18-20 mitogen-activated-protein kinase (MAPK) 20, and ERK/Akt 21 pathway activation have all been implicated with TUDCA administration. TUDCA and UDCA also stabilize the mitochondrial membrane. They directly inhibit mitochondrial permeability transition, inhibit cytosolic Bax translocation and suppress mitochondrial release of cytochrome c 22, 23. Bile acids block reactive oxygen intermediate (ROI) production 22-24 and may themselves be antioxidants 23. They block caspase activation, including caspase-3 12, 22, 23, 25 and also prevent inactivation of the nuclear enzyme PARP 12, 22, 23.
Due to the prominent role of apoptosis in retinal degenerative disease, TUDCA was tested in the Pde6brd10 (hereafter, rd10) mouse. The rd10 mouse has a missense point mutation in exon 13 of the β-subunit of the rod cGMP phosphodiesterase (β-PDE) 25-27. Thus, the rd10 mouse is similar to the popular Pde6brd1 or rd1 mouse, which also shares a mutation in β-PDE gene 28, 29. The rate of degenerate in the rd10 mice is similar to the rd1 mutant model with a delayed onset; both have rapid rod degeneration followed by cone degeneration25-27, 30.
In a previous study, TUDCA treatments of rd10 mice showed significant preservation of photoreceptor function and morphology at P1825. In the rd10 mice, maximal retinal cell loss on a C57BL/6J background occurs at approximately postnatal day 28 (P28) 30. Rods degenerate faster than cones in rd10 mice, with rod function decreased by ∼70% under dark-adapted conditions while cone isolating light-adapted conditions show a 50% decline at P30 26. Furthermore, while rod degeneration appears nearly complete by P40 in rd10 mice 26, cones have been identified until 9 months of age 30.
The current study tests the hypothesis that TUDCA preserves rods and cones to P30, the stage at which photoreceptor cell loss peaks30. At this stage of degeneration in the rd10 model, the majority of rods have degenerated and only some cones remain26. This stage of degeneration represents the endstage of RP for most patients since only 0.5% of RP patients are estimated to develop complete blindness (no light perception)31. Thus, these experiments test the efficacy of TUDCA at a critical stage of degeneration. To test this hypothesis, rd10 mice were treated with TUDCA or vehicle from P6 to P30 and rod and cone function and morphology assessed by electroretinography, histology, immunohistochemistry, and TUNEL labeling. While TUDCA and UDCA have been shown to be very well tolerated in animals8-12 and humans32 (UDCA is FDA approved), these studies have all been performed in adult animals. Thus, in this study we also determine if the anti-apoptotic affects of TUDCA had an effect on early retinal development by treating age-matched C57BL/6J wild-type mice at the same timepoints. These studies demonstrate that TUDCA is effective in preserving photoreceptors in the rd10 mice at P30.
Materials and Methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee at the Atlanta VA Medical Center and conform to the standards of the Association for Research in Vision and Ophthalmology. To establish a breeding colony, Pde6brd10 mice on a C57BL/6J background (or rd10) were obtained from Jackson Laboratories (Bar Harbor, Maine). All mice were housed under controlled lighting conditions on a 14:10 light:dark cycle (25-200 lux). Each litter was randomly divided at P6 to receive TUDCA treatment (500 mg/kg)(Calbiochem)(n =16) or vehicle (0.15 M NaHCO3 1ml/kg, pH 7.0) (Sigma)(n=10). A breeding colony of wildtype (WT) C57BL/6J mice (Jackson Laboratories) received the same treatments as the rd10 mice (n=5 for TUDCA, n=2 for vehicle treatment) starting at P6. TUDCA and vehicle solutions were made up fresh prior to every injection and pH adjusted to 7.4 25. Mice were weighed and injected once every 3 days beginning at P6 and ending at P30, resulting in 8 total administrations per animal for the treated and vehicle groups. All injections were made subcutaneously at the nape of the neck.
Electroretinographic (ERG) Methods
Mice were dark adapted overnight and ERGs were performed using a commercial recording system at P30 (UTAS 3000, LKC Technologies)25, 33, 34. After anesthesia induction (ketamine, 80 mg/kg; xylazine, 16mg/kg), the cornea was anesthetized (1% tetracaine) and the pupils dilated (1% tropicamide, 1% cyclopentolate). Body temperature was maintained at 37° C by placing the mice on a heating pad inside a Faraday cage. The active electrode consisted of a silver wire loop that was positioned on the cornea using 1% methylcellulose. Needle electrodes placed in the cheek and tail served as reference and ground, respectively. A desktop Ganzfeld was used to administer a series of increasing intensity light flashes ranging from −3.0 to 2.1 log cd s/m2. Dark-adapted ERG recordings were averaged over 5-10 separate flashes per light intensity with the inter-stimulus time increasing from 10 to 60 seconds as the flash intensity increased. Animals were then light adapted for 10 minutes using a steady background light (30 cd/m2). Cone-isolating responses were recorded to a seven step intensity series (−0.82 to 1.88 log cd s/m2) presented at 3 Hz in the presence of same light-adapting background. Light-adapted ERGs were averaged over 25 separate flashes per intensity.
ERG a-wave amplitudes were measured from baseline to the first negative wave. The b-wave was measured from the trough of the a-wave to the peak of the first positive wave, or when the a-wave was not present, from baseline to the peak of the first positive wave. Implicit time measurements were made from flash onset to the trough or peak of the a- or b-wave, respectively. Statistical analysis across flash intensity and between treatment groups was conducted by using repeated measures ANOVA tests (SPSS, Chicago, IL)25, 33.
Histological Methods
Following ERG recordings, deeply anesthetized mice were sacrificed by cervical dislocation. Eyes were immediately enucleated. Fixative was injected into the superior limbus to mark orientation and to aid in the rapid fixation of the retina. All left eyes were immersion fixed in 4% paraformaldehyde for 30 minutes for TUNEL labeling and all right eyes were fixed overnight in 2.5% glutaraldehyde/2% paraformaldehyde for light microscopy. After fixation, right eyes were dehydrated through a graded alcohol series, infiltrated with propylene oxide, and embedded in Epon 812/ Der 736 resin (Electron Microscopy Sciences, Hatfield, PA). Sections (0.5 μm) bisecting the optic disc superiorly to inferiorly were then cut on an ultramicrotome (Reichert UltraCut IL, USA) using a histo-diamond knife and collected on glass slides. Slides were stained with 1% aqueous toluidine blue (Sigma, St. Louis, MO).
Left eyes were processed through a graded series of alcohols and embedded in paraffin. Sections (5 μm thick) were cut on a rotary microtome, bisecting the optic disc superiorly to inferiorly. Paraffin sections were used for cone opsin immuno-labeling and TUNEL stain.
Total photoreceptor cell counts
Plastic sections were analyzed using light microscopy (Leica DMRB, Bannockburn, IL) to determine photoreceptor cell counts. Four retinal regions in the vertical meridian (0.5mm in width) were photographed at 20X magnification. The locations in reference to the optic nerve head were 2.0-1.5 mm superior, 1.0-0.5 mm superior, 0.5-1.0 mm inferior, and 1.5-2.0 mm inferior. Photoreceptor nuclei counts were performed using image software (ImagePro Plus 5.0, Silver Spring, MD). A total of 3 sections were counted for each of the four retinal areas in each eye. These values were then averaged and Analysis of Variance (ANOVA), (SPSS 8.0, Chicago, IL) performed.
Cone photoreceptor labeling
Paraffin sections were de-paraffinized with xylenes followed by a graded series of alcohol rinses. After an initial blocking step with 5% goat serum (Chemicon, Temecula, CA) made with Superblock (Pierce, Rockford, IL) , sections were incubated in anti-opsin green/red and blue (1:500; Chemicon, Temecula, CA) for 48 hours at room temperature. The primary antibody was visualized by labeling with Alexa-fluor 488 goat anti-rabbit IgG secondary (1:500: Abcam, Cambridge, MA) for 1 hour, after optimization with a titrated series for both the primary and secondary antibody. Each slide contained a negative control by eliminating primary antibody from one section per slide. Sections were then coverslipped with an aqueous mounting medium (Gel/Mount, Biomeda, Foster City, CA). Digital micrographs were captured of images at 20X magnification using a confocal microscope. All micrographs were taken from sections stained the same day with the same camera settings.
Cone photoreceptor nuclei counts
Rod and cone photoreceptor nuclei have differently shaped heterochromatin such that rods have a dense central clump and cones have irregular shaped heterochromatin that can appear as one to three clumps in tissue sections 35, 36. Thus, cone photoreceptor nuclei were quantified by counting all photoreceptor nuclei with two or more clumps of heterochromatin 35 in toluidine blue stained plastic sections (see figure 5 for examples of cone versus rod photoreceptor nuclei). Using the same plastic sections and retina regions described above, three independent observers counted cone nuclei at 20X. Different treatment groups and strains were compared using ANOVA analysis (SPSS 8.0, Chicago, IL)
Figure 5.

Quantitative assessment of cones after TUDCA treatment at P30. Cone nuclei in WT (A) and rd10 (B) mice were counted based on heterochromatin pattern. Heterochromatin that formed two or more clumps was counted as a cone (arrows) according to the criteria established by Carter-Dawson and LaVail35. This contrasts with rod nuclear staining, which appears dark and uniformly stained. C) Cone counts for each 500 μm retinal region did not show any differences between treatment groups for rd10 or WT mice. However, WT mice had two to three times more cone nuclei per retinal area than rd10 mice (Student’s t-test, p<0.001). The error bars indicate standard error of the mean.
TUNEL labeling
Paraffin sections were stained by terminal deoxynucleotidyl transferase-mediated 2′deoxyuridine 5′triphosphate-biotin nick end labeling (TUNEL) using a DeadEnd Fluorometric TUNEL kit (Promega, Madison, WI) following the manufacturer's kit instructions and counter-stained with propidium iodide 25. Images of TUNEL-stained sections were captured by computer-aided confocal microscopy and photoreceptor nuclei counted per field at 20X magnification as described above. For each field, ANOVAs were used to compare the means between the treatment groups (SPSS 8.0, Chicago, IL).
Results
The results are broken into two broad sets of experiments: I) those dealing with the efficacy of TUDCA in the rd10 mouse model of RP and II) those testing TUDCA in age-matched WT mice with the same doses.
I. Efficacy of TUDCA in the rd10 mouse
TUDCA preserves retinal function at P30
To determine if TUDCA treatments preserved rod and cone function in rd10 mice, we performed dark and light-adapted ERGs at P30. We found that TUDCA treatments significantly preserved both rod and cone function in rd10 mice. Comparing TUDCA- and vehicle-treated mice, dark-adapted ERGs from TUDCA-treated animals showed significantly larger waveforms (Figure 1A). At the highest flash intensity, the a-wave amplitude was approximately five times larger in TUDCA treated mice than the control treatment groups, while the b-wave amplitude was twice as large as the other treatment groups (Figure 1). Furthermore, mice treated with TUDCA showed significant preservation of mean a- and b-wave amplitudes (Figure 1B) over a range of ERG flash intensities as assessed by Repeated measures ANOVA [a-wave: F(4, 88) = 2.66, p = 0.038; b-wave: F(4, 96) = 2.55 p = 0.044; n=16 for TUDCA-treated group and n=10 for vehicle-treated group].
Figure 1.

Dark-adapted retinal function in rd10 mice after TUDCA treatments at P30. ERG recordings showing rod-dominated retinal function in P30 rd10 mice treated with TUDCA or vehicle. A) Representative ERG waves to a series of flash intensities from a TUDCA- or vehicle-treated mouse. At every light level, the TUDCA-treated mice had a larger response than the vehicle-treated rd10 mouse. The a-wave is much larger in the TUDCA-treated animals. B) Plot of average ERG amplitude (±SEM) for a- and b-waves from the two treatment groups. TUDCA (N=16) and vehicle (N=10). TUDCA-treated animals had significantly greater a- and b-wave amplitudes than the vehicle groups [Repeated measures ANOVA; a-wave: F(4, 88) = 2.66, p = 0.038; b-wave: F(4, 96) = 2.55 p = 0]. We conclude that TUDCA is efficacious in preventing the loss of ERG signals in response to a range of scotopic flashes.
Cone mediated, light-adapted ERG waveforms were significantly larger in TUDCA treated rd10 mice compared to vehicle (Figure 2A). In the representative waveforms shown in Figure 2A, the ERG from the TUDCA-treated animal was clearly larger in amplitude at all intensities, and the b-wave threshold was measurable at −0.8 log cd s/m2 compared to −0.4 log cd s/m2 for the vehicle-treated animal. When amplitude was plotted across flash intensity, it was seen that there are significant differences in b-wave amplitude between the treatment groups at the brightest flashes (n=15 TUDCA; n=10 vehicle; Repeated ANOVA F (6, 138) = 6.364, p <0.001). No significant differences were detected between the treatment groups for the small light-adapted a-wave (Repeated ANOVA F(6, 120) = 2.075, p = 0.061)
Figure 2.

Light-adapted retinal function after TUDCA treatment in rd10 mice at P30. A) ERG waves recorded in response to a series of Ganzfeld flashes in the presence of an adapting background light (30 cd/m2). The vehicle-treated mouse shows a slight decrease in sensitivity with the first recordable response at −0.4 log cd s/m2. At the brightest flashes, the TUDCA-treated mouse had ERG b-wave amplitudes that were twice the size of a vehicle-treated control. B) Average ERG amplitudes (± SEM) for light-adapted a- and b-waves are plotted across intensity. The cone-mediated b-wave in the TUDCA-treated group (n=15) is significantly different compared to the vehicle group (n=10) [F (6, 138) = 6.364, p < 0.001]. The a-wave amplitude is not quite statistically significantly different between the groups [Repeated ANOVA; F(6, 120) = 2.075, p = 0.061]. We conclude that TUDCA is efficacious in preserving cone-mediated electrical responses in the retina.
Preservation of photoreceptor nuclei
Rod and cone photoreceptors
To determine if TUDCA treatments preserved photoreceptors, we examined photoreceptor structure and numbers in rd10 mice. TUDCA treatments significantly preserved photoreceptor numbers and the inner and outer segments in the rd10 mice (Figure 3C) compared to vehicle-treated controls (Figure 3D). The number of photoreceptor nuclei in a 0.5 mm field at four locations from inferior to superior in TUDCA-treated rd10 mice at P30 showed significant preservation of photoreceptors (Figure 3F; ANOVA F (3, 87) = 3.013, p <0.034). TUDCA treated rd10 retinas contained an average of 333 ±44 photoreceptor nuclei per region compared to only 68 ±9 photoreceptor nuclei/0.5 mm region in the vehicle-treated rd10 retina, (mean ±SEM; Figure 3C, 3D, 3F), a fivefold preservation.
Figure 3.


Retinal morphology following TUDCA treatment at P30. A and B) Retinal micrographs from P30 WT C57BL/6 mice treated with TUDCA (A) or vehicle (B). All retinal layers are intact and the photoreceptor layers are normal. Qualitatively, panels A and B show no changes between treatment groups in any of the layers of the retina of WT mice. This finding suggests that TUDCA treatment at the given dose and for the indicated duration is safe. C and D) Retinal micrographs from rd10 mice treated with TUDCA (C) or vehicle (D). The photoreceptor layer has been reduced to about 1 row of nuclei in the vehicle-treated mouse (D), while the TUDCA-treated retina retains 3 to 4 rows of nuclei (C). Panels C and D show clear differences in the thicknesses of the ONL, outer segments (OS), and inner segments (IS), with TUDCA treatment demonstrating an efficacious delay of retinal degeneration. E and F) Plots of the total number of photoreceptors at each retinal location from WT and rd10 mice with reference to the optic nerve. The inset shows a schematic diagram of the retina and optic nerve. Each shaded square indicates a sampling region as indicated on the graphs. E). Photoreceptor cell counts in the WT mice from the two treatment groups show no differences across the retina. (n=5 TUDCA, n=2 vehicle, ANOVA F (3, 12) = 0.397, p = 0.757) F). Photoreceptor cell counts from rd10 mice treated with TUDCA (n=17) or vehicle (n=14). The TUDCA-treated mice have significantly more photoreceptors across all areas sampled than vehicle-treated mice (ANOVA F (3, 87) = 3.013, p = 0.034). We conclude that TUDCA treatment is efficacious in preventing or slowing photoreceptor degeneration up to P30 in the rd10 mouse model of RP.
Cone photoreceptors
To identify the population of cone photoreceptors, paraffin sections were labeled with anti-cone opsin. Immuno-labeled WT retinas showed distinct labeling of cone photoreceptor outer segments (Figure 4A), as previously reported 37, 38. The labeled cone outer segment of WT mice always appeared long with a distinct tip (Figure 4A). In TUDCA-treated rd10 retinas, distinct cone labeling was still visible (Figure 4B-C). The labeled segments correlated with the extent of preservation. Retinas with the most preservation of photoreceptors and longer outer segments had the most cone labeling (Figure 4B). In rd10 mice with less preservation from the TUDCA treatments, the cone outer segments were smaller and the cone opsin labeling appeared as small round structures or punctuate in the region of the outer segments (Figure 4C). In vehicle-treated rd10 mice, the outer segments were not visible and cone opsin labeling was only seen as small punctuate labeling (Figure 4D, E). While some variability in cone opsin labeling was present in all treatment groups, TUDCA-treated retinas more frequently presented with a continuous line of outer segments and longer cone outer segments compared to vehicle-treated retinas (Figure 4B, C vs D, E ).
Figure 4.
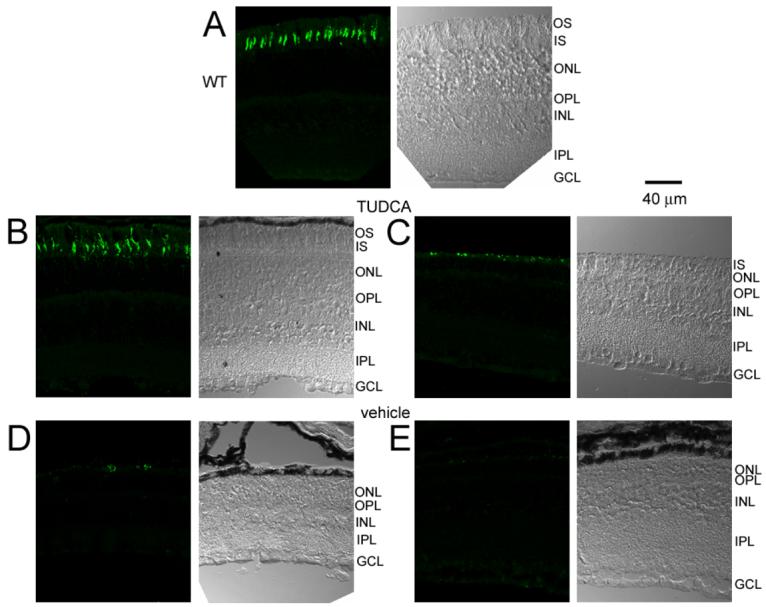
Cone opsin labeling in WT and rd10 mice after TUDCA treatments at P30. Each micrograph is presented as a pair with the DIC image on the right and the cone opsin labeling on the left. A) Cone opsin immunohistochemistry in a WT retina showing distinct outer segments of cones. B and C) TUDCA-treated rd10 retinas showing long cone outer segments, similar to WT retina (B) and shorter cone outer segments with more punctuate labeling (C). D and E) Cone labeling in vehicle-treated rd10 retinas appeared as sparse punctuate labeling. Note that no outer segments are visible in the DIC images of the vehicle-treated mice. These results suggest that TUDCA treatments preserve cone outer segments.
Cone nuclei were quantified by counting in toluidine blue plastic-embedded sections. All photoreceptor nuclei with two or more clumps of heterochromatin were classified as cones 35. Figure 5A (arrows) shows cone nuclei in a WT retina, while Figure 5B (arrows) shows cone nuclei identified in an rd10 TUDCA-treated retina. Figure 5C shows the average cone nuclei counts across the retinal areas from TUDCA-, and vehicle-treated rd10 retinas were very similar, 2.92 ±0.27 vs 4.08 ±0.49 cones/500 μm (TUDCA, n=14; vehicle, n=11; ANOVA F(3, 69) = 1.25, p = 0.298). In addition, no differences in cone counts in WT retinas were found between treatment groups (Student's t-test, p = 0.93). WT mice had 10.5 ±1.1 nuclei/500 μm; significantly more cone nuclei than rd10 mice, as expected (Student's t-test, p <0.001).
Apoptosis in the rd10 retina at P30
We have previously shown that rd10 retinas at P18 have numerous TUNEL positive nuclei, but TUDCA-treated retinas have very few apoptotic nuclei 25, 26. Thus, at P30, we expect to see fewer TUNEL positive nuclei in vehicle-treated retinas due to the prior massive loss of photoreceptors before this stage. In contrast, due to the delay in degeneration produced by TUDCA treatments, we expect to observe some apoptotic nuclei present in TUDCA-treated retinas.
Our results show that TUDCA-treated rd10 retinas had similar numbers of apoptotic nuclei to vehicle-treated retinas. The average number of TUNEL positive nuclei in TUDCA-treated retinas per microscope field was 6.8 ±1.1 (mean ±sem; n=10) compared to 11.2 ±2.9 (n=3) in vehicle- treated retinas. These differences were not significant (Student's t-test, p = 0.29).
II. TUDCA Studies in WT mice
TUDCA treatment does not affect normal retinal function
To test whether TUDCA has adverse affects on normal retinal function when given early in development, we recorded ERGs from TUDCA- and vehicle-treated mice. Figure 6A shows the typical dark-adapted ERG waveform to a series of flash intensities. The waveforms from TUDCA-treated retina are larger than those from vehicle-treated retina. The mean dark-adapted ERG amplitudes for the a- and b-waves were nearly identical for the TUDCA- and vehicle-treated mice (Figure 6B, C; Repeated ANOVA F(4, 20) = 2.020, p = 0.130, and F(4, 20) = 3.110, p = 0.138, respectively; n=5 for TUDCA-treated and n=2 for vehicle-treated). Light-adapted b-wave amplitudes were also similar between treatment groups (Figure 6D; Repeated measures ANOVA F(6, 30)=0.532, p = 0.779).
Figure 6.

Studies in WT mice: Retinal function measurements were recorded from WT C57BL/6J mice treated with TUDCA or vehicle from P6 to P30. A) Representative dark-adapted ERG waveforms for WT TUDCA- and vehicle-treated mice. Dark-adapted a-wave (B), dark-adapted b-wave (C) or light-adapted b-wave (D) amplitudes from WT mice treated with TUDCA (n=5) or vehicle (n=2) show no differences in amplitude or timing across different flash intensities [Repeated measures ANOVA F(4, 20) = 2.020, p = 0.130; F(4, 20) = 3.110, p = 0.138; F(6, 30)=0.532, p = 0.779; respectively]. We concluded that TUDCA treatment from P6 to P30 has no deleterious effect on retinal function, as measured with the ERG.
TUDCA treatment does not affect normal retinal morphology
Retinal morphology appeared normal in WT mice from all treatment groups (Figure 3A, B, E). The average number of photoreceptor nuclei was similar with 795.4 ±27.4, and 754.2 ± 46.9, nuclei/region in TUDCA-treated, and vehicle-treated retinas, respectively. The number of nuclei across retinal areas was not significant between treatment groups in the WT mice (Repeated ANOVA F(3, 12) = 0.397, p = 0.757; Figure 3E).
TUDCA treatment alters body weight in rd10 and WT mice
Animal weights were also compared across time with TUDCA versus vehicle treatment. TUDCA-treated WT mice were found to have significantly lower body weights than vehicle injected mice (Figure 7A; Repeated measures ANOVA; F(5, 25) = 4.276, p = 0.006). As treatments progressed, the TUDCA-treated mice gained less weight than vehicle-treated. The largest differences were not reached until P24. TUDCA-treated rd10 mice also showed a significant decrease in body weight over the treatment period (Figure 7B, Repeated measures ANOVA; F(7, 245) = 10.973, p < 0.001). In rd10 mice, the effect of TUDCA on body weight was apparent by P9, much sooner than in the WT mice.
Figure 7.

Body weights in WT and rd10 mice after TUDCA treatments from P6 to P30. A) WT mice treated with TUDCA (n=5), showed a reduction in body weight compared to vehicle (n=2; Repeated ANOVA F(5, 25) = 4.276, p = 0.006). B) TUDCA-treatment of rd10 mice (n=24) resulted in significantly lower body weight than vehicle (n=13) from P15 to P27 [Repeated ANOVA F(7, 245) = 10.973, p <0.001]. We conclude that TUDCA treatments suppressed body weight in the WT and rd10 mice. Symbols represent mean ±SD.
Discussion
The present study demonstrates that TUDCA treatment, through the advanced degenerative timepoint of P30 in the rd10 mouse, is remarkably effective in sustaining photoreceptor cells and their function. We have previously shown successful preservation of rod function and structure through P18 in this model 25. Though slightly delayed compared to rd1 mice, the retinal degeneration in rd10 mice is rapid and aggressive. In untreated rd10 mice, the outer nuclear layer (ONL) is reduced to a single nuclear layer by P30 25-27. Functionally, the dark-adapted a-wave is largely undetectable (only 3% of WT response) and the b-wave is greatly diminished (14% of WT response)25-27. At this stage, rods are almost completely absent and cones are degenerating. This study shows that TUDCA treatments starting at P6 sustain retinal function and morphology through this critical stage from 30% (dark-adapted a-wave) to 45% (light-adapted and dark-adapted b-waves and photoreceptor numbers) of WT responses. These results show significant preservation of the retina by a systemic agent at this stage of degeneration.
A pan-retinal preservation of retinal function and photoreceptor nuclei was found in the TUDCA-treated rd10 mice at P30. Rod function and rod photoreceptors were significantly greater in TUDCA-treated rd10 mice (Figures 1, 2, 3). In addition, the number of total photoreceptor nuclei was fivefold greater in TUDCA-treated mice compared to vehicle -treated mice (Figure 3).
Interestingly, TUDCA treatments appeared to preserve cone outer segment morphology compared to vehicle-treated (Figure 4), which correlates well with the increased amplitudes in the light-adapted TUDCA-treated retinas (Figure 2). However, no differences were found in the number of cone nuclei between the treatment groups (Figure 5). These data may suggest that TUDCA causes an overall increase in preservation of the photoreceptors. Since cone degeneration occurs secondarily in this model, the significant preservation of rods from TUDCA treatments may sustain the cones. A later timepoint may be needed to exclusively evaluate the effect of TUDCA treatments on cone preservation. However, since humans rely most on cone vision, the finding of greater cone function and healthier cone outer segments after TUDCA treatments suggests that this drug might be useful for preserving cone vision in people.
The similarity in TUNEL labeling between TUDCA- and vehicle-treated rd10 retinas may suggest that TUDCA is delaying the death of photoreceptors in the rd10 model. In our analysis of rd10 retinas at P18, vehicle-treated retinas showed a vastly greater number of TUNEL-positive nuclei compared to TUDCA-treated retinas 25. At P30, as might be expected, the vehicle-treated rd10 retinas do not have many nuclei remaining to undergo apoptosis. Conversely, the TUDCA-treated retinas have many remaining nuclei, some of which are apoptotic. WT mice showed no significant differences between the treatment groups.
Similar to other studies which showed TUDCA to be well-tolerated in adult animals 8-12, we found no evidence at the functional or morphological level that TUDCA itself had any significant adverse effects on the retina in WT mice when given from P6 to P30. However, TUDCA treatments did produce a decrease in body weight in both WT and rd10 mice (Figure 7). The body weight of TUDCA-treated WT mice dropped 22% while TUDCA-treated rd10 mice decreased 23%. These values are slightly below the 25% loss of body weight that is considered an endpoint criterion by our IACUC. Other studies have not reported a reduction in body weight after TUDCA treatment, perhaps due to the fact that other animal models were treated as adults 8-12 and not during early development, as in this study. Future studies will determine whether a different vehicle would have less effect on body weight. Nevertheless, the lack of toxic effects on the retina complement other studies that showed the safety of UDCA treatment in human patients with liver disease 32, 39, of TUDCA treatment in rodent models of Huntington's disease 8, 9, and TUDCA treatment in rodent models of stroke 11, 12. If TUDCA continues to show effects on body weight in young animals, drug delivery approaches could be pursued to administer TUDCA exclusively to the retina.
Comparing the preservation of the rd10 retina between P18 and P30, TUDCA appears to delay degeneration in rd10 mice by about 12 days, or some 35% over the course of the degenerative period. Since it has been reported that different species have surprisingly similar rates of degeneration based on maximal life expectancy 40 and that typical RP patients have a linear rate of retinal function loss 41, a prediction of TUDCA's ability to preserve vision in RP patients can be made: TUDCA might preserve retinal function for about 18 years in a patient who had photoreceptor loss beginning in their 20s and ending in their 70s. Coupling this possibility of efficacy with the demonstrated lack of toxicity of hydrophilic bile acids in several animal models 8-12 and humans 32, 39, suggests that hydrophilic bile acids may be relevant to the ophthalmic clinic.
Acknowledgments
Grant Support: Supported by Department of Veterans Affairs, Foundation Fighting Blindness, Research to Prevent Blindness, Knights Templar Education Foundation, and NIH grants R01 EY014026, P30 EY06360, R01 EY012514, P30 AT000609, R24EY017045 and R01 EY016470.
References
- 1.Berson EL. Retinitis pigmentosa: unfolding its mystery. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:4526–4528. doi: 10.1073/pnas.93.10.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reme CE, Grimm C, Hafezi F, Marti A, Wenzel A. Apoptotic cell death in retinal degenerations. Prog Retin Eye Res. 1998;17:443–464. doi: 10.1016/s1350-9462(98)00009-3. [DOI] [PubMed] [Google Scholar]
- 3.Cidian ZYD. Dictionary of Traditional Chinese Medicine. Shanghai Science and Technology Press; Shanghai: 2004. [Google Scholar]
- 4.Williamson DF, Phipps MJ. Proceedings in the Third Annual Symposium on the Trade in Bear Parts 1999; TRAFFIC East Asia; Hong Kong. 2001. [Google Scholar]
- 5.Hofmann AF. Medical treatment of cholesterol gallstones by bile desaturating agents. Hepatology (Baltimore, Md. 1984;4:199S–208S. doi: 10.1002/hep.1840040837. [DOI] [PubMed] [Google Scholar]
- 6.Crosignani A, Battezzati PM, Setchell KD, et al. Tauroursodeoxycholic acid for treatment of primary biliary cirrhosis. A dose-response study. Digestive diseases and sciences. 1996;41:809–815. doi: 10.1007/BF02213140. [DOI] [PubMed] [Google Scholar]
- 7.Schoemaker MH, Gommans WM, Conde de la Rosa L, et al. Resistance of rat hepatocytes against bile acid-induced apoptosis in cholestatic liver injury is due to nuclear factor-kappa B activation. Journal of hepatology. 2003;39:153–161. doi: 10.1016/s0168-8278(03)00214-9. [DOI] [PubMed] [Google Scholar]
- 8.Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10671–10676. doi: 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keene CD, Rodrigues CM, Eich T, et al. A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-nitropropionic acid model of Huntington's disease. Exp Neurol. 2001;171:351–360. doi: 10.1006/exnr.2001.7755. [DOI] [PubMed] [Google Scholar]
- 10.Duan WM, Rodrigues CM, Zhao LR, Steer CJ, Low WC. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson's disease. Cell Transplant. 2002;11:195–205. [PubMed] [Google Scholar]
- 11.Rodrigues CM, Sola S, Nan Z, et al. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6087–6092. doi: 10.1073/pnas.1031632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodrigues CM, Spellman SR, Sola S, et al. Neuroprotection by a bile acid in an acute stroke model in the rat. J Cereb Blood Flow Metab. 2002;22:463–471. doi: 10.1097/00004647-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Castro RE, Amaral JD, Sola S, Kren BT, Steer CJ, Rodrigues CM. Differential regulation of cyclin D1 and cell death by bile acids in primary rat hepatocytes. American journal of physiology. 2007;293:G327–334. doi: 10.1152/ajpgi.00093.2007. [DOI] [PubMed] [Google Scholar]
- 14.Castro RE, Sola S, Ramalho RM, Steer CJ, Rodrigues CM. The bile acid tauroursodeoxycholic acid modulates phosphorylation and translocation of bad via phosphatidylinositol 3-kinase in glutamate-induced apoptosis of rat cortical neurons. J Pharmacol Exp Ther. 2004;311:845–852. doi: 10.1124/jpet.104.070532. [DOI] [PubMed] [Google Scholar]
- 15.Ramalho RM, Borralho PM, Castro RE, Sola S, Steer CJ, Rodrigues CM. Tauroursodeoxycholic acid modulates p53-mediated apoptosis in Alzheimer's disease mutant neuroblastoma cells. J Neurochem. 2006;98:1610–1618. doi: 10.1111/j.1471-4159.2006.04007.x. [DOI] [PubMed] [Google Scholar]
- 16.Ramalho RM, Ribeiro PS, Sola S, Castro RE, Steer CJ, Rodrigues CM. Inhibition of the E2F-1/p53/Bax pathway by tauroursodeoxycholic acid in amyloid beta-peptide-induced apoptosis of PC12 cells. J Neurochem. 2004;90:567–575. doi: 10.1111/j.1471-4159.2004.02517.x. [DOI] [PubMed] [Google Scholar]
- 17.Sola S, Castro RE, Kren BT, Steer CJ, Rodrigues CM. Modulation of nuclear steroid receptors by ursodeoxycholic acid inhibits TGF-beta1-induced E2F-1/p53-mediated apoptosis of rat hepatocytes. Biochemistry. 2004;43:8429–8438. doi: 10.1021/bi049781x. [DOI] [PubMed] [Google Scholar]
- 18.Sola S, Castro RE, Laires PA, Steer CJ, Rodrigues CM. Tauroursodeoxycholic acid prevents amyloid-beta peptide-induced neuronal death via a phosphatidylinositol 3-kinase-dependent signaling pathway. Mol Med. 2003;9:226–234. doi: 10.2119/2003-00042.rodrigues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beuers U, Denk GU, Soroka CJ, et al. Taurolithocholic acid exerts cholestatic effects via phosphatidylinositol 3-kinase-dependent mechanisms in perfused rat livers and rat hepatocyte couplets. The Journal of biological chemistry. 2003;278:17810–17818. doi: 10.1074/jbc.M209898200. [DOI] [PubMed] [Google Scholar]
- 20.Marzioni M, Francis H, Benedetti A, et al. Ca2+-dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. The American journal of pathology. 2006;168:398–409. doi: 10.2353/ajpath.2006.050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dent P, Fang Y, Gupta S, et al. Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology (Baltimore, Md. 2005;42:1291–1299. doi: 10.1002/hep.20942. [DOI] [PubMed] [Google Scholar]
- 22.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. 1998;4:165–178. [PMC free article] [PubMed] [Google Scholar]
- 23.Rodrigues CM, Ma X, Linehan-Stieers C, Fan G, Kren BT, Steer CJ. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999;6:842–854. doi: 10.1038/sj.cdd.4400560. [DOI] [PubMed] [Google Scholar]
- 24.Colell A, Coll O, Garcia-Ruiz C, et al. Tauroursodeoxycholic acid protects hepatocytes from ethanol-fed rats against tumor necrosis factor-induced cell death by replenishing mitochondrial glutathione. Hepatology (Baltimore, Md. 2001;34:964–971. doi: 10.1053/jhep.2001.28510. [DOI] [PubMed] [Google Scholar]
- 25.Boatright JH, Moring AG, McElroy C, et al. Tool from ancient pharmacopoeia prevents vision loss. Molecular vision. 2006;12:1706–1714. [PubMed] [Google Scholar]
- 26.Chang B, Hawes NL, Pardue MT, et al. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision research. 2007;47:624–633. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision research. 2002;42:517–525. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- 28.Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- 29.Pittler SJ, Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:8322–8326. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. The Journal of comparative neurology. 2007;500:222–238. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grover S, Fishman GA, Anderson RJ, et al. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology. 1999;106:1780–1785. doi: 10.1016/S0161-6420(99)90342-1. [DOI] [PubMed] [Google Scholar]
- 32.Angulo P, Jorgensen RA, Lindor KD. Incomplete response to ursodeoxycholic acid in primary biliary cirrhosis: is a double dosage worthwhile? The American journal of gastroenterology. 2001;96:3152–3157. doi: 10.1111/j.1572-0241.2001.05270.x. [DOI] [PubMed] [Google Scholar]
- 33.Pardue MT, Phillips MJ, Yin H, et al. Neuroprotective effect of subretinal implants in the RCS rat. Investigative ophthalmology & visual science. 2005;46:674–682. doi: 10.1167/iovs.04-0515. [DOI] [PubMed] [Google Scholar]
- 34.Pardue MT, Phillips MJ, Yin H, et al. Possible sources of neuroprotection following subretinal silicon chip implantation in RCS rats. J Neural Eng. 2005;2:S39–47. doi: 10.1088/1741-2560/2/1/006. [DOI] [PubMed] [Google Scholar]
- 35.Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. The Journal of comparative neurology. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 36.Daniele LL, Lillo C, Lyubarsky AL, et al. Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Investigative ophthalmology & visual science. 2005;46:2156–2167. doi: 10.1167/iovs.04-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bemelmans AP, Kostic C, Crippa SV, et al. Lentiviral gene transfer of RPE65 rescues survival and function of cones in a mouse model of Leber congenital amaurosis. PLoS medicine. 2006;3:e347. doi: 10.1371/journal.pmed.0030347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW, Crouch RK. Cone opsin mislocalization in Rpe65−/− mice: a defect that can be corrected by 11-cis retinal. Investigative ophthalmology & visual science. 2005;46:3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- 39.Hempfling W, Dilger K, Beuers U. Systematic review: ursodeoxycholic acid--adverse effects and drug interactions. Alimentary pharmacology & therapeutics. 2003;18:963–972. doi: 10.1046/j.1365-2036.2003.01792.x. [DOI] [PubMed] [Google Scholar]
- 40.Wright AF, Jacobson SG, Cideciyan AV, et al. Lifespan and mitochondrial control of neurodegeneration. Nature genetics. 2004;36:1153–1158. doi: 10.1038/ng1448. [DOI] [PubMed] [Google Scholar]
- 41.Berson EL. Long-term visual prognoses in patients with retinitis pigmentosa: The Ludwig von Sallmann lecture. Experimental eye research. 2007;85:7–14. doi: 10.1016/j.exer.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]