

Abstract
MCM7 is a critical component of the DNA replication licensing complex that controls DNA replication in both yeast and Xenopus. Our previous studies have indicated that MCM7 is both amplified and overexpressed in metastatic prostate cancer. In this study, we found that MCM7 interacts with the androgen receptor (AR) with high affinity both in vitro and in vivo. We identified the AR-binding motif for MCM7, comprised of amino acids 221 to 248, and the MCM7-binding motif for the AR, comprised of amino acids 426 to 475. AR stimulation with high doses of the synthetic androgen R1881 led to a decrease in MCM7 binding to genomic DNA, a reduction of DNA synthesis, decreases in the number of cells progressing through S phase and cell proliferation, whereas low doses produced an increase in the DNA licensing activity of MCM7 and higher levels of cell proliferation. In addition, the MCM7/AR interaction down-regulated MCM7 expression. The gene transcription or repressor activity of AR is dependent on its interaction with MCM7 because either a mutant AR defective in its interaction with MCM7 or a MCM7 knockdown primarily eliminated AR effects on gene expression. Thus, this study reveals a novel mechanism by which AR and MCM7 facilitate each other’s function, suggesting that AR-independent activation of MCM7 may be a mechanism by which prostate cancers bypass therapeutically induced AR blockade.
Androgen receptor (AR) plays a critical role in the development of the prostate gland. It has long been known that growth of the prostate gland is dependent on androgens (testosterone and dihydroxytestosterone). Studies in the 1980s and 1990s demonstrated that castration of animals resulted in rapid atrophy of the prostate gland,1,2 whereas introduction of exogenous androgen restored most of the prostate gland size in castrated animals. In addition, prostate cancer does not occur in eunuchs. Many of the current hormonal therapies used to treat prostate cancer remove androgens or inhibit AR. It appears that AR activation is critical for both prostate gland development and for tumorigenesis in the prostate. There are some contradictory observations, however, related to AR function and growth of prostatic and other epithelia. In prostate cancer cell lines devoid of AR expression, restoration of AR expression resulted in suppression of cell growth.3,4 These findings, along with the observation that dihydroxytestosterone inhibits the growth of thyroid epithelial and adrenocortical cells,5,6 suggest that AR plays the role of tumor suppressor under certain physiological conditions.
MCM7 is a critical component of the DNA replication licensing complex.7 To ensure that DNA replication is initiated only once per cell division cycle, the synthesis of the MCM complex is temporally regulated. For example, MCM7 expression is shut down during S, G2, and early M phase to prevent re-initiation of DNA synthesis. Our previous study, along with others, indicated that MCM7 is amplified and overexpressed in prostate cancers that relapse.8,9 Continuous expression of MCM7 in the prostate cancer cell line Du145 resulted in a higher level of DNA synthesis, cell proliferation, and tumor invasiveness. A transgenic mouse model with a knockin MCM7 expression under the control of a keratin promoter generated a strain of mouse prone to the development of multiple malignancies.10 It appears that high level of MCM7 expression is one of the determining factors for abnormal cell growth and tumorigenesis. In this study, we determined that MCM7 interacts directly with AR. Excessive activation of AR, however, leads to decrease of MCM7 expression, inhibition of DNA synthesis, and growth arrest in both LNCaP and PC3 cells with forced expression of wild-type AR. Mutant AR that does not interact with MCM7 has no such effect. Surprisingly, the transcriptional activity of AR is dependent on its interaction with MCM7.
Materials and Methods
Constructions of pBD-MCM7 Fusion Proteins
A mutagenic primer set (5′-CAGCCA AGCTCAACATATGGCACTGAAGGACTACGCG-3′ and 5′-TTCATTTCAAGCAAAGT CGACGGAGTAAGTGCAGCATGGGAG-3′) was designed to create two restriction sites (NdeI and SalI) for the full length of MCM7, so that the polymerase chain reaction (PCR) product could be ligated into a pGBKT7 vector. PCR was performed on a cDNA template of donor prostate cDNA, and the PCR conditions were as follows: 94°C for 1 minute followed by 35 cycles of 94°C for 30 seconds, 68°C for 3 minutes, and a final 10-minute extension step at 68°C. The PCR product was restricted with NdeI and SalI, gel-purified, and ligated into a similarly restricted pGBKT7 vector. The fusion protein contained 719 amino acids from MCM7 and 219 amino acids from the bait domain. The construct was transformed into One Shot competent cells (Invitrogen, Carlsbad, CA). Plasmid DNA was extracted from selected transformed cells and digested with NdeI and SalI to detect the presence of the insert. The coding frame was confirmed by automated sequencing. The pBD-MCM7n and pBD-MCMc were similarly constructed except the primers used for pBD-MCM7n were 5′-CAGCCAAGCTCAACATATGGCACT GAAGGACTACGC G-3′/5′-ATTTCCCACAGGCACCTGATCACT-3′, and for pBD-MCMc 5′-CAGCCAGCTCAACATATGATCCCTCGTAGTATCACGGTG-3′/5′-TTCAT TTCAAGCAAAGTCGACGGAGTAAGTGCAGCATGGGAG-3′.
For construction of the glutathione S-transferase (GST)-MCM7n fusion protein, a mutagenic primer set (5′-TGGGATCCCACTGAAGGACTACGCGCTAGA-3′/5′-CGCTCG AGATTTCCCACAGGCACCTGATCACT-3′) was designed to create a BamHI and XhoI restriction site within the MCM7 coding region that encodes a 233-amino acid region of the 5′ terminal of MCM7. PCR was performed using these primers under the following conditions: 94°C for 1 minute followed by 35 cycles of 94°C for 30 seconds, 68°C for 3 minutes, and a final 10-minute extension step at 68°C. The PCR product was subsequently gel-purified and ligated into a pCR2.1 TA cloning vector. The DNA was transformed into Escherichia coli. DNA from the selected transformants was restricted with BamHI and XhoI, and ligated into a similarly restricted pGEX-5T vector in frame. A series of deletions, including 5′ or 3′ deletions of pGST-MCM7n, pGST-AR fragments, and pET-MCM7n were performed using the primer sets listed in Table 1 (also see Figure 1 for amino acid sequences in the constructs). The procedures for generating these mutants were similar to those described for pGST-MCM7n. The pGST-MCM7n, pGST-ARs, pET-MCM7n, and their mutants were transformed into E. coli BL21 cells for recombinant protein production.
Table 1.
Primers for Generating pGST-MCM7, pGST-AR, and pET-MCM7n Constructs
| Primer names | Sequence |
|---|---|
| MCM7n1a: | 5′-TGGGATCCCACTGAAGGACTACGCGCTAGAG-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| MCM7n2a: | 5′-GGGGATCCCCGAGTTGGTGGACTCAATTTGT-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| MCM7n3a: | 5′-AGGGATCCTGGACGTTTACATTGAGCATCGG-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| MCM7n4a: | 5′-CCGGATCCCTGCTGAACTCATGCGCAGATTT-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| MCM7n5a: | 5′-AAGGATCCGTGGAATCGTCACTCGTGTCTCT-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| MCM7n6a: | 5′-TTGGATCCCTCTGATCATGTGCCCAAGCCAG-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| MCM7n7a: | 5′-ATCTGCGGATCCGGGGCTCCAGATTCATC-3′ |
| 5′-CGCTCGAGATTTCCCACAGGCACCTGATCACT-3′ | |
| ARI | 5′-ATCGAAGGTCGTGGGATCCAAGTGCAGTTAGGGCTGGGAAGG-3′ |
| 5′-CCTTGAAAGGGGAATACTCGAGCAGTATCTTCAGTGCTCTTG-3′ | |
| ARII | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-CTTCCGGGCTCCTCGAGTCATCCCTGCTTCATAACATTTCCG-3′ | |
| ARIII | 5′-ATCGAAGGTCGTGGGATCCCTCTGGGAGCCCGGAAGCTGAAG-3′ |
| 5′-ACATCTGAAAGGGGGCTCGAGCTGGGGTGGGGAAATAGG-3′ | |
| ARII1b: | 5′-ATCGAAGGTCGTGGGATCCCTGGCGGCATGGTGAGCAGAGT-3′ |
| 5′-CTTCCGGGCTCCTCGAGTCATCCCTGCTTCATAACATTTCCG-3′ | |
| ARII2b: | 5′-TAATAGGATCCCGGGAGCTGTAGCCCCCTACGGCTAC-3′ |
| 5′-CTTCCGGGCTCCTCGAGTCATCCCTGCTTCATAACATTTCCG-3′ | |
| ARII3b: | 5′-AGTTGTATGGATCCCGTGTGGTGGTGGTGGGGGTGG-3′ |
| 5′-CTTCCGGGCTCCTCGAGTCATCCCTGCTTCATAACATTTCCG-3′ | |
| ARII4b: | 5′-GTTCTGGGATCCCCTCAGCCGCCGCTTCCTCATCC-3′ |
| 5′-CTTCCGGGCTCCTCGAGTCATCCCTGCTTCATAACATTTCCG-3′ | |
| ARII5b: | 5′-ATAACCCGCTGGATCCACGGCAGCGCCTGGGCGGCTG-3′ |
| 5′-CTTCCGGGCTCCTCGAGTCATCCCTGCTTCATAACATTTCCG-3′ | |
| ARII1a: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-CGGTCCGGCTCGAGCCAGTGGAAAGTTGTAGTAGTCGCGACT-3′ | |
| ARII2a: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-ATGAGGAAGCGGCGGCTCGAGGGTGACCCAGAAC-3′ | |
| ARII3a: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-TCACCATGCCTCGAGGGTACCACACATCAGGTGCGGTGAAG-3′ | |
| ARII4a: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-ACACCCAGAAGCTTCATCTCGAGAGATCAGGCAGGTCTTCT-3′ | |
| ARII3a1: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-ACCACCACCACTCGAGTCCATACAACTGGCCTTCTTCG-3′ | |
| ARII3a2: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-AATATATTCCTCGAGCCTCGCCGCCGCCGC-3′ | |
| ARII3a3: | 5′-ATCGAAGGTCGTGGGATCCCTGCTGAGTATTCCCCTTTCAAG-3′ |
| 5′-ATAAATTTTCTCGAGCCCCTGAGGGGG-3′ | |
| ET-MCM7n: | 5′-GGCACATATGGCACTGAAGGACTACGCGCT-3′ |
| 5′-ACGAGGGATCCTTCCCACAGGCACCTGATC-3′ | |
| MCM7 RT-PCR | 5′-TCAATTTGTGAGAATGCCAGGCGC-3′ |
| 5′-CACAGTTACCAACTTCCCCACAGA-3′ | |
| b-actin RT-PCR | 5′-TCAAGATCATTGCTCCTC CTGAGC-3′ |
| 5′-TGCTGTCACCTTCACCGTTCCAGT-3′ |
Figure 1.

MCM7 interaction with AR. A: Schematic diagram of pBD-MCM7, pBD-MCM7n, and pBD-MCM7c. B: Growth in SD-4 high stringency agar plate and α-galactosidase activity of co-transfection isolates of yeast harboring pAD-AR+pBD-MCM7, pAD-AR+pBD-MCM7n, pAD-AR+pBD-MCM7c, and pAD-T-antigen+pBD-p53 (positive control). C: MCM7 and AR co-localization in nuclei of LNCaP cells induced with or without R1881. These cells were stained with antibodies against MCM7 (mouse monoclonal and FITC-conjugated donkey anti-mouse antibodies) and AR (rabbit polyclonal and rhodamine-conjugated donkey anti-rabbit antibodies). D: Expression of AR and co-immunoprecipitation of MCM7 and AR. Immunoblot analysis of AR from LNCaP (lane 1), DU145 (lane 2), and PC3 (lane 3), PC3 clone 1 transfected with pCMVscript (lane 4), PC3 clone 11 transfected with pCMV-AR (lane 5), PC3 clone 5 transfected with pCMVscript (lane 6), PC3 clone 19 transfected with pCMV-AR (lane 7). Co-immunoprecipitation was performed on protein extracts from LNCaP cells induced with R1881 (lanes 8 to 13). AR (lane 8) or MCM7 (lane 12) was immunoprecipitated and blotted with antibodies against MCM7 (lanes 8 to 10) or AR (lanes 11 to 13). Lysate input (lanes 9 and 11) was used as positive control for blotting, whereas pre-immune serum (PreIm) precipitation (lanes 10 and 13) was used as negative control for precipitation. Co-immunoprecipitation of AR and MCM7 from PC3 cells transfected with pCMV-AR (clone 11) was similarly performed using MCM7 (lane 17) or AR (lane 20) antibodies for immunoprecipitation, and blotted with AR (lanes 14 to 17) or MCM7 (lanes 18 to 21) antibodies. MCM7/B denotes the primary immunocomplexes of MCM7 incubation with plain Sepharose instead of protein G-conjugated Sepharose.
For construction of pLenti-mcs-GCNT3-bla vector, a mutagenic primer set (5′-TTGTAATC GATATCATGGCCAAAACC-3′/5′-TTTCTGGAATTCATAAGATGTCCAAGG-3′) was designed to create an EcoRV and EcoR1 site in the GCNT3 promoter and to encompass the ARE element within the promoter. PCR was performed using these primers under the following conditions: 94°C for 1 minute followed by 35 cycles of 94°C for 30 seconds, 68°C for 3 minutes, and a final 10-minute extension step at 68°C. The PCR product was restricted with EcoRV and EcoR1, and ligated into similarly restricted pLenti-mcs-bla vector. pLenti-mcs-COX2-bla and pLenti-mcs-PSA-bla vectors were similarly constructed using primers set 5′-GACCCGTGGATATC ACATTAACTATT-3′/5′-TATAGGTAGAATTCGCTGTCTGAGGGCGT-3′ (EcoRV and EcoR1) for COX2 and 5′-TGATCCTCGTACGTTGGCCTCGGAAAG-3′/5′-ACATG GTGATATCTCTCCGGGTGCAGG-3′ (BsiW1 and EcoRV) for PSA.
Yeast Transformation and Library Screening
The yeast competent cell preparation was described previously.11 One hundred μl of freshly prepared competent AH109 cells were mixed with plasmid DNA (0.25 to 0.50 μg) plus 0.5 μg of DNA from prostate yeast two-hybrid cDNA library constructed in pACT2 in 0.5 ml of PEG/LiAc, incubated at 30°C for 30 minutes. After this initial incubation with plasmid DNA, the cell solution was combined with 20 μl of dimethyl sulfoxide and subjected to a 15-minute incubation at 42°C. The cells were pelleted, resuspended in 1 ml of YPD medium, and shaken at 30°C for 40 minutes. The transformed cells were then pelleted, resuspended in 0.5 ml of 0.9% NaCl, and plated onto the appropriate SD agar plate. The transformants were first plated on low- and medium-stringency plates of SD-Leu/-Trp and SD-Leu/-Trp/-His, respectively. The grown colonies were subjected to the colony-lift filter β-galatosidase assay as described previously11 and allowed to grow further in the high-stringency plate (SD-Ade/-His/-Leu/-Trp).
Validation of Protein Interactions in AH109
Plasmid DNA samples from positive clones were isolated from yeast, transformed into E. coli, and selected with ampicillin (100 μg/ml) to obtain genes interacting with the bait-domain fusion protein. The purified AD/library plasmid DNA was then co-transformed with pBD-MCM7 into AH109 yeast cells and grown in a SD-Ade/-His/-Leu/-Trp high-stringency medium. α-Galactosidase activity was assayed on cells grown in this medium.
Immunoprecipitation
Protein extracts of PC3 cells transformed with pCMV-AR or LNCaP cells were incubated with MCM7 (1:500) or AR (1:1000) antibodies for 16 hours, then with protein G Sepharose for 3 hours. The complex was washed five times with RIPA buffer, and the bound proteins were eluted with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. The bound AR or MCM7 was electrophoresed in 8% SDS-PAGE and immunoblotted with anti-AR antibodies or MCM7 antibodies (1:2000).
GST Fusion Protein Pull Down to Examine AR/MCM7 Binding
The cells were grown in 100 ml of LB medium supplemented with ampicillin (100 μg/ml) overnight and induced by IPTG (final concentration of 1 mmol/L) for 3 hours. The cells were then pelleted, resuspended in 1× phosphate-buffered saline (PBS), and sonicated for 2 minutes. The proteins were solubilized in 1% Triton X-100. The supernatant was collected after centrifugation at 15,000 × g for 5 minutes. The GST and GST-MCM7n fusion protein were purified through a glutathione Sepharose 4B column (Amersham Bioscience, Arlington Heights, IL). The LNCaP, PC3 transfected with pCMV-AR, or WI-38 cell protein extracts were precleared with the column for 15 minutes at 4°C. The flow-through was collected after spinning at 3000 × g for 1 minute. The precleared cell lysates were then incubated with GST fusion protein-packed glutathione Sepharose 4B at 4°C for 2 hours. The column was spun at 3000 × g at room temperature for 1 minute, and further washed twice with PBS. The proteins were eluted from the column with 40 μl of SDS-PAGE gel sample loading dye. SDS-PAGE and Western blot analyses were subsequently conducted.
Construction of AR-Expressing Cell Lines
An AR cDNA clone was generated from total RNA from normal donor prostate tissue by extended long PCR12 with primers specific for the 5′ and 3′ ends of AR. The 3.4-kb PCR product was ligated into a TA cloning vector (Invitrogen) and subsequently cloned into a pCMVscript vector (Clontech, Palo Alto, CA) with HindIII and XhoI (New England Biolab, Ipswich, MA). The final pCMV-AR construct was sequenced to confirm that no mutation had been introduced. This construct was transfected into PC-3 cells. Colonies containing pCMV-AR were selected with medium that included G418 (400 μg/ml).
For siRNA assays, a pool of siRNA specific for MCM7 (5′-UCCAGGAGA UGAAGAUGCAUU-3′/5′-UGCAUCUUCAUCUCCUGGA-3′) or scramble siRNA (5′-UAA UGUAUUGGAACGCAUAUU-3′/5′-UAUGCGUUCCAAUACAUUA-3′) was transfected into cultured cells using lipofectamine 2000 (Invitrogen). The detailed procedure follows the manufacturer’s manual.
Immunofluorescence Staining
LNCaP or PC3 cells that were transfected with pCMV-AR were cultured with 10 nmol/L R1881 on chamber slides for 24 hours. The slides were washed with PBS three times. The cells were fixed with 4% paraformaldehyde for 1 hour at room temperature as described previously.13 After washing the slides with PBS twice, the cells were blocked with 10% donkey serum with 0.4% Triton X-100. The cells were then incubated with mouse monoclonal antibody against MCM7 (1:500) and rabbit antisera against AR (1: 500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at room temperature for 1 hour. The slides were washed with PBS twice. Secondary antibodies from donkey against mouse (fluorescein conjugated, 1:2000) and against rabbit (rhodamine conjugated, 1:2000) were added and incubated at room temperature for 1 hour. The slides were then washed with PBS twice before addition of 4′-6-diamidino-2-phenylindole (DAPI). After additional washes with PBS, slides were mounted with immuno-mounting buffer. Immunofluorescence staining was examined using a confocal microscope (Fluoview 1000; Olympus, Tokyo, Japan).
MTT Assay
Three thousand LNCaP, PC3/AR, or PC3/ΔAR cells were seeded per well in a 96-well plate and incubated (37°C, 5% CO2) overnight. R1881 at various concentration (0, 0.01, 0.1, 1.0, and 10.0 nmol/L) was added for 24 hours or 96 hours. One hundred μl of 1.2 mol/L MTT/medium solution were added to each well and incubated at 37°C for 4 hours to allow the MTT to be metabolized. After removal of medium, cells were lysed with 200 μl of dimethyl sulfoxide and placed on a rotating table for 5 minutes to thoroughly mix the formazan into the solvent. The formazan concentration was quantified at 595 nm in a spectrophotometer.
Bromo-Uridine Labeling Analysis
To label LNCaP, PC3/AR, or PC3/ΔAR cells, 10 μl of BrdU solution (1 mmol/L BrdU in 1× PBS) was added directly to each ml of tissue culture media. The treated cells were then incubated for 3 hours at 37°C. Cells were then resuspended with 100 μl of BD Cytofix/Cytoperm buffer per sample (BD PharMingen, San Diego, CA) and incubated for 30 minutes at room temperature. The cells were then pelleted and washed with 1 ml of 1× BD Perm/Wash buffer. The cells were then incubated with Cytoperm Plus buffer for 10 minutes on ice. The permeation procedure was repeated twice. The cells were resuspended with 100 μl of diluted DNase (diluted to 300 μg/ml in DPBS) per tube, (ie, 30 μg of DNase to each tube), and incubated for 1 hour at 37°C. The cells were washed with 1× BD Perm/Wash buffer, and incubated with 50 μl of BD Perm/Wash buffer containing diluted fluorescent anti-BrdU and propidium iodide for 20 minutes at room temperature. The incubation was washed with 1 ml of 1× BD Perm/Wash buffer. The results of the stains were analyzed in a LSC-II flow cytometer.
β-Lactamase Assay
LNCaP cells were transfected with siMCM7 and scramble siRNA for 6 hours using Lipofectamine 2000 (Invitrogen). R1881 was added into transfected cells in a final concentration of 10 nmol/L for 48 hours. The growth medium was removed, and these cells were washed once with Hanks’ balanced salt solution. These cells were treated with 1 μmol/L CCF2-AM for 1 hour at room temperature in the dark. This treatment was followed by washing cells with calcium- and magnesium-free Hanks’ balanced salt solution. The cells were then pelleted and resuspended in sorting buffer at a density of 3 to 5 × 106 cells/ml before being analyzed in a LSC-II flow cytometer.
Chromatin Association Assay
LNCaP, PC3+AR, or PC3+ΔAR cells were cultured to 75% confluence, synchronized in serum-free medium for 24 hours, and then treated with R1881 for 24 or 48 hours. These cells were washed with PBS and trypsinized. These cells were resuspended in 1 ml of buffer A (110 mmol/L KC2H3O2, 15 mmol/L NaC2H3O2, 2 mmol/L MgC2H3O2, 0.5 mmol/L EGTA, 20 mmol/L HEPES, pH 7.3). The cell suspension was treated to the final concentration of 2 mmol/L dithiothreitol and 50 μg/ml digitonin. The cells were incubated at 4°C for 10 minutes in a rotator. Nuclei were pelleted by centrifugation at 1500 × g for 10 minutes. They were resuspended in hypotonic buffer (buffer B: 1 mmol/L HEPES, pH 7.5, 0.5 mmol/L EDTA supplemented with 0.5% Nonidet P-40). The nuclear suspensions were then incubated at 4°C for 15 minutes in a rotator, laid on top of a 10-ml sucrose cushion (100 mmol/L sucrose, 0.5 mmol/L Tris-HCl, pH 8.5), and centrifuged at 3500 × g for 15 minutes at 4°C. The chromatin pellets were suspended in 0.25 mmol/L EDTA, pH 8.0, and sonicated 10 seconds for three times each sample. The chromatin suspensions were centrifuged twice at high speed for 10 minutes at 4°C, and the supernatants were analyzed in SDS-PAGE.
Results
To investigate what proteins regulate the function of MCM7 and how such interaction results in enhanced invasion in prostate cancer cells, we performed a yeast two-hybrid screen using GAL4 DNA binding domain-MCM7 fusion proteins, using MATCHMAKER system 3 from Clontech. Three BD-MCM7s were constructed (Figure 1A). All were demonstrated with proper expression in the yeast (data not shown). Using pBD-MCM7, we identified 24 positive colonies after three rounds of metabolic screening of a prostate yeast two-hybrid cDNA library. These colonies were subsequently isolated. After several restriction enzyme digestions, several redundant clones were eliminated. Three unique clones were identified and sequenced. One of these clones contained a cDNA encoding AR.
To validate the yeast two-hybrid screening results, pAD-AR and pBD-MCM7 were co-transfected into yeast AH109 cells, grown in high stringency medium, and tested for α-galactosidase activity. Both pBD-MCM7 (full length) and pBD-MCM7n (N-terminus) showed positive galactosidase activity, whereas the C-terminus of MCM7 was negative, suggesting that the AR binding activity is mediated by a region located in the N-terminus of MCM7 (Figure 1B). Among prostate cancer cell lines, AR is abundantly expressed in LNCaP cells, but is absent in PC3 and DU145 cells (Figure 1D, left). To verify the interaction, an in vivo MCM7-AR binding analysis was performed in protein extracts of LNCaP cells. As shown in Figure 1D, co-immunoprecipitation of MCM7 and AR was readily apparent. Similar results were obtained in PC3 cells transfected with pCMV-AR (Figure 1D). To visualize whether MCM7 and AR co-localize in nucleus, double-immunofluorescence staining using antibodies against MCM7 and AR were performed in LNCaP cells and stimulated with 1 nmol/L R1881 (a synthetic testosterone). As demonstrated in Figure 1C, MCM7 and a significant amount of AR co-localized in the nucleus after the testosterone stimulation. Similar co-localization results were obtained with PC3 cells transfected with pCMV-AR (data not shown).
To validate the interaction between MCM7 N-terminus and AR in vitro, a fragment of 247 amino acids from the N-terminus of MCM7 was constructed into pGEX-5T to create a GST-MCM7n fusion protein. The results of the binding assays indicate that GST-MCM7n binds with AR in a cell-free system (Figure 2, A and B). To examine whether such binding is cell-type-specific, we also performed binding assays using protein extracts from PC3 cells transfected with pCMV-AR and the primary human fibroblast cell line WI-38. Similar binding results were obtained (data not shown). To rule out potential bridging proteins between AR and MCM7 interaction, AR was truncated into the N-terminus, middle, and C-terminus. These DNA fragments were ligated into pGEX-5T vector. The binding analysis with the recombinant HisTag-MCM7 N-terminus (2 to 248 amino acids) shows that the middle segment of AR binds the MCM7 N-terminus directly (Figure 2, C and D). These results indicate that the interaction between MCM7 and AR is direct and does not require a bridge protein. A series of deletion mutants of GST-MCM7n were constructed to identify the motifs that were required to interact with AR. A stretch of 30 amino acids located in 221 to 248 of MCM7 was found crucial for MCM7 binding with AR because the fusion proteins with deletions in this sequence did not bind AR, whereas all proteins containing this sequence bound AR (Figure 2B). To define the AR’s motif that is required for AR interaction with MCM7, a series of GST-ARII deletion mutants were made (Figure 2C). These mutant fusion proteins were analyzed for their interaction with a HisTag-MCM7 N-terminus. As shown in Figure 2D, a stretch of 50 amino acids located between amino acids 426 to 475 of the AR was found essential for AR and MCM7 interaction. The function of this sequence, however, has not been previously defined.
Figure 2.

Identification of sequence motifs required for AR and MCM7 interaction. A: Schematic diagram of GST-MCM7n and its deletion mutants. B: Binding of AR with GST-MCM7n deletion mutants. The GST-MCM7n fusion protein or its mutants were purified from glutathione columns, and binding assays with AR were performed with protein extracts from LNCaP cells. After extensive washes, the bindings were visualized with SDS-PAGE and immunoblotted with antibodies against AR. Top: Immunoblot with anti-AR antibodies. Bottom: Coomassie staining of glutathione column-purified GST-MCM7n fusion proteins. The names of fusion proteins and MCM7 coding sequences are indicated. C: Schematic diagram of GST-AR fusion proteins. D:In vitro binding assays of bacterial expressed HisTag-MCM7n and GST-AR fusion proteins. The HisTag-MCM7n protein was purified through a histidine tag column. The purified HisTag-MCM7n protein was applied to GST-AR fusion protein to perform binding assays as described in B. The bindings of GST-ARs and HisTag-MCM7n were visualized through immunoblots with antibodies against MCM7. Top: Immunoblot with anti-MCM7 antibody. Bottom: Coomassie staining of glutathione column-purified GST-AR fusion proteins. Names of fusion proteins and AR coding sequences are indicated. These binding assays have been repeated seven times. Similar results were obtained.
To investigate whether the interaction between the AR and MCM7 stimulates the DNA replication licensing activity of MCM7, LNCaP cells were synchronized with serum starvation for 24 hours and stimulated with two different concentrations of R1881. As demonstrated in Figure 3A, the majority of MCM7 was associated with chromatins of LNCaP and PC3 cells whereas a significant amount of MCM7 was found nonchromatin binding. A low dosage (10 pmol/L) of R1881 increased nuclear localization of MCM7, whereas a high level (10 nmol/L) of testosterone did not. Similarly, low-dose R1881 enhanced MCM7 and chromatin association. High-dose R1881, however, increased the association of AR with chromatin but decreased the binding of MCM7 with the genome dramatically (Figure 3, B and C). To rule out whether this effect is only specific to LNCaP cells, the pCMV-AR construct was then transfected into PC3 cells that do not express AR protein. Stimulation of these cells with R1881 resulted in nuclear translocation of AR. Similar to our findings for LNCaP cells, there was stimulation of MCM7 chromatin association with low R1881 dosage but inhibition with high dosage (Figure 3B, middle section). To investigate whether these effects on MCM7 binding with genome DNA were dependent on AR/MCM7 interaction, we construct a pCMV-ΔAR in which 50 amino acids (amino acids 426 to 475) of MCM7 binding motif were deleted. We transfected PC3 cells with this vector and found that chromatin association of MCM7 was essentially unchanged with or without R1881 stimulation, even though the mutant AR was similarly translocated to the nucleus and bound with chromatin on R1881 stimulation.
Figure 3.

Interaction of AR and MCM7 leading to increase or decrease of MCM7-chromatin association. A: Immunofluorescence staining of MCM7 in PC3 cells transfected with pCMV-AR, or PC3 cells transfected with mutant AR, or LNCaP cells, untreated or treated with a low dosage or a high dosage of R1881. B: Chromatin association of MCM7 and AR induced by R1881. Chromatin extracts form LNCaP, PC3+AR, and PC3+ΔAR cells treated with indicated dosage of R1881 for 0.5, 24, and 48 hours. The chromatin associations of AR or MCM7 or nonchromatin MCM7 were visualized on immunoblots with antibodies against AR or MCM7. Antibodies against histone 3 were used as normalization controls. C: Densitometry analysis of the ratios of chromatin/nonchromatin MCM7 of B.
To investigate whether the MCM7/AR interaction also has an impact on the recruitment of other licensing factors to chromatin, pCMV-AR-transfected PC3 was induced with or without 0.01 or 10 nmol/L R1881. MCM7, MCM6, Cdt1, and geminin were subsequently examined for their association with chromatin 48 hours after the stimulation. In contrast to MCM7 chromatin association, MCM6, Cdt1, and geminin showed a dose-dependent increase of association with chromatin (Figure 4). However, the AR-stimulated chromatin association of MCM6, Cdt1, and geminin virtually disappeared when AR was mutated to prevent interaction with MCM7. These results indicate a critical role of MCM7/AR interaction in facilitating AR-induced DNA replication licensing factor recruitment.
Figure 4.
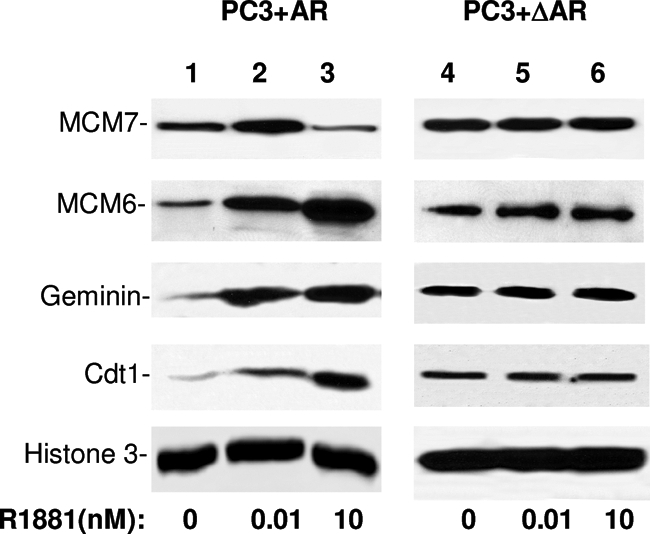
Interaction of AR and MCM7 leading to increase of chromatin association of MCM6, Cdt1, and geminin. PC3+AR or PC3+ΔAR cells were treated with R1881 at 0, 0.01, and 10 nmol/L. Chromatin from these cells was purified. The chromatin-associated proteins were analyzed in immunoblots using antibodies specific for MCM7, MCM6, geminin, and Cdt1. Antibodies against histone 3 were used as normalization controls.
Subsequently, we performed bromo-uridine labeling analysis to investigate whether MCM7 genome association correlates with DNA replication in these cells. As shown in Figure 5A, 10 pmol/L of R1881 induced a mild increase of DNA synthesis in LNCaP cells and PC3 cells that were transfected with pCMV-AR, but higher dosages of R1881 produced dosage-dependent inhibition of incorporation of bromo-uridine in both cell types. In contrast, cells with mutant AR that lacks the MCM7 binding motif did not respond to R1881 stimulation. Increase of DNA synthesis and cell entry into S phase were observed in LNCaP cells within 48 hours of low-dose androgen stimulation (Figure 5B), but 10 nmol/L R1881 induced growth arrest as indicated by a 10-fold decrease of cells in S phase with concomitant increase of cells in G0/G1 phase (>97%). Wild type AR in PC3 cells increased the number of cells in S phase in low dosage of R1881, but decreased in high dosage, albeit less dramatically than found in LNCaP cells. On the other hand, PC3 cells containing mutant AR that does not bind MCM7 had no discernable changes in cell distribution in these cell cycle phases. To detect cell proliferation, MTT analysis was performed. The results indicate that up to a 20% increase in LNCaP cell proliferation was observed in low dosage R1881-treated cells at 4 days but up to a 50% decrease of cell proliferation after 4 days of high-dose R1881 (Figure 6). Similar findings were identified for PC3 cells expressing wild type AR. PC3 cells expressing mutant AR, however, appeared unaffected by R1881 stimulation. These results indicated that the function of AR and MCM7 interaction is bidirectional: Inhibition of cell proliferation at high levels of stimulation but induction of proliferation at low levels of AR stimulation. A mutation of AR that abrogates AR/MCM7 interaction eliminated the AR’s ability to regulate cell growth.
Figure 5.

Interaction of AR and MCM7 leading to increase or decrease of DNA synthesis. A: Incorporation of bromo-uridine into DNA of LNCaP cells, or PC3 cells transfected with pCMV-AR, or PC3 cells transfected with pCMV-ΔAR. B: Induction of cell proliferation or cell growth arrest by R1881 in LNCaP, PC3+AR, and PC3+ΔAR cells. Cells were treated with indicated amounts of R1881 and pulsed labeled with bromo-uridine for 24 hours. The cells were subsequently stained with FITC-conjugated antibodies against bromo-uridine and propidium iodide. FACS analyses were performed to determine the cell cycle distribution of these cells. Standard deviations are indicated.
Figure 6.

AR and MCM7 interaction regulating cell proliferation. MTT assays were performed to determine cell proliferation of LNCaP, PC3, PC3+AR, and PC3+ΔAR cells when they were treated with 0, 0.01, 1, and 10 nmol/L R1881 for 24 hours (top) and 96 hours (bottom).
To investigate the mechanism of the bidirectional effect of AR stimulation on cell growth, immunoblotting on total protein extract using antibodies against MCM7 was performed. Time- and dosage-dependent decrease of MCM7 was found in PC3 cells transfected with wild type AR, with a critical decrease of MCM7 starting at 0.1 nmol/L R1881, whereas the MCM7 level at dosage 0.01 nmol/L was virtually unchanged (Figure 7A). In contrast, mutant AR that does not interact with MCM7 did not decrease MCM7 levels at either a low or high dosage of R1881. Pulse-chase analysis of S35-methionine labeled MCM7 in R1881 treated or untreated cells indicated that there was no significant difference in MCM7 half-life (data not shown). The mRNA level for MCM7, however, was significantly decreased in response to R1881 in LNCaP and PC3 transfected with wild-type AR but not PC3 cells with mutant AR (Figure 7B). These results suggest that suppression of MCM7 expression is an important contributing factor in AR-induced reduction of DNA synthesis and cell growth arrest with a high dosage of R1881.
Figure 7.

MCM7 and AR interaction essential for AR transcriptional activity. A: Immunoblot analysis of MCM7 protein expression after 24 or 48 hours of treatment of R1881 at different dosages. Left: PC3+AR cells. Right: PC3+ΔAR cells. B: Semiquantitative RT-PCR on MCM7 (top) and β-actin (bottom) mRNA in LNCaP, PC3+AR, or PC3+ΔAR cells treated with or without 10 nmol/L R1881. cDNA templates of 2 μg of total RNA were diluted at the indicated titrations. PCRs were subsequently performed using primers specific to MCM7 or β-actin. C: Immunoblot analysis of COX-2 and GCNT3 expression in PC3+AR or PC3+ΔAR cells treated with (T) or without (U) 10 nmol/L R1881 (18 hours) and with siRNA specific for MCM7 (M7) or scramble (Scr). D: Knocking down MCM7 reducing AR-dependent promoter activities of PSA, GCNT3, and COX2 promoters in LNCaP cells. LNCaP cells were transfected with pLenti-mcs-GCNT3-bla or pLenti-mcs-COX2-bla or pLenti-mcs-PSA-bla vectors, treated with the indicated siRNA, and induced with or without 10 nmol/L R1881. β-Lactamase activities were quantified as percent of scramble controls. Five experiments were performed for each treatment. SD is indicated. siM7, siRNA for MCM7; scr, scramble siRNA. β-Lactamase activities of these cells were quantified and measured as the fold change of untreated controls. Inset: immunoblots of MCM7 and β-actin on LNCaP cells; U, untreated; Scr, scramble siRNA; siM7, siRNA specific for MCM7.
To investigate whether binding of MCM7 functionally contributes to AR-mediated gene transcription activity, the R1881-stimulated AR-expressing PC3 cells were transfected with siRNAs specific for MCM7 or a scramble control. The cells were examined for induction of expression of androgen-responsive genes. As shown in Figure 7C, induction of AR induced a mild increase in expression of GCNT3 and COX-2 proteins. However, such induction was inhibited when cells were treated with siRNA specific for MCM7. Furthermore, an AR mutant that does not interact with MCM7 did not have GCNT3 and Cox-2 induction activity, supporting a role of MCM7 in AR-mediated transcription. To validate these findings, promoters of PSA, Cox2, and GCNT3 were ligated into plenti-mcs-bla vector (β-lactamase reporter vector) to analyze AR-responding transcription activity from these promoters. The results showed that MCM7 knocked-down in LNCaP cells dramatically reduced reporter gene activities driven by these three androgen-responding promoters (Figure 7D). These results indicate that MCM7 may act as a co-transcription factor for AR.
Discussion
AR plays a critical role in the development of the prostate gland. Elimination of dihydroxytestosterone aborts the development of the prostate gland.14 Mutation of AR in mesenchyme cells from urogenital sinus abrogates the formation of the prostate gland in mice.15 The presence of androgen is therefore critical for the differentiation and growth of prostate gland. Interaction between AR and MCM7 provides a direct link to cell growth control mediated by androgen. Low-dosage androgen enhances MCM7 chromatin binding, and thus increases DNA synthesis and cell proliferation. A high dosage of androgen suppresses the expression of MCM7. This leads to a decrease in MCM7 licensing and inhibition of DNA replication and cell growth arrest. These findings provide new insight into the previous finding that high levels of testosterone inhibit cell growth,3 whereas low dosages have a pleiotrophic effect.16
MCM7 is a critical component of the DNA replication licensing complex in the yeast and Xenopus.17,18,19,20 Some studies suggest that MCM4, -6, and -7 complex contains DNA helicase activity.21,22 Previous studies link overexpression and amplification of MCM7 to several human malignancies.8,10,23 The DNA replication licensing complex is multimeric. In yeast, DNA replication licensing proteins include MCM2 to MCM7 and several replication origin binding proteins such as Cdc6 and Cdt1. This raises the question as to whether AR and MCM7 interaction requires the presence of other proteins. Our study, however, supports the hypothesis that MCM7 interacts with AR independently without the need of other co-binding factors or even ligand binding of AR. This is because in the binding assay, AR and MCM7 interact in a manner free of other eukaryotic proteins and free of AR ligand. To our knowledge, this is the first report indicating that AR interacts with a member of DNA replication complex. Our results provide a novel example in which a transcription factor interacts with a DNA replication complex and acts as a co-replication factor in mammalian cells. This finding also supports and explains a recent finding that AR functions similar to a licensing factor.24 The bidirectional effect of AR on MCM7 in LNCaP and PC3 cells with forced expression of AR is consistent with previous findings that a low dosage of androgen induces cell proliferation, whereas a high dosage induces cell growth arrest.3 It appears that at a high dosage of testosterone, the cell growth arrest results from both MCM7 expression suppression and its displacement from chromatin association, whereas at a low dosage, AR/MCM7 interaction results in enhancement of MCM7 DNA licensing. As a result, our findings provide new insight into testosterone control of cell proliferation or cell growth inhibition.
The dependence of AR transcription activity on MCM7 is an unexpected finding. Several lines of evidences support that MCM7 acts as a co-transcription factor for AR. First, down-regulation of MCM7 using siRNA specific for MCM7 dramatically suppressed the expression of AR-responsive genes. Second, a mutant of AR that does not interact with MCM7 failed to induce the expression of AR-responsive genes, indicating that expression of AR-responsive genes is dependent on AR/MCM7 interaction. Third, AR-responsive promoter-driven reporter gene assays showed a dramatic decrease in expression when MCM7 expression level was suppressed. The significance of the AR/MCM7 transcription complex is underlined by the feedback suppression of MCM7 expression that leads to growth suppression with a high dosage of testosterone. A potential interpretation of these biological effects of AR/MCM7 interaction is the presence of different thresholds for MCM7-dependent DNA synthesis and MCM7-dependent transcription activity for activated AR. It appears that AR-induced MCM7-dependent DNA replication has a lower quantitative threshold for the AR/MCM7 complex, whereas MCM7-dependent AR transcription has higher one. When the quantity of AR/MCM7 complex is low, it induces MCM complex licensing activity but does not enhance AR-mediated transcription or transcription suppression activity. However, if the AR/MCM7 complex reaches the critical threshold for transcription, the AR/MCM7 complex is displaced from replication origin and functions as a transcription complex to induce MCM7-dependent AR transcription or transcription suppression activity that suppresses the expression of MCM7 and abrogates the DNA replication. As a result, AR/MCM7 interaction is operated in a quantitative balance to control cell growth and differentiation.
Footnotes
Address reprint requests to Jian-Hua Luo, Department of Pathology, University of Pittsburgh, 3550 Terrace St., Scaife Hall S-760, Pittsburgh, PA 15090. E-mail: luoj@msx.upmc.edu.
Supported by the National Cancer Institute (grant RO1 CA098249 to J.H.L.), the development fund from the Department of Urology, and American Cancer Society (RSG-08-157-01-CNE to YPY).
References
- Furuya Y, Isaacs JT. Differential gene regulation during programmed death (apoptosis) versus proliferation of prostatic glandular cells induced by androgen manipulation. Endocrinology. 1993;133:2660–2666. doi: 10.1210/endo.133.6.8243289. [DOI] [PubMed] [Google Scholar]
- English HF, Kyprianou N, Isaacs JT. Relationship between DNA fragmentation and apoptosis in the programmed cell death in the rat prostate following castration. Prostate. 1989;15:233–250. doi: 10.1002/pros.2990150304. [DOI] [PubMed] [Google Scholar]
- Heisler LE, Evangelou A, Lew AM, Trachtenberg J, Elsholtz HP, Brown TJ. Androgen-dependent cell cycle arrest and apoptotic death in PC-3 prostatic cell cultures expressing a full-length human androgen receptor. Mol Cell Endocrinol. 1997;126:59–73. doi: 10.1016/s0303-7207(96)03970-6. [DOI] [PubMed] [Google Scholar]
- Litvinov IV, Antony L, Dalrymple SL, Becker R, Cheng L, Isaacs JT. PC3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006;66:1329–1338. doi: 10.1002/pros.20483. [DOI] [PubMed] [Google Scholar]
- Rossi R, Zatelli MC, Franceschetti P, Maestri I, Magri E, Aguiari G, Cavazzini P, degli Uberti EC, del Senno L. Inhibitory effect of dihydrotestosterone on human thyroid cell growth. J Endocrinol. 1996;151:185–194. doi: 10.1677/joe.0.1510185. [DOI] [PubMed] [Google Scholar]
- Rossi R, Zatelli MC, Valentini A, Cavazzini P, Fallo F, del Senno L, degli Uberti EC. Evidence for androgen receptor gene expression and growth inhibitory effect of dihydrotestosterone on human adrenocortical cells. J Endocrinol. 1998;159:373–380. doi: 10.1677/joe.0.1590373. [DOI] [PubMed] [Google Scholar]
- Blow JJ, Hodgson B. Replication licensing—defining the proliferative state? Trends Cell Biol. 2002;12:72–78. doi: 10.1016/s0962-8924(01)02203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, Michalopoulos G, Yu YP, Luo JH. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25:1090–1098. doi: 10.1038/sj.onc.1209134. [DOI] [PubMed] [Google Scholar]
- Levesque MH, El-Alfy M, Berger L, Labrie F, Labrie C. Evaluation of AIbZIP and Cdc47 as markers for human prostatic diseases. Urology. 2007;69:196–201. doi: 10.1016/j.urology.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Honeycutt KA, Chen Z, Koster MI, Miers M, Nuchtern J, Hicks J, Roop DR, Shohet JM. Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis. Oncogene. 2006;25:4027–4032. doi: 10.1038/sj.onc.1209435. [DOI] [PubMed] [Google Scholar]
- Yu YP, Luo JH. Myopodin-mediated suppression of prostate cancer cell migration involves interaction with zyxin. Cancer Res. 2006;66:7414–7419. doi: 10.1158/0008-5472.CAN-06-0227. [DOI] [PubMed] [Google Scholar]
- Jing L, Liu L, Yu YP, Dhir R, Acquafondada M, Landsittel D, Cieply K, Wells A, Luo JH. Expression of myopodin induces suppression of tumor growth and metastasis. Am J Pathol. 2004;164:1799–1806. doi: 10.1016/S0002-9440(10)63738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yu YP, Tseng GC, Wu C, Chen K, Rao UN, Nelson J, Michalopoulos GK, Luo JH. Analysis of integrin alpha7 mutations in prostate cancer, liver cancer, glioblastoma multiforme, and leiomyosarcoma. J Natl Cancer Inst. 2007;99:868–880. doi: 10.1093/jnci/djk199. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Wilson JD. Genetic and hormonal control of male sexual differentiation. J Cell Physiol. 1975;85:365–377. doi: 10.1002/jcp.1040850405. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Lung B. The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J Exp Zool. 1978;205:181–193. doi: 10.1002/jez.1402050203. [DOI] [PubMed] [Google Scholar]
- Lin MF, Meng TC, Rao PS, Chang C, Schonthal AH, Lin FF. Expression of human prostatic acid phosphatase correlates with androgen-stimulated cell proliferation in prostate cancer cell lines. J Biol Chem. 1998;273:5939–5947. doi: 10.1074/jbc.273.10.5939. [DOI] [PubMed] [Google Scholar]
- Kearsey SE, Maiorano D, Holmes EC, Todorov IT. The role of MCM proteins in the cell cycle control of genome duplication. Bioessays. 1996;18:183–190. doi: 10.1002/bies.950180305. [DOI] [PubMed] [Google Scholar]
- Chong JP, Thommes P, Blow JJ. The role of MCM/P1 proteins in the licensing of DNA replication. Trends Biochem Sci. 1996;21:102–106. [PubMed] [Google Scholar]
- Coxon A, Maundrell K, Kearsey SE. Fission yeast cdc21+ belongs to a family of proteins involved in an early step of chromosome replication. Nucleic Acids Res. 1992;20:5571–5577. doi: 10.1093/nar/20.21.5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton S, Whitbread L. Cell cycle-regulated nuclear import and export of Cdc47, a protein essential for initiation of DNA replication in budding yeast. Proc Natl Acad Sci USA. 1995;92:2514–2518. doi: 10.1073/pnas.92.7.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimi Y. A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J Biol Chem. 1997;272:24508–24513. doi: 10.1074/jbc.272.39.24508. [DOI] [PubMed] [Google Scholar]
- You Z, Komamura Y, Ishimi Y. Biochemical analysis of the intrinsic Mcm4-Mcm6-mcm7 DNA helicase activity. Mol Cell Biol. 1999;19:8003–8015. doi: 10.1128/mcb.19.12.8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brake T, Connor JP, Petereit DG, Lambert PF. Comparative analysis of cervical cancer in women and in a human papillomavirus-transgenic mouse model: identification of minichromosome maintenance protein 7 as an informative biomarker for human cervical cancer. Cancer Res. 2003;63:8173–8180. [PubMed] [Google Scholar]
- Litvinov IV, Vander Griend DJ, Antony L, Dalrymple S, De Marzo AM, Drake CG, Isaacs JT. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci USA. 2006;103:15085–15090. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]