

Abstract
Background
Cardiovascular development is vital for embryonic survival and growth. Early gestation embryo loss or malformation has been linked to yolk sac vasculopathy and congenital heart defects (CHDs). However, the molecular pathways that underlie these structural defects in humans remain largely unknown hindering the development of molecular-based diagnostic tools and novel therapies.
Methodology/Principal Findings
Murine embryos were exposed to high glucose, a condition known to induce cardiovascular defects in both animal models and humans. We further employed a mass spectrometry-based proteomics approach to identify proteins differentially expressed in embryos with defects from those with normal cardiovascular development. The proteins detected by mass spectrometry (WNT16, ST14, Pcsk1, Jumonji, Morca2a, TRPC5, and others) were validated by Western blotting and immunoflorescent staining of the yolk sac and heart. The proteins within the proteomic dataset clustered to adhesion/migration, differentiation, transport, and insulin signaling pathways. A functional role for several proteins (WNT16, ADAM15 and NOGO-A/B) was demonstrated in an ex vivo model of heart development. Additionally, a successful application of a cluster of protein biomarkers (WNT16, ST14 and Pcsk1) as a prenatal screen for CHDs was confirmed in a study of human amniotic fluid (AF) samples from women carrying normal fetuses and those with CHDs.
Conclusions/Significance
The novel finding that WNT16, ST14 and Pcsk1 protein levels increase in fetuses with CHDs suggests that these proteins may play a role in the etiology of human CHDs. The information gained through this bed-side to bench translational approach contributes to a more complete understanding of the protein pathways dysregulated during cardiovascular development and provides novel avenues for diagnostic and therapeutic interventions, beneficial to fetuses at risk for CHDs.
Introduction
The foundations of the primitive heart form between gastrulation and early organogenesis, as critical specification, differentiation, and morphogenic events occur. During this period, the embryo is highly susceptible to environmental insults and genetic lesions that perturb cardiovascular development. These disturbances lead to congenital heart defects (CHDs), a significant cause of spontaneous abortions (20%) and infant mortality (10% of all infant deaths) [1]. Of live births, approximately 1% experience a spectrum of CHDs including transposition of the great arteries, double outlet right ventricle, tetralogy of Fallot, and septal defects [2]. The majority of cardiac malformations (30%) are ventricular septal defects, an abnormal communication between the left and right ventricles [3]. These cardiac anomalies, and several others, arise in part due to morphogenesis defects in the primitive valve tissues (endocardial cushions) which reside in the atrioventricular canal and outflow tract [4]. Within these specific regions of the primitive heart, endothelial cells undergo a key differentiation event, epithelial to mesenchymal transition (EMT) [4]. Lineage tracing experiments and serial anatomical dissection/reconstruction have demonstrated that these mesenchymal cells contribute to the mature mitral and tricuspid valve leaflets as well as the interatrial and interventricular septa [5], [6], [7], [8], [9]. Therefore miscues in the endocardial cushion early in development would affect the ultimate formation of these and other cardiac structures later in development.
Clinical strategies to diagnose cardiac structural anomalies rely on imaging the visible structural defect. Indications for fetal echocardiography include risk factors based on family history, exposures, or other fetal findings [10], [11], [12]. At this point, however, the structural defect has developed and in the current standard of care the clinical intervention is postnatal surgery. Approaching the diagnosis problem from a bed-side-to-bench manner, we believe a molecular-based detection method has the potential to revolutionize the diagnosis and treatment of cardiovascular defects. Therefore a study was designed in which biomarker discovery was performed in a controlled murine model, and then validated for biological significance in human amniotic fluid (AF). A biomarker approach utilizing biochemical analyses of maternal or fetal fluids would provide clinicians with information regarding the pathophysiological condition of the fetus and potentially identify biological deficiencies in the fetal environment. Thus, molecular-based detection provides additional information to guide clinical interventions and may pave the way for the development of novel treatments based on dietary supplementation [13].
The development of such interventions has been hindered by our incomplete knowledge of human cardiac development. A number of genes that drive cardiovascular development have been described in mouse, zebrafish, and chicken model systems, although only a handful of genes such as PTPN11, TBX5 GATA4, and Nkx2.5 have been definitively identified in the etiology of human CHDs [4]. Furthermore, little is known about defects at the protein level. Thus this project aimed to identify novel proteins involved in both cardiovascular development and CHDs.
Identification of proteins involved in specific cardiac developmental events using whole embryonic hearts is difficult because it requires distinguishing and deconvoluting candidate proteins expressed concurrently during endocardial cushion morphogenesis, cardiomyocyte differentiation, looping and/or chamber specification. Therefore, our strategy utilized yolk sac vascular tissue rather than cardiac tissue for proteomic studies. Our hypothesis was that proteins involved in cardiac development and CHDs could be isolated and characterized from the vascular tissue dataset for the following reasons. Yolk sac tissue at the primary capillary plexus stage provides a relatively simple, homogeneous tissue consisting of few cell types in which the primary developmental event is endothelial differentiation and assembly into a mature vascular bed. In the heart, endothelial cell differentiation via EMT is a key event in endocardial cushion morphogenesis. Additionally, trabeculation and coronary system development both also rely on endothelial transdifferentiation or endothelial cell reorganization and assembly. Thus, during development of both the yolk sac and heart, endothelial biology plays a central role. The overlapping developmental pathways in the vasculature and heart have been revealed by the generation of numerous knockout mice that have described both vascular and cardiac phenotypes. This pattern may be a reflection of a common endothelial/cardiac progenitor (though a controversial theory), non-cell autonomous interactions between endothelial and cardiac progenitor lines which are intermingling during migration through the splanchnic mesoderm, or the intimate juxtaposition of differentiated endothelial cells and cardiomyocytes in the embryonic heart [14], [15], [16], [17], [18]. Thus although there is heterogeneity in the type of heart defects that present in the humans there will be a set of markers which will be inherently common to all heart defects because endothelial cell differentiation is a key process in heart development.
From our vascular proteomic dataset, we assembled a list of differentially expressed proteins which were validated by Western blot. The data accrued through these studies highlight the importance of adhesion/migration, differentiation, transport, and insulin/insulin like growth factor (IGF) pathways in cardiovascular development. Immunofluorescence microscopy studies demonstrated the presence of many of these proteins (including Morca2a, TRPC5, Enolase 1, Fbxo42, ST14, and Jumonji) in the embryonic heart. Subsequent functional analysis in an ex vivo cardiac culture model revealed novel roles for Collagen IV, NOGO-A/B, WNT16 and ADAM15 in heart development. Together with the immunofluorescence data, the functional analysis data confirmed that our approach did identify proteins relevant to cardiac development. In our dataset, molecules that may be useful as biomarkers in human AF were targeted, in particular secreted proteins and protein ectodomains. The successful application of a cluster of proteins (WNT16, ST14 and Pcsk1) as biomarkers of CHDs was confirmed in a study of human AF samples from women carrying normal fetuses and those with CHDs. This study adds new information to our understanding of the molecular pathways underlying normal and abnormal cardiovascular development, paving the way for the development of novel prenatal screens and treatments for human fetuses with CHDs.
Materials and Methods
Mice
CD1 mice (Charles River) were maintained under standard conditions. Timed-matings were detected by the presence of a vaginal plug. NOGO-A/B mice were provided by Steve Strittmatter (Yale University). The Yale University Animal Care and Use Committee approved all animal protocols and experimentation was performed in accordance with National Institute of Health regulations.
Whole Embryo Culture
Conceptuses were harvested at 7.5 dpc and cultured for 48 hours in pooled rat serum supplemented with 20 µm of the nitric oxide donor NOC-18 (2,2′-(hydroxynitrosohydrazino)bis-ethanamine) (Calbiochem), 10 pg/ml rVEGF (Chemicon), 20 mM L-glucose (control) and/or 20 mM D-glucose. The developmental stage was determined using Downs and Davies' criteria for staging morphological landmarks of the primitive streak, neural plate and head fold [19]. Embryos from head fold stage to somite stage 27–30 correspond to human gestational weeks 3–6 [20], [21], [22]. Embryos were examined under a dissecting microscope for gross defects (neural tube closure, axial rotation completion, yolk sac circulation and heart beat) and processed for mass spectrometry.
Atrioventricular Canal Explant Assay
Atrioventricular canals were isolated from E9.0 embryos and placed onto a 1 mg/ml Type I Rat Tail collagen gel (BD Biosciences). The explants were allowed to adhere prior to the addition of media [Medium 199, 1% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, and 0.1% each of insulin, transferrin, and selenium (GIBCO)] [23]. Cultures were stopped at 48–72 hours, fixed with 4% paraformaldehyde and stained with F-actin (Invitrogen) to evaluate cellular morphology. All experiments were repeated at least five times.
Liquid Chromatography Tandem Mass Spectrometry
Individual 8.5 dpc yolk sacs were lysed in RapiGest SF (Waters), digested with trypsin (Princeton Separations), and desalted using C-18 columns (VivaScience). LC-MS and LC-MS-MS analysis were performed on a Q-TOF API-US quadrupole time of flight tandem mass spectrometer (Waters) equipped alternatively with electrospray and nonospray sources. Peptides were eluted into the source following chromatography on a capillary HPLC CapLC system equipped with a dual wave length UV detector (Waters). For both standard ESI (flow set at 10 uL/min) and nonoflow (flow set at 100 nL/min) ionization methods a linear 30 min gradient of increasing acetonitrile concentration was applied to fractionate and elute the samples. The mass spectrometer's source voltage for this application was set at 3.5 kV with scan time duration of 1 s/scan and spectra were acquired using both survey and parent scan MS methods with data dependent acquisition (DDA) setting defined by MassLynx software instrument interface. Following acquisition, data was preprocessed automatically by MassLynx and the resulting MS-MS spectra was submitted to MZmine for post-acquisition processing (peak picking, data alignment, and normalization) to generate the peak intensity matrix [24], [25]. Data analysis was performed with Matlab and Excel. First the intensity matrix was filtered by dropping away peaks which were detected in less than 5 samples. This left 12908 rows to intensity matrix. T-test (unpaired; two-tailed; equal variances) and log ratio values were calculated for each peak between sample and control independently for all three sample/control pairs: HG vs. control, HG+rVEGF vs. control, and HG+nitric oxide donor vs. control. Log ratios were calculated as average peak intensity in the sample divided by average peak intensity in the control (Table S1). To find peaks with differential intensity between sample and control, threshold levels p-value<0.05 and absolute (log ratio)≥1 were used in each pair. These peaks were targeted (in an aliquot of the original yolk sac lysate sample) for MS-MS fragmentation and their spectra were searched against the nonredundant protein sequence database (NCBI, National Center for Biotechnology) using the Mascot search engine.
Immunofluorescence
Conceptuses were harvested from timed-pregnant mice at the indicated time points, frozen in OCT medium and cryo-sectioned (10 µm). Immunoreactivity was visualized with Alexa Fluor 594 or 488 secondary antibodies (Invitrogen). Nuclei were stained with DAPI. Digital images were captured on an Olympus IX71 Inverted Microscope.
Western Blot
Yolk sacs were harvested from culture conditions at the indicated time points and lysed in RIPA Buffer (Upstate) supplemented with 1% SDS, Complete Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktails (Calbiochem). The samples were homogenized and soluble extracts obtained by centrifugation. Protein concentrations were determined by BCA Assay (Bio-Rad) and 25 µg was electrophoresed on SDS-PAGE gels (Bio-Rad). Luminescence was performed using the Western Lightening Chemiluminescence Reagent (PerkinElmer) and densitometry using Quantity One Software (Bio-Rad).
Antibodies
Primary antibodies included NOGO-A (gift from Steve Strittmatter, Yale), PECAM (J.A.M), Laminin 1 (J.A.M.), α smooth muscle cell actin (Sigma), ADAM15 ectodomain (R&D Systems), WNT16 (BD Pharmigen), SCVT (Slc23a1) (Alpha Diagnostic International), Collagen IV (Abcam and USBiological), PIASγ (Abcam), TRPC5 (Chemicon), KIF21B (Upstate), Erk2 (Santa Cruz), NOGO-A/B (Santa Cruz), Enolase 1 (USBiological), and the following from Novus: Fbxo42, Jumonji, Pcsk1, ST14, CHST3 and ZCWCC1 (Morca2a).
Zymography
Yolk sacs were lysed in 50 mM Tris/1% Triton-X and homogenized with a motorized pestle. 25 µg of protein was mixed with non-reducing Laemmli buffer and loaded onto a 10% Zymogram Gel (Bio-Rad) for electrophoresis. The gels were washed in 2.5% Triton X-100, followed by water, and incubated for 48–72 h at 37°C in a 50 mM Tris-HCl buffer, pH 8.0, containing 5 mM calcium chloride or 10 mM EDTA (negative control for MMP activity). Gels were stained in 50% methanol, 10% acetic acid containing 0.1% Coomassie Blue R250 and imaged on a BioRad Chemidoc XRS.
Human Samples
Via ultrasound-guided amniocentesis, 16–22 week gestation amniotic fluids (AFs) were obtained from healthy women (average age 27.7±6.6) carrying fetuses with (n = 17) and without (n = 16) cardiac defects from 2004–2007 at Yale New Haven Hospital. The Human Investigation Committee of Yale University approved our study protocol, and written informed consent for research purpose was obtained from all participants before the procedure. The purpose of the amniocentesis was clinical: to rule out chromosomal abnormalities. Criteria for the “diseased group” were heart defect visualized on at least two ultrasound exams. In cases resulting in live newborns, the defects were confirmed postnatally. In the remaining cases, the pregnancy course was terminated and the defect was confirmed in elected autopsies. CHDs included ventricular septal defects, valvular stenosis, complete atrioventricular septal defects, bilateral ventriculomegaly, hypoplastic ventricle, tetralogy of Fallot, and transposition of the great arteries. Criteria for the “non-diseased group” were AF samples from women undergoing genetic testing who had a normal course of pregnancy including term delivery. All fetuses in the non-diseased group were euploid and had normal heart ultrasound examinations during pregnancy and a normal physical exam at birth.
Frozen AFs were centrifuged and aliquoted prior to assay. Western Blot was performed as described above on 25 µg of AF. A “standard” control sample was chosen to run on each gel. Coomassie staining of gels confirmed equal protein load. Films were scanned and analyzed in Quantity One as follows. Bands were selected for quantification using the automated volume contour tool. Band densities were calculated by computer algorithm as the volume, the sum of pixel intensities multiplied by pixel area, adjusted by global background subtraction. Blotting and analysis was performed by a blinded observer.
Univariate logistic regression was performed using the design packet in R (GNU S). The AF measurements were log-transformed using log2 and the regression coefficient and odds ratio (OR) were calculated. The OR can be interpreted as the increased risk of developing a cardiac defect per doubling of the AF measurement. The mean classification error was calculated using 10 by 10 cross-validation. An attempt to perform multiple regression analysis did result in inflated regression coefficient estimates due to strong multicollinearity. After normalization and centering of all biomarker data, Principle Component Analysis (PCA), a clustering tool that reduces multidimensional data sets to identify new meaningful underlying variables, was performed using the SVD function in Matlab.
Results
Tissue Samples from a Murine Model of Induced Cardiovascular Defects
One premise of our study is that proteomic profiling of hyperglycemia exposed vascular tissue will enrich for factors involved in endothelial differentiation or dysfunction. Hyperglycemia was used as an “inducer” because increased vascular and cardiac defects are observed in the offspring of diabetic rodents (streptozotocin-induced) and the infants of diabetic women [26], [27], [28]. Interestingly, pregnant women presenting with altered diurnal gylcemic profiles not related to diabetes also have a higher incidence of preterm labor, fetal malformations and perinatal mortality similar to diabetic patients, underscoring the devastating developmental effects of hyperglycemia [29]. The addition of 20 mM D-glucose (a plasma concentration of glucose often observed in both human and rodent diabetics during a hyperglycemic episode) to ex vivo embryo cultures results in vasculopathy in the yolk sac and inhibition of EMT in the cardiac endocardium [23], [30], [31], [32], [33], [34]. These studies contribute to accumulating evidence that transient hyperglycemia directly affects the endothelium, and highlight the importance of endothelial cell homeostasis during embryonic development [35]. Identification of candidate differentially expressed proteins between normal and hyperglycemic groups will provide insight into currently unrecognized proteins in cardiovascular development and may allow for the discovery of biomarkers of CHDs applicable to the general population.
Under normoglycemic conditions, control embryos exhibit normal organogenesis and vascular development of the yolk sac (Fig. 1A, whole embryo inside a functional yolk sac containing circulating red blood cells). Hyperglycemic embryos display malformation of the embryo proper and yolk sac vasculature (Fig. 1B, abnormal embryo with a nonfunctional vitelline circulation, evidenced by pooling of blood at one pole of the yolk sac). Whole mount staining of control yolk sacs for the endothelial marker PECAM-1 confirms the formation of a mature, remodeled vasculature with a hierarchy of arborizing large and small vessels (Fig. 1C) while hyperglycemic yolk sacs appear arrested at the primary capillary plexus stage of vascular development (Fig. 1D). By E9.5, endocardial cells (cardiac endothelial cells) of the endocardial cushions are actively undergoing EMT and invading the cardiac jelly in control embryos (Fig. 1E). Hyperglycemia results in an acellular endocardial cushion due to inhibition of EMT, which prematurely arrests cushion morphogenesis (Fig. 1F).
Figure 1. Experimental Model: Applying Proteomic Profiling to a Hyperglycemia Induced Model of Cardiovascular Defects.

Embryos cultured ex vivo exhibit normal organogenesis and vascular development (A, normal embryo inside a functional yolk sac containing circulating red blood cells). Embryos treated with 20 mM D-glucose display malformation of the embryo and vasculature (B, abnormal embryo and pooling of blood at one pole). PECAM staining of control yolk sacs reveals a mature, remodeled vasculature (C) while hyperglycemic yolk sacs appear immature and mal-developed (D). Normally by E9.5, EMT has begun within the cardiac endocardial cushions (E). Treatment with hyperglycemia results in an acellular endocardial cushion (F). E–F: α-Smooth muscle cell actin (green) labeling of the myocardium (Myo) and mesenchymal cells (Mes); PECAM (red) staining of the endocardium (En); CJ (Cardiac Jelly). Volcano plots (G–I)) depicting each peptide peak detected by LC-MS as a blue dot and plotting the log2 ratio (of treatment to control) against −log10 p value (G, HG vs Control; H, HG+rVEGF vs Control; I, HG+ nitric oxide donor vs Control). The red dots represent the 143 peptide peaks that were dysregulated by hyperglycemia but returned to control levels by both rVEGF (H) and nitric oxide donor (I). The red horizontal lines are drawn at p = 0.05 and the red vertical lines are drawn at log2 ratio = 1 and −1. Scale bars = 100 µm. (A–B is reprinted from Am J Pathol 2001, 158:1199-206 with permission from the American Society for Investigative Pathology.)
We hypothesized that the hyperglycemic insult may be used to enrich for factors involved in endothelial differentiation/dysfunction. Due to the global biological effects of hyperglycemia, we expected a large number of proteins to exhibit expression changes, but only a subset to be relevant to endothelial differentiation/dysfunction. Therefore two additional groups, “rescue conditions”, were included in the experimental design. Our previous studies demonstrated that exogenous rVEGF-A165 or a nitric oxide donor supplementation reverses the morphological, functional and biochemical changes induced by hyperglycemia in the cardiovascular system [23], [36], [37]. Thus, we hypothesized that the proteins dysregulated by hyperglycemia but returned to normal levels by both rVEGF and nitric oxide donor represent a set of proteins that are important for normal vascular development.
Liquid Chromatography Mass Spectrometry (LC-MS) of Yolk Sac Tissue
Groups of ten 7.5 dpc conceptuses were cultured in normoglycemia, hyperglycemia, hyperglycemia plus rVEGF-A165, or hyperglycemia plus nitric oxide donor. Yolk sacs were harvested at 24 hours, at the end of the primary capillary plexus stage and beginning of the vascular remodeling stage, and processed individually for mass spectrometry. LC-MS detected 39,120 peaks which were subsequently analyzed using MZmine software [24], [25]. To increase confidence, a data reduction step discarded peaks detected in less than 5 samples resulting in 12,908 peptide peaks in the intensity matrix (Table S1). Data alignment, normalization and statistical treatment revealed 651 differentially regulated peaks between normal and hyperglycemic samples. Among those, 143 were dysregulated (upregulated or downregulated) by hyperglycemia and returned to control levels by both the rVEGF and nitric oxide donor groups. These 143 peaks were targeted (in an aliquot of the original yolk sac lysate sample) for MS-MS fragmentation and subsequent protein identification using the Mascot search engine (www.matrixscience.com) and NCBInr Database.
To globally visualize the dataset, volcano plots were generated depicting each of the 12,908 peptide peaks as a point and plotting the log2 ratio (of treatment group to control) against −log10 p value (Fig. 1G–I). The red dots in the volcano plots represent the 143 peptide peaks that are dysregulated by hyperglycemia [inclusion criteria: both p<0.05 and log2 (fold change)>1] but returned to control levels by both rVEGF and nitric oxide donor [inclusion criteria: p>0.05 or p<0.05 and log2 (fold change)<1]. Significant peptide peaks were ranked by fold change and those greater than or equal to 2 [log2 (fold change)≥1] were included. Of the 143 peptide peaks, approximately 75% were upregulated. The majority of peptide peaks display a 2–4 fold change [log2 (fold change)≥1–2]. Though these changes appear minor, in developmental contexts slight changes in growth factors or signaling pathways disrupt cardiovascular development. For instance, a one fold increase or half fold decrease in VEGF leads to lethal cardiac defects [38].
Protein Identification of the Subset of Peptide Peaks Dysregulated by Hyperglycemia But Returned to Normal Levels by Both rVEGF and Nitric Oxide Donor
Candidate proteins with the highest Mowse score [39] were mapped onto Gene Ontology classes in order to group proteins according to biological processes (Table 1) [40]. Candidates included distinct collagen and laminin chains which are known to surround vessels and provide environmental cues to endothelial cells [41]. Extracellular matrix (ECM) and provisional matrices, including the cardiac jelly, contain laminin, collagen and other ECM molecules many of which are also known to be expressed in human amniotic fluid (AF). Interestingly, patients with diabetic retinopathy secrete into the circulation a detectable increase in the level of laminin, suggesting that it may be a useful biomarker [42]. Proteoglycans are also present in the vasculature and the endocardial cushions, therefore it was not surprising to find chondroitin 6 sulfotransferase 3 (CHST3), an enzyme that sulfates proteoglycans, as one of the dysregulated proteins in our dataset.
Table 1. Candidate Proteins Associated with the Subset of Statistically Significant Peptide Peaks Dysregulated by Hyperglycemia But Returned to Normal Levels by Both rVEGF and Nitric Oxide Donor.
| Category | Protein Name | Entrez GeneID | Gene Ontology Processes |
| Adhesion/ Migration | Chemokine (C-X-C motif) ligand 15 (Cxcl15) | 20309 | Chemotaxis; hemopoiesis; signal transduction; immune response |
| Chondroitin 6-sulfotransferase (Chst3) | 53374 | Carbohydrate metabolic process | |
| Collagen, type IV, alpha 2 (Col4a2) | 12827 | Cell adhesion; phosphate transport | |
| Collagen, type VII, alpha 1 (Col7a1) | 12836 | Cell adhesion; phosphate transport | |
| Laminin, alpha 4 chain (Lama4) | 16775 | Cell adhesion; blood vessel development; regulation of embryonic development | |
| Matrix metalloprotease 2 (Mmp2) | 17390 | Blood vessel maturation; collagen catabolic process; proteolysis | |
| NOGO-A (Rtn4) | 68585 | Regulation of cell migration; angiogenesis; nervous system development | |
| Proprotein convertase 1 (Pcsk1) | 18548 | Peptide biosynthetic process; proteolysis | |
| Suppressor of tumorigenicity 14 (St14) | 19143 | Cell migration; proteolysis | |
| Cytoskeleton | Dishevelled associated activator of morphogenesis 2 (Daam2) | 76441 | Actin cytoskeleton organization and biogenesis |
| Filamin C, gamma (actin binding protein 280) (Flnc) | 68794 | Actin filament-based process | |
| Kinesin family member 21B (Kif21b) | 16565 | Microtubule-based movement | |
| Tubulin, alpha 1B (Tuba1b) | 22143 | Microtubule-based movement | |
| Differentiation/ Development | E74-like factor 5 (Elf5) | 13711 | Ectoderm development; transcription |
| Hairy enhance of spilt 6 (Hes6) | 55927 | Cell differentiation; transcription | |
| Jumonji, AT rich interactive domain 2 (Jarid2) | 16468 | Multicellular organismal development; transcription | |
| Laminin, alpha 4 chain (Lama4) | 16775 | Blood vessel development; cell adhesion; regulation of embryonic development | |
| Notch1 | 18128 | Cell differentiation; heart development; EMT; angiogenesis | |
| NOGO-A (Rtn4) | 68585 | Nervous system development; angiogenesis; regulation of cell migration | |
| Protein inhibitor of activated STAT, 4 (Pias4) | 59004 | Multicellular organismal development; JAK-STAT cascade; transcription; protein sumoylation | |
| Single-minded 2 (Sim2) | 20465 | Cell differentiation; nervous system development; transcription | |
| WNT16 | 93735 | Multicellular organismal development | |
| Insulin Signaling | Proprotein convertase 1 (Pcsk1) | 18548 | Peptide biosynthetic process; proteolysis |
| Protein tyrosine phosphatase, non- receptor type 1 (Ptpn1) | 19246 | Insulin receptor signaling pathway; Dephosphorylation | |
| Zinc finger protein 106 (Zfp106) | 20402 | Insulin receptor signaling pathway | |
| Adaptor-related protein complex 3, beta 1 subunit (Ap3b1) | 11774 | Antigen processing/presentation; protein transport; endocytosis | |
| Chemokine (C-X-C motif) ligand 15 (Cxcl15) | 20309 | Immune response; chemotaxis; hemopoiesis; signal transduction | |
| Interleukin 25 (Il25) | 140806 | Inflammatory response | |
| Proteolysis | A disintegrin and metalloproteases 15 (Adam15) | 11490 | Proteolysis; integrin-mediated signaling |
| Proprotein convertase 1 (Pcsk1) | 18548 | Proteolysis; peptide biosynthetic process | |
| Matrix metalloprotease 2 (Mmp2) | 17390 | Proteolysis; blood vessel maturation; collagen catabolic process | |
| Suppressor of tumorigenicity 14 (St14) | 19143 | Proteolysis; cell migration | |
| Signal Transduction | A disintegrin and metalloproteases 15 (Adam15) | 11490 | Integrin-mediated signaling; proteolysis |
| Chemokine (C-X-C motif) ligand 15 (Cxcl15) | 20309 | Signal transduction; chemotaxis; hemopoiesis; immune response | |
| Protein inhibitor of activated STAT, 4 (Pias4) | 59004 | JAK-STAT cascade; multicellular organismal development; transcription; protein sumoylation | |
| Protein kinase C binding protein 1 (Prkcbp1) | 228880 | Phosphorylation | |
| Protein tyrosine phosphatase, non- receptor type 1 (Ptpn1) | 19246 | Dephosphorylation; insulin receptor signaling pathway | |
| Transcription/ Translation | DEAD (Asp-Glu-Ala-Asp) box polypeptide 31 (Ddx31) | 227674 | RNA processing |
| E74-like factor 5 (Elf5) | 13711 | Transcription; Ectoderm development | |
| Hairy enhance of spilt 6 (Hes6) | 55927 | Transcription; Cell differentiation | |
| Jumonji, AT rich interactive domain 2 (Jarid2) | 16468 | Transcription; Multicellular organismal development | |
| Protein inhibitor of activated STAT, 4 (Pias4) | 59004 | Transcription; JAK-STAT cascade; multicellular organismal development; protein sumoylation | |
| Ribosomal protein L18 (Rpl18) | 19899 | Translation | |
| Serine/arginine repetitive matrix 2 (Srrm2) | 75956 | RNA splicing | |
| Single-minded 2 (Sim2) | 20465 | Transcription; cell differentiation; nervous system development | |
| SRY-box containing gene 19 (Sox19) | 20673 | Regulation of transcription | |
| Transport | Adaptor-related protein complex 3, beta 1 subunit (Ap3b1) | 11774 | Protein transport; endocytosis; antigen processing/presentation |
| ATPase type 13A1 (Atp13a1) | 170759 | Cation transport; metabolic process | |
| ATP-binding cassette transporter, sub- family A, member 13 (Abca13) | 268379 | Transport; protein targeting | |
| ATP-binding cassette, sub-family A, member 7 (Abca7) | 27403 | Transport; phagocytosis | |
| Collagen, type IV, alpha 2 (Col4a2) | 12827 | Phosphate transport; cell adhesion | |
| Collagen, type VII, alpha 1 (Col7a1) | 12836 | Phosphate transport; cell adhesion | |
| Down syndrome critical region gene 3 (Dscr3) | 13185 | Vacuolar transport | |
| Solute carrier family 23 (nucleobase transporters), member 1 (Slc23a1) | 20522 | Ion transport | |
| Transcobalamin 2 (Tcn2) | 21452 | Ion transport | |
| Transient receptor potential protein 5 (Trpc5) | 22067 | Ion transport | |
| Others/Unknown | Enolase 1, alpha non-neuron (Eno1) | 13806 | Glycolysis |
| F-box protein 42 (Fbxo42) | 213499 | Ubiquitin cycle | |
| TNF receptor-associated protein 1 (Trap1) | 68015 | Protein folding; response to stress | |
| Microrchidia 2a (Morca2a) | 74522 | ? | |
| T complex expressed gene 4 (Cramp1l) | 57354 | ? | |
| T complex protein-10 (Tcp10) | 21458 | ? | |
| Tetratricopeptide repeat domain 15 (Ttc15) | 217449 | ? | |
| Tripartite motif containing 68 (Trim68) | 101700 | ? |
Several proteins in our dataset mapped to the Gene Ontology term “Proteolysis”. These proteins function in varied biological processes involving proteolytic activity such as cell-cell adhesion, migration/invasion, matrix remodeling, growth factor release/activation, receptor shedding and cytokine signaling. The candidate proteases detected, included ADAM15, MMP2, Suppressor of tumorigenicity 14 (ST14) and Proprotein convertase 1 (Pcsk1).
A number of candidate proteins that directly affect differentiation and embryonic development were discovered in our dataset, including Hairy and enhancer of split 6 (HES6), Notch1, NOGO-A, Jumonji, and WNT16. Several other candidate proteins identified in our screen mapped to the Gene Ontology term “Transport” including Transient receptor potential protein (TRPC5), a calcium channel, Sodium/vitamin C transporter (SVCT or Slc23a1), a sodium/ascorbate cotransporter, and ATP binding proteins (Atp13a1, Acca13, and Abca7). Other proteins of note found in our database include Fbxo42 (part of the ubiquitin ligase complex), Protein inhibitor of activated STAT4 (PIASγ) (a SUMO ligase), Kinesin-like protein KIF21b (a microtubule binding motor protein), Enolase 1 (an enzyme in the glycolysis pathway) and Microrchidia 2a (Morc2a or ZCWCC1).
Several candidate factors in our dataset have been correlated with disease states including the Ets family member Elf5, which is upregulated in breast cancer [43]. Interestingly, the bHLH transcriptional repressor Single-minded 2 which is thought to contribute to the etiology of Down syndrome [44] and Down syndrome critical region gene 3 (DSCR3), located on human chromosome 21, were present in our dataset. Approximately, 40–50% of infants with Down syndrome have CHDs. Finally, protein tyrosine phosphatase, non-receptor type 1 (PTPN1) was identified in our dataset. PTPN1 plays a role in obesity and metabolism. Further, gene variants have been identified in obese people and type 2 diabetics [45], [46], [47].
In order to assess the biological significance of the dysregulation of these candidate proteins and validate our MS-MS results (Fig. 2), Western blotting was performed on lysates of yolk sacs harvested from embryos cultured under the four experimental conditions. ADAM15, Pcsk1, and NOGO-A were confirmed to be downregulated while Laminin, TRPC5, WNT16, Morc2a, and CHST3 were confirmed to be upregulated. Additionally, a reduction in the proteolytic activity of MMP2 was observed by zymography. After hyperglycemia treatment, the change in expression of most proteins was 2 fold: densitometry of the blots displayed in Figure 2 detected a fold change decrease of 1.9 for MMP2, decrease of 2.7 for ADAM15, increase of 2 for ST14, and decrease of 1.9 for Pcsk1 in the HG group. These differences are consistent with the mass spectrometry analysis (Figure 1G–I and Table S1) which revealed that the majority of peptide peaks display a 2–4 fold change. Additional proteins that displayed the expected fold change included WNT16, Jumonji, TRPC5, SVCT, NOGO-A, Fbxo42, Enolase1, and Morca2a. The only protein that validated but did not demonstrate the predicted fold change was Laminin. These data contribute to the accumulating evidence of the accuracy of label-free LC-MS measurements to predict relative protein changes [48], [49]. Several identified proteins did not validate by Western blot and were not pursued further. Several other proteins were not pursued because no commercially available antibodies were available for validation and subsequent investigation.
Figure 2. Validation of Differential Expression of Candidate Proteins Identified in the Proteomic Profiling Study.

To validate candidate proteins detected by LC-MS-MS, Western blotting and zymography was performed on lysates of yolk sacs harvested from embryos cultured under the four experimental conditions. Erk2 was used as a loading control. Green arrows represent protein levels that increased with hyperglycemia (HG) treatment but returned to control levels in both rescue conditions (HG/V and HG/NO). Red arrows represent protein levels that decreased with hyperglycemia treatment but returned to control levels in both rescue conditions (HG/V and HG/NO). The Phi symbols represent protein levels that did not vary across treatment groups or did not vary in the expected pattern (increased or decreased by HG and returned to control levels in both rescue conditions).
We discovered a number of unnamed and predicted proteins with significant dysregulation. Continued curation of the public databases will reveal the identities and functions of unnamed proteins identified from cDNA initiatives; however, as little information is currently available on such proteins these were not further pursued. Many proteins were identified from multiple different significant peptide peaks (i.e. different fragments within the same protein). The consolidated list consisting of 44 proteins is presented in Table 1 (excluding unnamed and predicted proteins).
Factors Involved in Yolk Sac Vascular Development Are Also Important for Cardiac Development
To validate our assumption that proteins relevant to cardiac development may be isolated from vascular tissue, proteins from the validated candidate list were tested by immunofluorescence microscopy for their presence in the endocardial cushion at E9.5 (a stage of cardiac development when EMT is actively occurring). This is a time and place during cardiogenesis that is widely recognized to be a “hot spot” of cardiac anomalies.
Laminin 1 was strongly present at the interface of the myocardial and mesenchymal zones of the cushion, consistent with basement membrane localization (Fig. 3A). TRPC5 and SVCT were highly expressed in the compact myocardium and weakly/absent throughout the rest of the myocardium and cushion (Fig. 3B, C). Potentially these factors play a role in contraction or function of the valves. NOGO-A localizes strongly within the myocardium while staining with a NOGO-A/B antibody reveals localization within the mesenchymal and endothelial cells, suggesting NOGO-B presence in these cell types (Fig. 3D, E). β-galactosidase staining of transgenic lacZ NOGO-A/B whole hearts harvested at E9.5 demonstrated localization to the outflow tract and atrioventricular canal (Fig. 3F). CHST3, WNT16, and ST14 were detected throughout the cell layers of the endocardial cushion and in the myocardium (Fig. 3G–I). Jumonji was present strongest in the myocardium and mesenchyme, and weakly in the endocardium (Fig. 3J). The expression pattern of Enolase 1 was localized to the endocardium, mesenchyme, and epicardium, but absent from the myocardium (Fig. 3K). ADAM15 was highly expressed in the endocardium and mesenchymal cells, but also present in the myocardium (Fig. 3K, L). NOGO-B, ST14, Enolase 1, and ADAM15 had a notable presence within the cellular projections of the mesenchymal cells. Immunoreactivity for Pcsk1 was strongest in the myocardium and weaker in the endocardium and mesenchyme (Fig. 3M). Fbxo42 displayed nuclear expression in a gradient pattern across the layers of the cushion to the myocardium (Fig. 3N). The zinc finger protein, Morc2a, was present in an increasing gradient from the endocardium to the mesenchymal cells to the myocardium, and most intensely in a nuclear/perinuclear pattern (Fig. 3O).
Figure 3. Spatial Expression Pattern of Validated Proteins in in vivo Embryonic Hearts.

Immunofluorescence (red or green) was performed on hearts excised at E9.5 to confirm that the validated proteins identified in the vascular dataset are also present in the heart (A–E, G–O). Insert contains the DAPI (blue) merged image. F: Whole mount β-gal staining of an E9.5 NOGO-A/B-lacZ heart. Outflow Tract (OFT); Ventricle (V); AVC (Atrioventricular Canal); A (Atrium); Myocardium (Myo); Endocardium (En); Mesenchyme (Mes); Scale bar = 50 µm.
Proteins were selected for further study in a functional assay, the ex vivo atrioventricular canal assay, that has yielded many insights into the molecular events that underlie endocardial cushion development [4]. The atrioventricular canal assay mimics the spatiotemporal, molecular and biochemical events that occur during in vivo valve development and provides a system amenable to experimental manipulation [50], [51]. In this model, the atrioventricular canal segment is excised prior to EMT; the cushion tissue is exposed and placed onto the surface of a collagen gel. Media containing preservative free antibodies directed against secreted proteins or the extracellular domain of membrane proteins is added to the cultures. The endothelial cells migrate onto the surface of the gel and away from the explants. In response to inductive signals secreted by the myocardium a subset of the endothelial cells undergo EMT and invade into the gel. Cells undergoing EMT follow a semi-linear progression of morphological changes including: 1) lateral cell-cell separation from epithelial sheets, 2) elongation of the single, rounded cells on the surface of the gel, and 3) invasion of elongated cells into the gel (Fig. 4K). Approximately 300 cells migrate out of each of the explants, however in order to observe the cellular morphology in detail, a high magnification image of few cells is displayed in Figure 4. Additionally, a cartoon representation of these cells is provided to highlight the varied and detailed cellular morphologies observed in this assay.
Figure 4. Analysis of the Potential Biological Function of Identified Proteins in an ex vivo Cardiac Assay.

The potential biological effects of identified proteins were characterized in the ex vivo atrioventricular canal assay to determine if proteins relevant to cardiac development may be isolated from the vascular dataset. Control IgG (A, C, E, G and I) or antibodies directed against WNT16 (B), Collagen IV (D), NOGO-A/B (F), ADAM15 (H), and SVCT (J) were added to the media bathing the explants. After 48 hours F-Actin (red) staining was performed and a high magnification image of a few representative cells was taken (blue = DAPI). Cartoons representing the various stages of EMT (K) and the morphology of the cells within the high magnification image from each treatment are displayed. Scale bar = 50 µm.
Normal cultures (IgG treatment) exhibited the characteristic phenotype of EMT, displaying elongated, invasive mesenchymal cells (Fig. 4A, C, E, G and I; images taken below the gel surface). However a dramatic inhibition of cardiac EMT resulting in the majority of cells maintaining an epithelial phenotype (rounded and non-invasive) was observed with WNT16 sequestration (Fig. 4B; gel surface image). In the yolk sac, an up regulation of WNT16 was associated with mal-development (abnormal increase in endothelial cells) while in the atrioventricular canal an inhibition of WNT16 was associated with decreased differentiation, suggesting WNT16 is a positive regulator of differentiation. Inhibition of cell adhesion using a collagen IV antibody led to the formation of epithelial cells with a rounded morphology and the presence of short cellular projections (Fig. 4D; gel surface image).
Blocking NOGO binding using a NOGO-A/B antibody directed against its extracellular domain revealed a novel role for NOGO in cardiac development. The treated cells appear to undergo a partial EMT or prematurely arrested EMT as evidenced by the formation of oval cells rather than fully elongated cells (Fig. 4F; gel surface image). However, given the known roles of NOGO in the neuronal system, a guidance or matrix interaction defect may underlie the observed phenotype. At this time it is unclear if this effect is due to NOGO-A or NOGO-B as the antibody used in these studies detects both isoforms. NOGO-A was the isoform identified in our MS dataset however our localization data suggest NOGO-B is expressed by the endocardium and mesenchyme. Further, a recent paper demonstrated a role for NOGO-B in vascular cell biology [52].
Atrioventricular canals cultured in the presence of an antibody directed against the ectodomain of ADAM15 resulted in blunted cardiac EMT (Fig. 4H; gel surface image) compared to IgG control. The cells begun to elongate and displayed multiple cellular extensions in a star-like pattern, but failed to complete elongation and invade the gel. Antibody blocking of the extracellular domain of SVCT failed to reveal a role for this transporter in EMT processes (Fig. 4J; image taken below the gel surface). Collectively, these results demonstrate that several proteins in our dataset are present in the embryonic heart and have novel roles at various steps of cardiac EMT, confirming that our approach utilizing vascular tissue did identify proteins relevant to cardiac development.
Identification of Biomarkers of Human Congenital Heart Defects
One of the aims of this study was to identify biomarkers that discriminate diseased from healthy fetuses using the proteomic dataset as a guide for candidate proteins that require follow up with more precise protein chemistry methods (Western blot, immunoflourescence microscopy, and explant assays). A pilot study was initiated to determine if the expression of these proteins vary in the AF of a cohort of healthy, non-diabetic pregnant women. AF was obtained from routine amniocentesis of women carrying normal fetuses (n = 17) and women carrying fetuses with CHDs (n = 16) (population details in Materials and Methods). The levels of candidate biomarkers in the AF were assessed by Western blot followed by densitometry.
CHST3 was significantly (p<0.01) increased in the AF of women carrying fetuses with CHDs (mean = 380) compared to controls (mean = 176) by Student's t-test (unpaired; two-tailed distribution; two-sample equal variance), but not significant by univariate logistic regression (p<0.1; misclassification error of 0.33) (Fig. 5A, B; Table 2). Morca2a demonstrated an increased trend in the AF of women carrying fetuses with CHDs (mean = 505) compare to controls (mean = 374); however, this finding was not significant by t-test (p<0.1) nor univariate logistic regression (p<0.2, misclassification error of 0.35).
Figure 5. Identification of Biomarkers of Human Congenital Heart Defects.
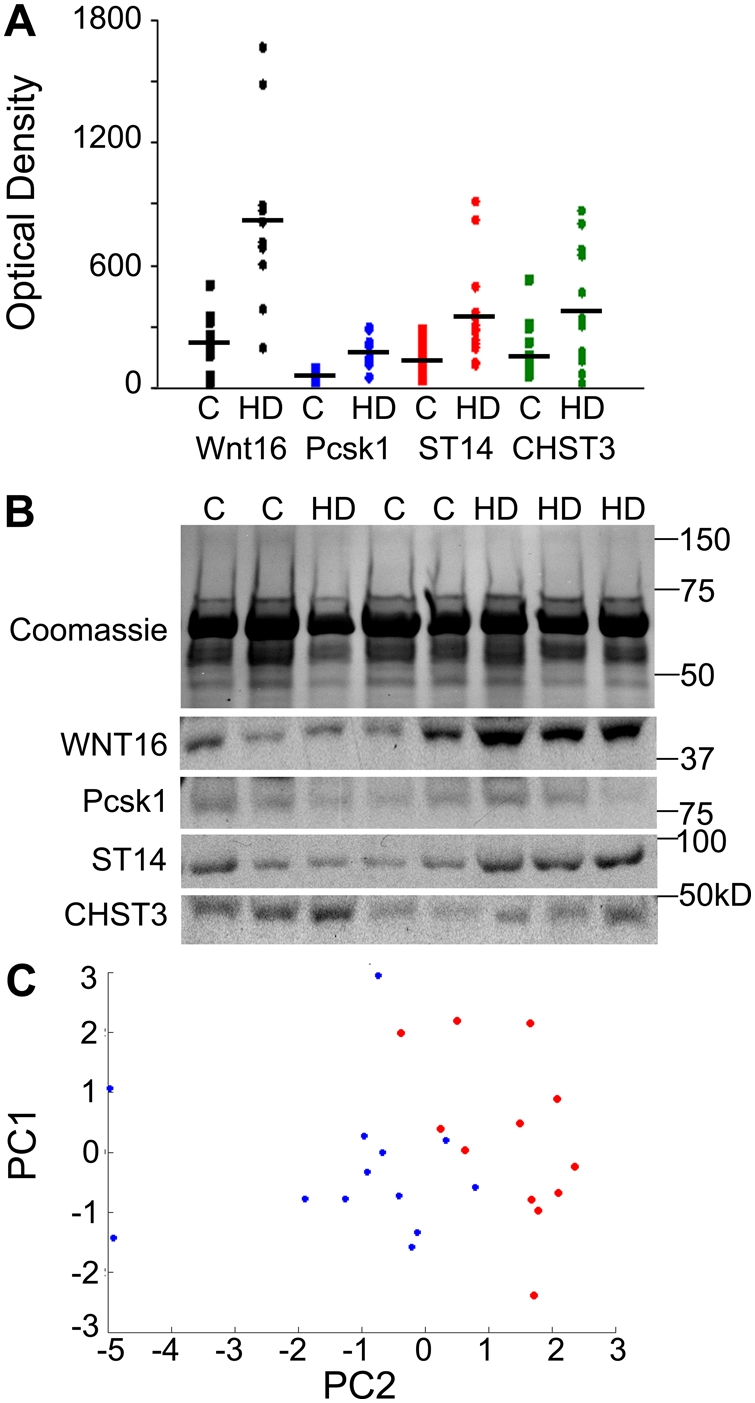
Densitometry data (A) from Western blots of AF [CHDs (HD) N = 17; Controls (C) N = 16]; black bars represent the mean. Coomassie stained gel of AF samples and representative Western blots for WNT16, Pcsk1, ST14, and CHST3 on a sampling of patients (B). PCA plot (C) for all biomarkers demonstrates separation of CHDs samples from controls. Red (Control); Blue (CHD).
Table 2. Univariate Logistic Regression and Misclassification Error for Amniotic Fluid Biomarkers.
| Marker | Coefficient | p-Value | OR | 95% CI | MC error |
| Wnt16 | 2.71 | 0.006 | 15.01 | 2.16–104.44 | 0.11 |
| SVCT | 0.62 | 0.252 | 1.86 | 0.64–5.35 | 0.41 |
| Pcsk1 | 3.29 | 0.012 | 26.95 | 2.07–350.70 | 0.12 |
| CHST3 | 0.51 | 0.129 | 1.67 | 0.86–3.22 | 0.33 |
| Enolase | −0.43 | 0.213 | 0.65 | 0.33–1.28 | 0.59 |
| PIAS | 0.46 | 0.209 | 1.58 | 0.77–3.21 | 0.63 |
| ST14 | 1.97 | 0.013 | 7.17 | 1.51–34.14 | 0.19 |
| Morca2a | 0.58 | 0.213 | 1.78 | 0.72–4.41 | 0.35 |
The biomarkers that showed significant association with CHDs in univariate logistic regression were WNT 16, Pcsk1 and ST14 (Table 2). WNT16 was significantly increased (p = 0.006) in the AF of women carrying fetuses with CHDs (mean = 818) compared to controls (mean = 220) as was Pcsk1 (p = 0.012; mean = 167 compared to control mean = 50), and ST14 (p = 0.013; mean = 356 compared to controls mean = 135) (Fig. 5A, B). In a cross-validation experiment, these markers were able to correctly classify controls from cases in up to 89% of cases (misclassification error of 0.11). Proteins that did not appear to be predictive for heart defect included SVCT, Enolase and PIASγ. PCA analysis using all biomarkers revealed clustering of controls and cases (Fig. 5C) demonstrating that the CHD samples segregate from the controls. Although a larger cohort is required to confirm the specificity and sensitivity of these markers for diagnosis, our pilot study of human AF revealed promising biomarkers able to detect a range of CHDs (ventricular septal defects, valvular stenosis, complete atrioventricular septal defects, tetralogy of Fallot, and transposition of the great arteries) that in part result from defects in cushion morphogenesis. Furthermore, dysregulation of WNT16, ST14 and Pcsk1 may reflect the molecular origins of CHDs.
Discussion
Applying a proteomic approach to embryonic vascular tissue generated from an ex vivo model of vasculopathy, we have identified proteins and pathways that are disrupted during cardiovascular development and lead to the observed pathological phenotype. Dysregulation of this set of proteins in a “normal” fetus may signal that the fetus is congenitally at risk or encountering an environmental stressor. Several of these protein targets (WNT16, Pcsk1 and ST14) provided the basis for a novel prenatal screen for the detection of CHDs, suggesting dysregulation of WNT16, ST14 and Pcsk1 may play a role in the etiology of human CHDs. Several forms of CHDs are known to have a genetic basis such as microdeletions (22q11) in DiGeorge Syndrome and single gene mutations (JAG1) in Alagille Syndrome. It would be of interest to determine if individuals with CHDs carry genetic mutations in WNT16, ST14 or Pcsk1. At the protein level, these markers are overexpressed therefore if sequence variants are found mutations would likely affect protein function and/or activity. In the case of ST14 and Pcsk1, both of which are enzymes, a hypomorphic or hypermorphic mutation in these genes has the potential to affect multiple substrates and hence several cellular processes.
Changes in cellular processes not only reflect the molecular origins of disease but also provide avenues for molecular therapies to ameliorate the defect. To this end, we identified several proteins, previously unknown to have a role in cardiovascular development, with diverse functions that cluster to adhesion/migration, differentiation, transport, IGF signaling pathways (Table 1). Although we discovered and characterized several proteins with currently unknown roles in cardiovascular development, we were able to place them in their relevant biological context by relating them to existing functional cascades (signaling, enzymatic, polarity, migration, and differentiation). Protein networking analysis revealed that several of the identified proteins interact upstream or downstream of each other and with established factors in cardiovascular development (Fig. 6). The presence of proteins known to have roles in cardiac development within our dataset further validates our approach. For example, our screen detected the transcriptional repressor Jumonji whose deletion is known to result in myocardial defects and ventricular septal defects [53]. However our data demonstrating decreased Jumonji during vasculopathy extends the role of Jumonji to vascular development.
Figure 6. Integration of Proteins Identified by Proteomic Profiling with Known Pathways and Biological Processes Involved in Cardiovascular Development.

We identified proteins (in red) known and previously unknown to have roles in cardiovascular development with diverse functions including migration, differentiation, IGF signaling, protease activity and transport (Table 1). Several of the proteins interact upstream or downstream of each other and with established factors in cardiovascular development. Dysregulation of these proteins/pathways not only reflect the molecular origins of CHDs but, in a normal fetus, may signal that the fetus is at risk or encountering an environmental stressor thus providing the basis for novel diagnostic tests.
Previously, we demonstrated that yolk sac vasculopathy is associated with increased nitric oxide, however the nitric oxide targets remained elusive [36]. Recently nitrosylation sites on the cytoplasmic domain of TRPC5 were identified and in response to nitric oxide, Ca(2+) entered bovine aortic endothelial cells [54]. In this study we observed increased TRPC5, raising the possibility that the observed yolk sac vasculopathy is mediated by nitric oxide driven over-activation of TRPC5. We speculate that the subsequent abnormal influx of calcium through TRPC5 activates the calcineurin/NFAT pathway [55]. In the developing heart, the NFAT pathway is tightly regulated and has complex interactions with DSCR (another protein identified in our screen) and VEGF to drive differentiation; low levels of VEGF are required for EMT while high levels terminate it [56], [57]. Therefore, it is tempting to speculate that TRPC5 is a novel player in cardiac development, acting upstream of NFAT, leading to dysregulation of VEGF expression and ultimately termination of heart development.
On the other hand, positive regulation of cardiac EMT relies on Wnt/β-catenin signaling however the specific Wnts involved are largely unknown with the exception of Wnt9a in the avian heart [58]. Variations in Wnt signaling are expected to have direct effects on valve plasticity as canonical Wnt/β-catenin signaling induces mesenchymal differentiation and modulates the amount of mesenchyme produced [59], [60]. One defect associated with vasculopathy is increased endothelial cell number per vessel, leading to a hypertrophic, nonfunctional capillary plexus. Thus the observed upregulation of WNT16 may lead to unregulated endothelial cell differentiation or proliferation, ultimately affecting the organization of vascular structures. Concomitantly, antibody neutralization of WNT16 blunts cardiac EMT in the atrioventricular canal assay. Though the WNT16 isoform is known to be expressed in the embryonic heart [61], a functional role for WNT16 has not been previously reported, thus our atrioventricular canal assay results describes a new function for a novel WNT member in the cardiac developmental cascade. Additionally, we observed increased WNT16 in human amniotic fluid (AF) of women carrying fetuses with CHDs and demonstrated that this is predictive of CHDs. Monitoring WNT16 levels and targeting WNT16 signaling may provide novel avenues for future diagnostic and pharmacological research.
Proteoglycans and extracellular matrix are important players in cell adhesion, migration, proliferation, and angiogenesis, processes important in both development and disease. CHST3 catalyzes the transfer of sulfate to position 6 of the N-acetylgalactosamine (GalNAc) residue of chondroitin. Human endothelial cells express CHST3 mRNA while endothelial cells overlying the porcine mitral valve produce chondroitin sulphate [62], [63]. Interestingly, gain of function experiments with the heart development regulatory gene Tbx20 in the avian endocardial cushion resulted in decreased chondroitin sulfate proteoglycans and increased mesenchyme [64]. Our data demonstrating CHST3 expression in the murine endocardial cushion suggests that this may be a conserved mechanism to regulate mesenchyme production, although further studies are required to confirm this hypothesis. Though not predictive of CHDs, CHST3 was dysregulated in the AF of woman carrying fetuses with CHDs, suggesting a role in cardiac development.
An additional factor involved in adhesion/migration identified in our study is the protease ADAM15. Multiple individual ADAM isoform KO mice display cardiac defects, including Adam 9, 17, and 19 [65], [66], [67]. ADAM15 is expressed in blood vessels and endocardium, and although ADAM15 mice are viable studies using retinopathy and tumor angiogenesis models demonstrate an inducible vascular phenotype (failure of remodeling of the primary capillary plexus) [68]. In conjunction, our data indicates that decreased ADAM15 enzymatic activity in the hyperglycemic yolk sacs disrupts growth factor/cytokine signaling of a yet unknown pathway, resulting in an inability of the endothelial cells to initiate remodeling, consistent with the previous report on an inducible vascular phenotype. Our data from the atrioventricular canal assay extends the known functions of ADAM15 to include cardiac EMT. Speculating on the substrates involved in its effects on differentiation to mesenchymal cells, ADAM15 proteolytic activity on cadherins is one plausible target [69]. Further, the strong immunofluorescent staining of ADAM15 in the cellular projections of cardiac mesenchymal cells suggests ADAM15 may be necessary for cell interaction with the matrix potentially via integrin binding. These findings warrant future studies to determine if ADAM15 is predicative of CHDs. Interestingly, ADAM12 is a known biomarker for Down syndrome, demonstrating the applicability of this family to biochemical assays for diagnosis [70]. The novel finding of a functional role for ADAM15 in the embryonic heart combined with its strong expression in the cardiovascular system make it an appealing target for future studies that isolate its downstream targets in the heart.
In addition to proteins involved in differentiation and adhesion/migration, several proteins involved in the IGF/insulin pathway were identified in our study. Early in embryogenesis the embryo does not produce insulin nor does maternal insulin cross the placental barrier. However, embryos express high levels of IGF receptors and early growth is mediated via IGF signaling through Akt. Factors that regulate IGF receptor engagement and its downstream signaling cascades were identified both on the extracellular face, including the proteases Pcsk1 and ST14, and the intracellular face, including ZFP106 (or son of insulin receptor mutant) and HES6. Pcsk1 is a secreted protease that cleaves precursors including neuropeptides, renin, somatostatin, and insulin/IGF [71]. Additionally substrates include adhesion molecules, proteases and receptors. Pcsk1 KO mice are viable, but growth retarded. Two strains of proprotein convertase KO mice, Furin and PACE4, exhibit defects in the vasculature and heart, while the other 8 proprotein convertase KO mice exhibit defects in hormone production, fertility, and lipid metabolism [71]. Our data demonstrating decreased Pcsk1 levels in the yolk sac after hyperglycemia insult suggests that Pcsk1 KO mice may have an unappreciated inducible vascular phenotype. We also demonstrated that Pcsk1 is predictive for CHDs, expressed in the embryonic myocardium, and increased in the AF of fetuses with CHDs. Pcsk1 downregulation in the yolk sac and upregulation in AF may reflect differences in developmental age, cell differentiation processes or clearance/accumulation of proteins in the AF versus tissue. Although proprotein convertases have been linked to Alzheimer's disease, tumorigenesis, and infections, they have not been previously reported to have a role in CHDs. Further given that gene knock out experiments have determined that individual PCs do not seem to be redundant in embryonic development, understanding the role of Pcsk1 in metabolism, growth, and development of cardiovascular system is an intriguing avenue of future investigation.
Another molecule in the IGF signaling pathway, ST14, a transmembrane serine protease, has been implicated in metastatic cancer, which utilizes a molecular program reminiscent of embryonic EMT [72]. Substrates for ST14 include hepatocyte growth factor, urokinase-type plasminogen activator, protease-activated receptor 2 (PAR-2), IGF binding protein-related protein-1, trask, laminin and fibronectin [73], [74]. Recently, VEGFR-2 was reported to be a putative substrate of ST14 in HUVECs, resulting in ectodomain cleavage and inactivation of cell signaling [75]. Here, we demonstrated for the first time that ST14 is present in the embryonic cardiovascular system and is predictive of CHDs. Given the known substrates of this protease, ST14 induction during vasculopathy and in AF of fetuses with CHDs may affect differentiation, adhesion/migration and growth.
Cell movements, growth and differentiation depend on environmental homeostasis; of particular importance during development is redox balance. According to current knowledge, SVCT transports ascorbate into cells and thereby participates in metabolic processes, matrix synthesis and antioxidant defense [76]. Interestingly, dehydroascorbate utilizes glucose transporters to enter the cell prior to conversion to ascorbate, and during a hyperglycemic episode glucose competitively inhibits the transport of dehydroascorbate, potentially compromising antioxidant defense and leading to reactive oxygen species induced damage [77], [78]. Consistent with the theory of ROS induced vasculopathy; our data demonstrating upregulation of SVCT may signify that the embryo is “at risk” and employing a defense to combat oxidative stress. Although SVCT was not predictive of CHDs, the fetal cardiovascular system is sensitive to oxidative stress and benefits from antioxidant supplements, such as folic acid [13], [79], [80]. Therefore understanding the upstream regulation, downstream targets, and/or activity of SVCT may shed light onto novel antioxidant therapies for fetuses “at risk”.
Molecular-based diagnostic tests that detect fetuses “at risk” provide clinicians with information to guide clinical interventions aimed at improving outcome. First-trimester biochemical screening is becoming a powerful tool, allowing clinicians to efficiently triage patients [81]. For example, increased pregnancy-associated plasma protein A (PAPP-A) and decreased free β-human chorionic gonatrophin (β-hCG) in maternal serum indicate increased risk of trisomy 21, while reduced PAPP-A in maternal serum is associated with an increased risk of trisomy 18 [81]. An obvious advantage of serum screening is that it is non-invasive and serum screening may be performed earlier than amniocentesis. In the present study, we utilized AF rather than maternal serum because of its low complexity and similarity to the fetus due to its origin. Cells in AF are fetal rather than maternal. Future studies involving early maternal serum samples are required to determine the merit of adding our biomarkers to existing screening panels. To this end, using an in vivo streptozocin-induced diabetic model in mice, we detected differences in specific Laminin chains, ADAM15 ectodomain, and MMP2 activity in maternal sera from mice carrying a high proportion of fetuses with heart defects compared to those with a high proportion of fetuses with normal hearts (unpublished data). These data suggest that protein fragments are released into the maternal circulation in sufficient quantities that allow detection early in pregnancy (E10.5 which is approximately equivalent to human developmental week 6). Experiments are currently underway to evaluate the levels of WNT16, ST14, and Pcsk1 in maternal sera from murine models and humans.
The creation of new biochemical diagnostic tools to assay proteins such as Pcsk1, ST14, and WNT16 may give insights into the well-being of the fetus by sensing an inability to combat an environmental insult, disruption in growth, or damage due to reactive oxygen species. Affordable, rapid, and accurate molecular-based diagnosis of potential problems in fetal cardiovascular development early in pregnancy will provide clinicians with information to guide clinical interventions and pave the way toward eradicating one of the major global causes of infant mortality in the world. In the future, dietary supplementation or molecular therapies based on Pcsk1, ST14 and WNT16 or their targets may ameliorate the deficiencies that ultimately lead to the formation of a CHD in the first trimester. Therefore isolating the specific substrate targets of ST14 and Pcsk1 that disrupt cardiac development and investigating the transcriptional or post-transcriptional regulatory mechanisms that lead to overexpression of ST14 and Pcsk1 will aid in deciphering the role of these proteins in the etiology of CHDs. Further, continued mining of the proteomic dataset coupled with analysis of novel factors in physiologically relevant models will continue to yield fruitful avenues for investigation into the biology of cardiovascular development and pathogenic mechanisms of CHDs in the hope of advancing prevention, diagnosis and treatment.
Supporting Information
(2.75 MB XLS)
Acknowledgments
We thank Michael Michaud (Yale) for mouse colony management, Christian Grote-Westrick (Ruhr University Bochum) for antibody optimization, and Ravinder Nath (Yale) for stimulating discussions and thoughtful comments on the manuscript.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This investigation was supported by NIHR37HL28373 to JAM, HG02357 to MS and 5 T32 GM07499 to AKN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hoffman JI. Incidence of congenital heart disease: II. Prenatal incidence. Pediatr Cardiol. 1995;16:155–165. doi: 10.1007/BF00794186. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 3.Wren C, Richmond S, Donaldson L. Temporal variability in birth prevalence of cardiovascular malformations. Heart. 2000;83:414–419. doi: 10.1136/heart.83.4.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335. doi: 10.1016/S0074-7696(05)43005-3. [DOI] [PubMed] [Google Scholar]
- 5.de la Cruz MV, Sanchez Gomez C, Arteaga MM, Arguello C. Experimental study of the development of the truncus and the conus in the chick embryo. J Anat. 1977;123:661–686. [PMC free article] [PubMed] [Google Scholar]
- 6.De la Cruz MV, Gimenez-Ribotta M, Saravalli O, Cayre R. The contribution of the inferior endocardial cushion of the atrioventricular canal to cardiac septation and to the development of the atrioventricular valves: study in the chick embryo. Am J Anat. 1983;166:63–72. doi: 10.1002/aja.1001660105. [DOI] [PubMed] [Google Scholar]
- 7.de Lange FJ, Moorman AF, Anderson RH, Manner J, Soufan AT, et al. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 8.Lamers WH, Viragh S, Wessels A, Moorman AF, Anderson RH. Formation of the tricuspid valve in the human heart. Circulation. 1995;91:111–121. doi: 10.1161/01.cir.91.1.111. [DOI] [PubMed] [Google Scholar]
- 9.McBride RE, Moore GW, Hutchins GM. Development of the outflow tract and closure of the interventricular septum in the normal human heart. Am J Anat. 1981;160:309–331. doi: 10.1002/aja.1001600308. [DOI] [PubMed] [Google Scholar]
- 10.Small M, Copel JA. Indications for fetal echocardiography. Pediatr Cardiol. 2004;25:210–222. doi: 10.1007/s00246-003-0587-z. [DOI] [PubMed] [Google Scholar]
- 11.Carvalho JS, Moscoso G, Ville Y. First-trimester transabdominal fetal echocardiography. Lancet. 1998;351:1023–1027. doi: 10.1016/S0140-6736(97)08406-7. [DOI] [PubMed] [Google Scholar]
- 12.Smrcek JM, Berg C, Geipel A, Fimmers R, Diedrich K, et al. Early fetal echocardiography: heart biometry and visualization of cardiac structures between 10 and 15 weeks' gestation. J Ultrasound Med. 2006;25:173––182; quiz 183–175. doi: 10.7863/jum.2006.25.2.173. [DOI] [PubMed] [Google Scholar]
- 13.Wilson RD, Johnson JA, Wyatt P, Allen V, Gagnon A, et al. Pre-conceptional vitamin/folic acid supplementation 2007: the use of folic acid in combination with a multivitamin supplement for the prevention of neural tube defects and other congenital anomalies. J Obstet Gynaecol Can. 2007;29:1003–1026. doi: 10.1016/S1701-2163(16)32685-8. [DOI] [PubMed] [Google Scholar]
- 14.Eisenberg CA, Bader D. QCE-6: a clonal cell line with cardiac myogenic and endothelial cell potentials. Dev Biol. 1995;167:469–481. doi: 10.1006/dbio.1995.1042. [DOI] [PubMed] [Google Scholar]
- 15.Linask KK, Lash JW. Early heart development: dynamics of endocardial cell sorting suggests a common origin with cardiomyocytes. Dev Dyn. 1993;196:62–69. doi: 10.1002/aja.1001960108. [DOI] [PubMed] [Google Scholar]
- 16.Yutzey KE, Kirby ML. Wherefore heart thou? Embryonic origins of cardiogenic mesoderm. Dev Dyn. 2002;223:307–320. doi: 10.1002/dvdy.10068. [DOI] [PubMed] [Google Scholar]
- 17.Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell. 2006;11:723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Hatzopoulos AK, Folkman J, Vasile E, Eiselen GK, Rosenberg RD. Isolation and characterization of endothelial progenitor cells from mouse embryos. Development. 1998;125:1457–1468. doi: 10.1242/dev.125.8.1457. [DOI] [PubMed] [Google Scholar]
- 19.Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118:1255–1266. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- 20.Mills JL, Baker L, Goldman AS. Malformations in infants of diabetic mothers occur before the seventh gestational week. Implications for treatment. Diabetes. 1979;28:292–293. doi: 10.2337/diab.28.4.292. [DOI] [PubMed] [Google Scholar]
- 21.Eriksson UJ, Borg LA. Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia. 1991;34:325–331. doi: 10.1007/BF00405004. [DOI] [PubMed] [Google Scholar]
- 22.Akazawa S. Diabetic embryopathy: studies using a rat embryo culture system and an animal model. Congenit Anom (Kyoto) 2005;45:73–79. doi: 10.1111/j.1741-4520.2005.00070.x. [DOI] [PubMed] [Google Scholar]
- 23.Enciso JM, Gratzinger D, Camenisch TD, Canosa S, Pinter E, et al. Elevated glucose inhibits VEGF-A-mediated endocardial cushion formation: modulation by PECAM-1 and MMP-2. J Cell Biol. 2003;160:605–615. doi: 10.1083/jcb.200209014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katajamaa M, Oresic M. Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics. 2005;6:179. doi: 10.1186/1471-2105-6-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katajamaa M, Miettinen J, Oresic M. MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 2006;22:634–636. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- 26.Loffredo CA, Wilson PD, Ferencz C. Maternal diabetes: an independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology. 2001;64:98–106. doi: 10.1002/tera.1051. [DOI] [PubMed] [Google Scholar]
- 27.Wren C, Birrell G, Hawthorne G. Cardiovascular malformations in infants of diabetic mothers. Heart. 2003;89:1217–1220. doi: 10.1136/heart.89.10.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen GL, Norgard B, Puho E, Rothman KJ, Sorensen HT, et al. Risk of specific congenital abnormalities in offspring of women with diabetes. Diabet Med. 2005;22:693–696. doi: 10.1111/j.1464-5491.2005.01477.x. [DOI] [PubMed] [Google Scholar]
- 29.Rudge MV, Calderon IM, Ramos MD, Abbade JF, Rugolo LM. Perinatal outcome of pregnancies complicated by diabetes and by maternal daily hyperglycemia not related to diabetes. A retrospective 10-year analysis. Gynecol Obstet Invest. 2000;50:108–112. doi: 10.1159/000010293. [DOI] [PubMed] [Google Scholar]
- 30.Hunter ES, 3rd, Sadler TW. The role of the visceral yolk sac in hyperglycemia-induced embryopathies in mouse embryos in vitro. Teratology. 1992;45:195–203. doi: 10.1002/tera.1420450213. [DOI] [PubMed] [Google Scholar]
- 31.Pinter E, Reece EA, Leranth CZ, Sanyal MK, Hobbins JC, et al. Yolk sac failure in embryopathy due to hyperglycemia: ultrastructural analysis of yolk sac differentiation associated with embryopathy in rat conceptuses under hyperglycemic conditions. Teratology. 1986;33:73–84. doi: 10.1002/tera.1420330110. [DOI] [PubMed] [Google Scholar]
- 32.Pinter E, Mahooti S, Wang Y, Imhof BA, Madri JA. Hyperglycemia-induced vasculopathy in the murine vitelline vasculature: correlation with PECAM-1/CD31 tyrosine phosphorylation state. Am J Pathol. 1999;154:1367–1379. doi: 10.1016/S0002-9440(10)65391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.New DA. The culture of postimplantation embryos. Hum Reprod. 1991;6:58–63. doi: 10.1093/oxfordjournals.humrep.a137259. [DOI] [PubMed] [Google Scholar]
- 34.Sadler TW, Hunter ES, 3rd, Balkan W, Horton WE., Jr Effects of maternal diabetes on embryogenesis. Am J Perinatol. 1988;5:319–326. doi: 10.1055/s-2007-999717. [DOI] [PubMed] [Google Scholar]
- 35.Joshua IG, Zhang Q, Falcone JC, Bratcher AP, Rodriguez WE, et al. Mechanisms of endothelial dysfunction with development of type 1 diabetes mellitus: role of insulin and C-peptide. J Cell Biochem. 2005;96:1149–1156. doi: 10.1002/jcb.20620. [DOI] [PubMed] [Google Scholar]
- 36.Nath AK, Enciso J, Kuniyasu M, Hao XY, Madri JA, et al. Nitric oxide modulates murine yolk sac vasculogenesis and rescues glucose induced vasculopathy. Development. 2004;131:2485–2496. doi: 10.1242/dev.01131. [DOI] [PubMed] [Google Scholar]
- 37.Pinter E, Haigh J, Nagy A, Madri JA. Hyperglycemia-induced vasculopathy in the murine conceptus is mediated via reductions of VEGF-A expression and VEGF receptor activation. Am J Pathol. 2001;158:1199–1206. doi: 10.1016/S0002-9440(10)64069-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- 39.Pappin DJ, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 40.Consortium GO. The Gene Ontology project in 2008. Nucleic Acids Res. 2008;36:D440–444. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hallmann R, Horn N, Selg M, Wendler O, Pausch F, et al. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev. 2005;85:979–1000. doi: 10.1152/physrev.00014.2004. [DOI] [PubMed] [Google Scholar]
- 42.Masmiquel L, Segura RM, Mateo C, Calatayud M, Marti R, et al. Serum laminin as a marker of diabetic retinopathy development: a 4-year follow-up study. Am J Ophthalmol. 2000;129:347–352. doi: 10.1016/s0002-9394(99)00361-x. [DOI] [PubMed] [Google Scholar]
- 43.Galang CK, Muller WJ, Foos G, Oshima RG, Hauser CA. Changes in the expression of many Ets family transcription factors and of potential target genes in normal mammary tissue and tumors. J Biol Chem. 2004;279:11281–11292. doi: 10.1074/jbc.M311887200. [DOI] [PubMed] [Google Scholar]
- 44.Chrast R, Scott HS, Madani R, Huber L, Wolfer DP, et al. Mice trisomic for a bacterial artificial chromosome with the single-minded 2 gene (Sim2) show phenotypes similar to some of those present in the partial trisomy 16 mouse models of Down syndrome. Hum Mol Genet. 2000;9:1853–1864. doi: 10.1093/hmg/9.12.1853. [DOI] [PubMed] [Google Scholar]
- 45.Cheyssac C, Lecoeur C, Dechaume A, Bibi A, Charpentier G, et al. Analysis of common PTPN1 gene variants in type 2 diabetes, obesity and associated phenotypes in the French population. BMC Med Genet. 2006;7:44. doi: 10.1186/1471-2350-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 47.Spencer-Jones NJ, Wang X, Snieder H, Spector TD, Carter ND, et al. Protein tyrosine phosphatase-1B gene PTPN1: selection of tagging single nucleotide polymorphisms and association with body fat, insulin sensitivity, and the metabolic syndrome in a normal female population. Diabetes. 2005;54:3296–3304. doi: 10.2337/diabetes.54.11.3296. [DOI] [PubMed] [Google Scholar]
- 48.Finney GL, Blackler AR, Hoopmann MR, Canterbury JD, Wu CC, et al. Label-free comparative analysis of proteomics mixtures using chromatographic alignment of high-resolution muLC-MS data. Anal Chem. 2008;80:961–971. doi: 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- 49.Cutillas PR, Vanhaesebroeck B. Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol Cell Proteomics. 2007;6:1560–1573. doi: 10.1074/mcp.M700037-MCP200. [DOI] [PubMed] [Google Scholar]
- 50.Bernanke DH, Markwald RR. Migratory behavior of cardiac cushion tissue cells in a collagen-lattice culture system. Dev Biol. 1982;91:235–245. doi: 10.1016/0012-1606(82)90030-6. [DOI] [PubMed] [Google Scholar]
- 51.Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Dev Biol. 1983;95:108–114. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- 52.Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, et al. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 53.Jung J, Mysliwiec MR, Lee Y. Roles of JUMONJI in mouse embryonic development. Dev Dyn. 2005;232:21–32. doi: 10.1002/dvdy.20204. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 55.Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, et al. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem. 2006;281:33487–33496. doi: 10.1074/jbc.M605536200. [DOI] [PubMed] [Google Scholar]
- 56.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, et al. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 57.Minami T, Miura M, Aird WC, Kodama T. Thrombin-induced autoinhibitory factor, Down syndrome critical region-1, attenuates NFAT-dependent vascular cell adhesion molecule-1 expression and inflammation in the endothelium. J Biol Chem. 2006;281:20503–20520. doi: 10.1074/jbc.M513112200. [DOI] [PubMed] [Google Scholar]
- 58.Person AD, Garriock RJ, Krieg PA, Runyan RB, Klewer SE. Frzb modulates Wnt-9a-mediated beta-catenin signaling during avian atrioventricular cardiac cushion development. Dev Biol. 2005;278:35–48. doi: 10.1016/j.ydbio.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 59.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, et al. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol. 2004;166:359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, et al. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003;425:633–637. doi: 10.1038/nature02028. [DOI] [PubMed] [Google Scholar]
- 61.Summerhurst K, Stark M, Sharpe J, Davidson D, Murphy P. 3D representation of Wnt and Frizzled gene expression patterns in the mouse embryo at embryonic day 11.5 (Ts19). Gene Expr Patterns. 2008;8:331–348. doi: 10.1016/j.gep.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X, Tu L, Murphy PG, Kadono T, Steeber DA, et al. CHST1 and CHST2 sulfotransferase expression by vascular endothelial cells regulates shear-resistant leukocyte rolling via L-selectin. J Leukoc Biol. 2001;69:565–574. [PubMed] [Google Scholar]
- 63.Flanagan TC, Black A, O'Brien M, Smith TJ, Pandit AS. Reference models for mitral valve tissue engineering based on valve cell phenotype and extracellular matrix analysis. Cells Tissues Organs. 2006;183:12–23. doi: 10.1159/000094902. [DOI] [PubMed] [Google Scholar]
- 64.Shelton EL, Yutzey KE. Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev Biol. 2007;302:376–388. doi: 10.1016/j.ydbio.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurohara K, Komatsu K, Kurisaki T, Masuda A, Irie N, et al. Essential roles of Meltrin beta (ADAM19) in heart development. Dev Biol. 2004;267:14–28. doi: 10.1016/j.ydbio.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 66.White JM. ADAMs: modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol. 2003;15:598–606. doi: 10.1016/j.ceb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 67.Kern CB, Twal WO, Mjaatvedt CH, Fairey SE, Toole BP, et al. Proteolytic cleavage of versican during cardiac cushion morphogenesis. Dev Dyn. 2006;235:2238–2247. doi: 10.1002/dvdy.20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, et al. Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol. 2003;23:5614–5624. doi: 10.1128/MCB.23.16.5614-5624.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Najy AJ, Day KC, Day ML. The ectodomain shedding of E-cadherin by ADAM15 supports ErbB receptor activation. J Biol Chem. 2008;283:18393–18401. doi: 10.1074/jbc.M801329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laigaard J, Christiansen M, Frohlich C, Pedersen BN, Ottesen B, et al. The level of ADAM12-S in maternal serum is an early first-trimester marker of fetal trisomy 18. Prenat Diagn. 2005;25:45–46. doi: 10.1002/pd.1029. [DOI] [PubMed] [Google Scholar]
- 71.Scamuffa N, Calvo F, Chretien M, Seidah NG, Khatib AM. Proprotein convertases: lessons from knockouts. Faseb J. 2006;20:1954–1963. doi: 10.1096/fj.05-5491rev. [DOI] [PubMed] [Google Scholar]
- 72.Satomi S, Yamasaki Y, Tsuzuki S, Hitomi Y, Iwanaga T, et al. A role for membrane-type serine protease (MT-SP1) in intestinal epithelial turnover. Biochem Biophys Res Commun. 2001;287:995–1002. doi: 10.1006/bbrc.2001.5686. [DOI] [PubMed] [Google Scholar]
- 73.Bhatt AS, Erdjument-Bromage H, Tempst P, Craik CS, Moasser MM. Adhesion signaling by a novel mitotic substrate of src kinases. Oncogene. 2005;24:5333–5343. doi: 10.1038/sj.onc.1208582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ahmed S, Jin X, Yagi M, Yasuda C, Sato Y, et al. Identification of membrane-bound serine proteinase matriptase as processing enzyme of insulin-like growth factor binding protein-related protein-1 (IGFBP-rP1/angiomodulin/mac25). Febs J. 2006;273:615–627. doi: 10.1111/j.1742-4658.2005.05094.x. [DOI] [PubMed] [Google Scholar]
- 75.Darragh MR, Bhatt AS, Craik CS. MT-SP1 proteolysis and regulation of cell-microenvironment interactions. Front Biosci. 2008;13:528–539. doi: 10.2741/2698. [DOI] [PubMed] [Google Scholar]
- 76.Takanaga H, Mackenzie B, Hediger MA. Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch. 2004;447:677–682. doi: 10.1007/s00424-003-1104-1. [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Jia RH, Qiu CJ, Ding G. Hyperglycemia inhibits the uptake of dehydroascorbate in tubular epithelial cell. Am J Nephrol. 2005;25:459–465. doi: 10.1159/000087853. [DOI] [PubMed] [Google Scholar]
- 78.Root-Bernstein R, Busik JV, Henry DN. Are diabetic neuropathy, retinopathy and nephropathy caused by hyperglycemic exclusion of dehydroascorbate uptake by glucose transporters? J Theor Biol. 2002;216:345–359. doi: 10.1006/jtbi.2002.2535. [DOI] [PubMed] [Google Scholar]
- 79.Moat SJ, Lang D, McDowell IF, Clarke ZL, Madhavan AK, et al. Folate, homocysteine, endothelial function and cardiovascular disease. J Nutr Biochem. 2004;15:64–79. doi: 10.1016/j.jnutbio.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 80.Moens AL, Vrints CJ, Claeys MJ, Timmermans JP, Champion HC, et al. Mechanisms and potential therapeutic targets for folic acid in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2008;294:H1971–1977. doi: 10.1152/ajpheart.91503.2007. [DOI] [PubMed] [Google Scholar]
- 81.Nyberg DA, Hyett J, Johnson JA, Souter V. First-trimester screening. Radiol Clin North Am. 2006;44:837–861. doi: 10.1016/j.rcl.2006.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(2.75 MB XLS)