

Abstract
Genistein, a widely consumed bioflavonoid with chemopreventative properties in adults, and etoposide, a commonly prescribed anticancer drug, are well-characterized topoisomerase II poisons. Although both compounds display similar potencies against human topoisomerase IIα and β in vitro and induce comparable levels of DNA cleavage complexes in cultured human cells, their cytotoxic and genotoxic effects differ significantly. As determined by assays that monitored viability or the phosphorylation of histone H2AX, etoposide was much more toxic in CEM cells than genistein. Further studies that characterized the simultaneous treatment of cells with genistein and etoposide indicate that the differential actions of the two compounds are not related to the effects of genistein on cellular processes outside of its activity against topoisomerase II. Rather, they appear to result from a longer persistence of cleavage complexes induced by etoposide as compared to genistein. Parallel in vitro studies with purified type II enzymes led to similar conclusions regarding cleavage complex persistence. Isoform-specific differences were observed in vitro and in cells treated with etoposide. To this point, the t1/2 of etoposide-induced DNA cleavage complexes formed with topoisomerase IIα in CEM cells was ∼5 times longer than those formed with topoisomerase IIβ. The cytotoxicity of etoposide following four treatment-recovery cycles was similar to that induced by continuous exposure to the drug over an equivalent time period. Taken together, these findings suggest that it may be possible to preferentially target topoisomerase IIα with etoposide by employing a schedule that utilizes pulsed drug treatment-recovery cycles.
Type II topoisomerases are enzymes that modulate the topological structure of DNA by generating transient double-stranded breaks in the backbone of the double helix (1-6). In order to maintain genomic integrity during this process, the enzymes form covalent bonds with the 5′-termini of the cleaved DNA (7-9).
Compounds that increase the concentration of covalent topoisomerase II-DNA complexes (i.e., cleavage complexes) are known as topoisomerase II poisons (3, 6, 10-13). These agents impact human health in a variety of ways. Some of the most successful anticancer drugs currently in clinical use, including etoposide and doxorubicin, are topoisomerase II poisons (3, 6, 10-13). However, 2–3% of patients treated with regimens that contain these drugs develop specific leukemias that involve translocations at chromosome band 11q23 (11, 13-17). Naturally occurring topoisomerase II-active compounds, such as genistein and other bioflavonoids, are widely consumed constituents of the human diet (18-22). While bioflavonoids are believed to be chemopreventative in adults, epidemiological studies link their consumption during pregnancy to the formation of infant leukemias (23-25). As seen with the anticancer drugs, many of these infant leukemias display 11q23 chromosomal rearrangements (17, 23-25).
The anticancer and leukemogenic effects of the above compounds are believed to result from their ability to poison topoisomerase II (6, 13, 17, 24-29). Humans express two isoforms of the enzyme, topoisomerase IIα and β (2-6). Although both appear to be targeted by anticancer and dietary topoisomerase II poisons, the relative contributions of the individual isoforms to cellular outcomes are not well-defined. However, recent studies imply that topoisomerase IIα may play an important role in the induction of cell death by these compounds (30, 31), while topoisomerase IIβ may play a disproportionate role in generating leukemogenic chromosomal translocations (30, 31).
Because of their importance to human health, many of the in vitro actions of etoposide and genistein against human topoisomerase IIα and β have been characterized (13, 24, 28, 32-35). In side-by-side comparisons, the two compounds display similar potencies against both enzyme isoforms and induce comparable levels of cleavage complexes in vitro and in cultured human cells (28). Although these findings predict that both agents would have similar physiological effects, the cytotoxic and genotoxic properties of etoposide and genistein have not been compared directly. Therefore, the effects of these drugs on cell viability and the generation of permanent double-stranded DNA breaks were evaluated.
Unexpectedly, etoposide was significantly more cytotoxic and genotoxic than genistein in human CEM cells. The differential actions of the two drugs were not related to the effects of genistein on cellular processes outside of its activity against topoisomerase II. Rather, they appear to be due to a longer persistence of cleavage complexes induced by etoposide as compared to genistein. Finally, differences in the persistence of etoposide-induced topoisomerase IIα- and β-DNA cleavage complexes suggest the potential for preferentially targeting the α isoform by employing a schedule that utilizes pulsed drug treatment-recovery cycles.
Experimental Procedures
Enzymes and Materials
Recombinant wild-type human topoisomerase IIα and β were expressed in Saccharomyces cerevisiae and purified as described previously (36-38). Negatively supercoiled pBR322 DNA was prepared from Escherichia coli using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Genistein was obtained from LKT Laboratories. Etoposide and the proteasome inhibitor MG132 (carbobenzoxyl-leucinyl-leucinyl-leucinal) were obtained from Sigma. All compounds were prepared as 20 mM stocks in 100% DMSO and stored at −20 °C.
Cell Culture
Human CEM leukemia cells were purchased from ATCC. Cells were grown under 5% CO2 at 37 °C in RPMI 1640 medium (Cellgro by Mediatech, Inc.), containing 10% heat-inactivated fetal bovine serum (Hyclone) and 2 mM glutamine (Cellgro by Mediatech, Inc.).
Cytotoxicity
Human CEM cells were plated in 96-well plates at 7.5 × 105 cells/mL and grown for 24 h prior to treatment. Cells were exposed to 0–200 μM genistein or etoposide for 8 h. After treatment, 10 μL of CCK-8 (Dojinodo) reagent was added to each well to measure cell viability. Plates were read in a VERSAmax tunable microplate reader at 450 nm. Viability was determined colorimetrically in comparison to cells treated in the absence of drug (set to 100%).
To examine the effects of genistein on etoposide-induced cytotoxicity, cells were incubated with 50 μM genistein starting 2 h prior to the further addition of 0–200 μM etoposide for 6 h. Following exposure, cell viability was assessed using the CCK-8 reagent.
In some experiments, a pulsed treatment schedule of etoposide in cultured human CEM cells was employed. Cultures were treated for 15 min with 200 μM etoposide followed by a 2 h recovery period in etoposide-free medium for 4 cycles. The CCK-8 proliferation assay described above was performed to monitor cell viability.
Induction of Histone H2AX Phosphorylation by Topoisomerase II Poisons
CEM cells were treated with 0–250 μM genistein or 50 μM etoposide for 1 h. Following treatment, CEM cells were harvested by centrifugation at 1500 rpm. Cells were resuspended in 50 mM Tris, pH 7.9, 150 mM NaCl, 0.5% (v/v) NP-40, and protease inhibitor cocktail (Roche), and sonicated. Protein concentrations were determined by the Bradford assay (39). Cell lysates were diluted 1:1 in Laemmili buffer and boiled for 15 min prior to loading into SDS-PAGE gels (Bio-Rad). Gels were subjected to electrophoresis for 1 h at 35 mA, 200 V in 25 mM Tris, 192 mM glycine, and 0.1% SDS (w/v), pH 8.3. Proteins were blotted overnight onto PDVF membranes (PerkinElmer) in 25 mM Tris, pH 8.3, 192 mM glycine at 30 V at 4 °C. Membranes were blocked with 5% (w/v) non-fat dried milk in 20 mM Tris, pH 7.6, 137 mM NaCl, 0.1% (v/v) Tween-20. To assess the levels of drug-induced permanent DNA breaks mediated by human type II topoisomerases, membranes were probed overnight at 4 °C with a monoclonal α-histone phospho-H2AX (Ser 139) antibody (Upstate) at a 1:1500 dilution. As a loading control, membranes were probed with a polyclonal antibody directed toward Cyclophilin A at a 1:2000 dilution. Membranes were incubated with a secondary antibody (α-rabbit or α-mouse) conjugated to horseradish peroxidase. The amount of protein was visualized using an ECL chemiluminescence kit (Amersham). Results were quantified using an Alpha Innotech digital imaging system.
To examine the effects of genistein on the ability of etoposide to induce permanent DNA damage (primarily double-stranded breaks), cells were incubated with 50 μM genistein starting 2 h prior to the further addition of 50 μM etoposide for 1 h. Following treatment, cells were processed as described above to measure levels of phosphorylated histone H2AX.
Neutral Comet Assay
The neutral comet assay was employed to detect double-stranded DNA breaks in cultured human CEM cells. Cells were seeded at 5x105 cells/mL for 24 h and were treated with 50 μM genistein or etoposide for 1 h. Following exposure, cells were harvested by centrifugation, resuspended in culture medium at 1x105 cells/mL, and mixed with LMAgarose (low-melting temperature agarose) at a 1:10 ratio of cells to LMAgarose. The mixture (75 μL) was added to comet slides and allowed to solidify in the dark for 10 min at 4 °C. Slides were incubated in cold lysis solution for 30 min at 4 °C and washed twice in TBE buffer (90 mM Tris, pH 7.9, 35 mM boric acid, 2.5 mM EDTA) for 5 min. Comet slides were subjected to electrophoresis in TBE buffer for 15 min at 20 V, gently rinsed with H2O, and immersed in 70% ethanol for 5 min. Samples were allowed to air dry and were stained with 50 μL of SYBR Green I. Cells were visualized using a green fluorescence filter on a Leica DM IRB inverted microscope equipped with a Nikon DXM1200C camera.
DNA Cleavage Mediated by Human Topoisomerase IIα and β in Cultured CEM Cells
The in vivo complex of enzyme (ICE) bioassay (as modified on the TopoGen, Inc. website) was utilized to determine the ability of genistein and etoposide to increase levels of topoisomerase II-DNA cleavage complexes in treated cells. Exponentially growing cultures were treated with 50 μM genistein or etoposide for 1 h. Cells (∼5x105) were harvested by centrifugation and lysed immediately with 3 mL of 1% sarcosyl. Following gentle homogenization with a dounce homogenizer, cell lysates were layered onto a 2 mL cushion of CsCl (1.5 g/mL) and centrifuged in a Beckman NVT90 rotor at 45000 rpm for 15 h at 20 °C. DNA pellets were isolated, resuspended in 5 mM Tris-HCl, pH 8.0, and 0.5 mM EDTA, normalized for the amount of DNA, and blotted onto nitrocellulose membranes using a Schleicher and Schuell slot blot apparatus at multiple DNA concentrations. Covalent complexes formed between human topoisomerase IIα or β and DNA were detected using polyclonal antibodies (Abcam) directed against either human topoisomerase IIα or β, respectively, at a 1:2000 dilution. Sub-saturating bands were used to quantify levels of topoisomerase II-DNA cleavage complexes.
ICE bioassays were performed to determine the persistence of drug-induced cleavage complexes mediated by human topoisomerase IIα or β in human CEM cells. Cells were treated with 50 μM genistein or etoposide for 15 min to 1 h. Following exposure, cells were transferred to drug-free medium for 0–2 h, and the loss of drug-induced covalent complexes formed between topoisomerase IIα or β and DNA was quantified.
To examine the role of the proteasome in the persistence of topoisomerase IIα- and β-DNA cleavage complexes induced by genistein and etoposide, CEM cells were incubated in the presence of 2 μM MG132 prior to and during exposure to 50 μM genistein or etoposide. Cells were treated under the conditions described above for monitoring the persistence of drug-induced cleavage complexes.
Persistence of Topoisomerase II-Mediated DNA Cleavage In Vitro
DNA cleavage reactions were performed using the procedure of Fortune and Osheroff (40). Assay mixtures contained 220 nM topoisomerase IIα or β and 5 nM negatively supercoiled pBR322 DNA in a total of 20 μL of cleavage buffer [10 mM Tris-HCl, pH 7.9, 5 mM MgCl2, 100 mM KCl, 0.1 mM EDTA, and 2.5% (v/v) glycerol] that contained 50 μM genistein or etoposide. After reactions reached DNA cleavage/ligation equilibria, mixtures were diluted 10–fold with cleavage buffer and were incubated up to 30 min. Enzyme-DNA cleavage complexes were trapped by adding 2 μL of 5% SDS and 1 μL of 375 mM EDTA, pH 8.0. Proteinase K was added (2 μL of a 0.8 mg/mL solution) and reaction mixtures were incubated for 30 min at 45 °C to digest topoisomerase II. Samples were precipitated with ethanol, resuspended in 20 μL of water, mixed with 2 μL of 60% sucrose in 10 mM Tris-HCl, pH 7.9, 0.5% bromophenol blue, and 0.5% xylene cyanol FF, heated for 2 min at 45 °C, and subjected to electrophoresis in 1% agarose gels in 40 mM Tris-acetate, pH 8.3, and 2 mM EDTA containing 0.5 μg/mL ethidium bromide. DNA cleavage was monitored by the conversion of negatively supercoiled plasmid DNA to linear molecules. DNA bands were visualized by ultraviolet light and quantified using an Alpha Innotech digital imaging system.
Results
Induction of Topoisomerase II-DNA Cleavage Complexes in Cultured Human CEM Leukemia Cells by Genistein and Etoposide
Genistein and etoposide induce similar levels of DNA cleavage mediated by topoisomerase IIα and β in vitro (28). To determine whether the two compounds generate comparable levels of cleavage complexes in cultured human cells, CEM cells were treated with 50 μM genistein or etoposide for 1 hr and covalent topoisomerase II-DNA complexes were monitored using the ICE bioassay (28). As seen in Figure 1, genistein and etoposide induce similar concentrations of covalent complexes between chromosomal DNA and both topoisomerase IIα and β. Therefore, at least at 50 μM, both compounds display equivalent effects on the type II enzymes in CEM cells.
FIGURE 1.
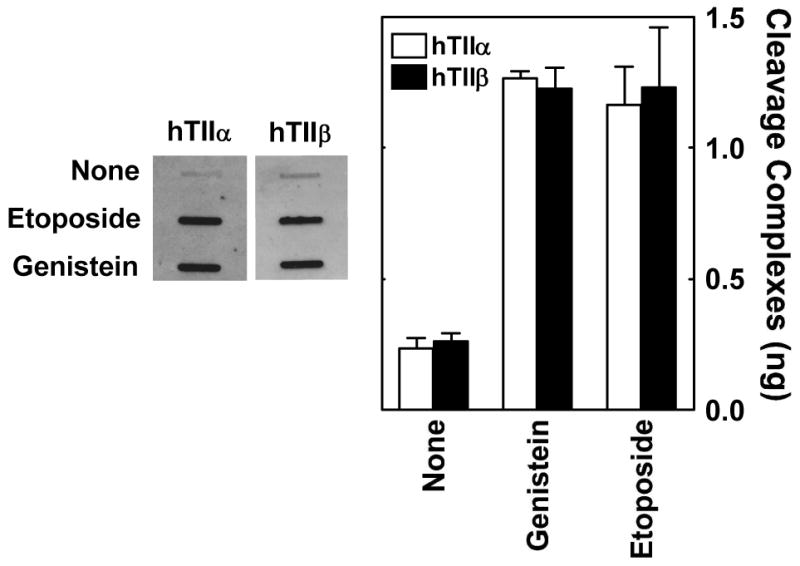
Ability of genistein and etoposide to enhance DNA cleavage mediated by topoisomerase IIα and β in cultured human CEM cells. The ICE bioassay was used to monitor levels of isoform-specific cleavage complexes in cells treated with genistein or etoposide. DNA from cell cultures treated for 1 h in the absence of compound (None) or in the presence of 50 μM genistein or etoposide was blotted onto a nitrocellulose membrane. Immunoblots were probed with polyclonal antibodies directed against either human topoisomerase IIα or β. A representative immunoblot is shown (left). The bar graph shows data for topoisomerase IIα (hTIIα; open bars) and β (hTIIβ; closed bars). Levels of covalently bound topoisomerase II are based on standards of purified human type II topoisomerases. Error bars represent the standard deviation for three independent experiments. Data were adapted from Ref. 34.
Cytotoxic and Genotoxic Effects of Genistein and Etoposide in Cultured Human CEM Leukemia Cells
Although genistein and etoposide have comparable effects on topoisomerase II-mediated DNA cleavage (28), they displayed markedly different effects on the viability of CEM cells (Figure 2, left panel). The IC50 values for the two compounds were ∼180 and ∼10 μM, respectively.
FIGURE 2.
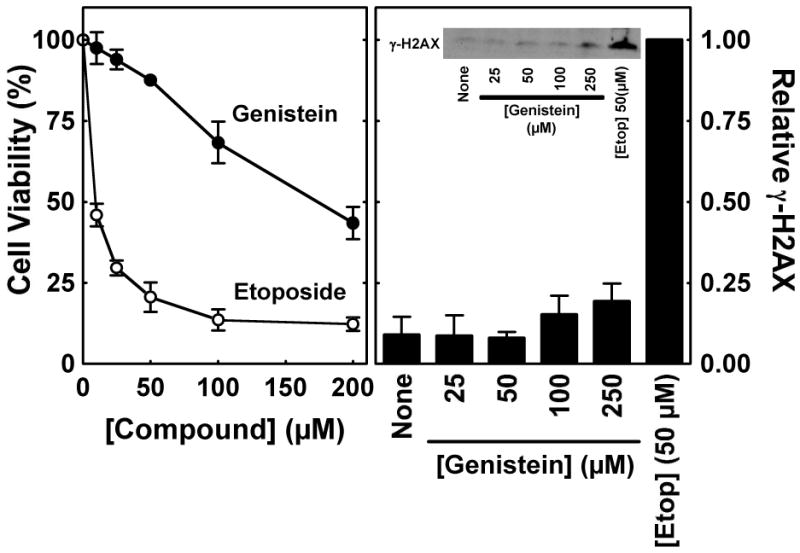
Cytotoxic and genotoxic effects of genistein and etoposide in cultured human CEM cells. Left Panel: The viability of cells in the presence of genistein or etoposide was monitored using the CCK-8 cell proliferation assay. Data are shown for cells treated with 0–200 μM genistein (closed circles) or etoposide (open circles) for 8 h. Cell viability was set to 100% in the absence of compounds. Right Panel: Phosphorylation of histone H2AX (i.e., γ-H2AX) was monitored as a marker for permanent DNA damage in CEM cells. Data are shown for cells treated in the absence of compound (None) or in the presence of 0–250 μM genistein or 50 μM etoposide for 1 h. The inset shows a representative immunoblot that was probed with a monoclonal antibody directed against γ-H2AX (Ser 139). Levels of permanent DNA damage are relative to those in the presence of 50 μM etoposide (set to 1.0). Error bars represent the standard deviation for three independent experiments.
To determine whether the differences in genistein- and etoposide-induced cytotoxicity reflect the conversion of topoisomerase II-DNA cleavage complexes to permanent DNA damage, levels of histone H2AX phosphorylation (γ-H2AX), were monitored in the presence of the compounds. Upon the generation of double-stranded breaks and other forms of DNA damage, histone H2AX is phosphorylated and recruited to the sites of DNA breaks (41, 42). Thus, γ-H2AX can be utilized as a barometer to monitor DNA damage in eukaryotic cells.
As seen in Figure 2 (right panel), levels of H2AX phosphorylation in cells incubated with 50 μM etoposide were ∼10 times higher than those treated with up to 250 μM genistein. Therefore, it appears that the covalent enzyme-DNA intermediates formed in the presence of the anticancer drug are converted to permanent DNA damage much more efficiently that those induced by the bioflavonoid.
Although etoposide primarily targets topoisomerase IIα and β (28, 32, 33, 43), genistein has numerous effects on mammalian cells that could potentially alter the ability of cleavage complexes to be converted to deleterious permanent DNA strand breaks. As an example, the bioflavonoid inhibits tyrosine kinases that are involved in cell cycle progression (44-46). Since collisions between cleavage complexes and replication or transcription enzymes represent the major pathways for transforming transient topoisomerase II-associated strand breaks into permanent DNA damage (3, 6, 11-13), the inhibitory effects of genistein on protein kinases could reduce its topoisomerase II-mediated cytotoxicity. Alternatively, genistein also could affect the pathways that are responsible for processing topoisomerase II from DNA strand breaks, thus allowing them to be recognized by cellular repair proteins.
Therefore, a global approach was employed to determine whether the decreased cytotoxicity and DNA damage formed in the presence of the bioflavonoid are due to its actions on protein kinases or other non-topoisomerase II targets. If these alternative effects are the underlying cause for the decreased levels of DNA damage triggered by genistein, the presence of the bioflavonoid should reduce the cytotoxicity and genotoxicity of other topoisomerase II poisons. Consequently, CEM cells were incubated with 50 μM genistein starting 2 h prior to treatment with etoposide (0–200 μM) for an additional 6 h in the continued presence of the bioflavonoid. As seen in Figure 3 (left panel), genistein had virtually no effect on the cytotoxicity of etoposide. Moreover, the presence of 50 μM genistein did not impact the ability of 50 μM etoposide to generate permanent DNA damage (as monitored by the generation of γ-H2AX) in treated cells (Figure 3, right panel). These findings provide strong evidence that the differential effects of the bioflavonoid and anticancer drug on human cells do not result from the non-topoisomerase II-directed activities of genistein.
FIGURE 3.
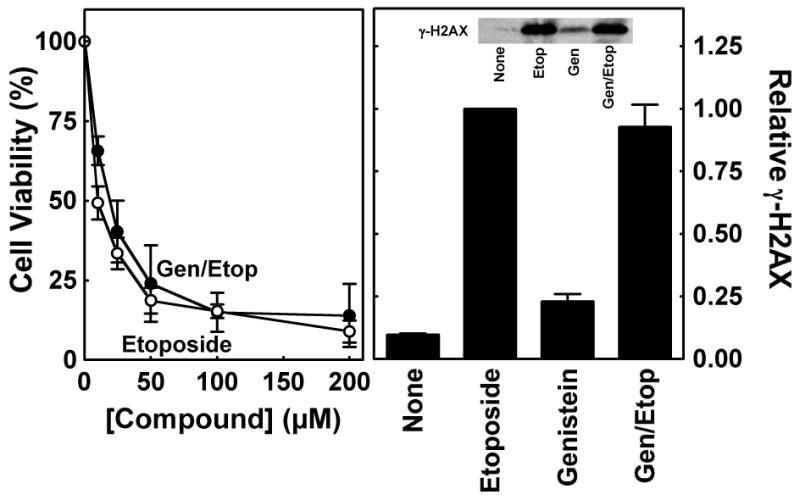
Effects of genistein on the cytotoxicity and genotoxicity of etoposide in cultured human CEM cells. Left Panel: The CCK-8 cell proliferation assay was used to monitor the viability of cells incubated with 0–200 μM etoposide alone (open circles) or 50 μM genistein for 2 h prior to and during a 6 h exposure to 0–200 μM etoposide (closed circles; Gen/Etop). Cell viability was set to 100% in the absence of compounds. Right Panel: Phosphorylation of histone H2AX (i.e., γ-H2AX) was monitored as a marker for permanent DNA damage to examine the effect of genistein on the genotoxicity of etoposide in CEM cells. Data are shown for cells treated in the absence of compound (None), in the presence of 50 μM etoposide alone for 1 h (Etoposide) or 50 μM genistein alone for 3 h (Genistein), or in the presence of 50 μM genistein for 2 h prior to and during a 1 h exposure to 50 μM etoposide (Gen/Etop). The inset shows a representative immunoblot that was probed with a monoclonal antibody directed against γ-H2AX (Ser 139). Levels of γ-H2AX are relative to those in the presence of etoposide (set to 1.0). Error bars represent the standard deviation for three independent experiments.
Differential Effects of Genistein and Etoposide on the Persistence of Topoisomerase IIα– and β–DNA Cleavage Complexes
The above findings suggest that the differential ability of genistein and etoposide to induce death and permanent DNA damage in CEM cells is related to the properties of the topoisomerase II-DNA cleavage complexes formed in the presence of these two compounds. Therefore, the persistence of cleavage complexes induced by genistein and etoposide were examined. This persistence could affect the generation of permanent DNA damage induced by topoisomerase II poisons by at least two different mechanisms. First, the longer a cleavage complex is maintained in a cell, the greater the probability that replication enzymes or other DNA tracking systems will collide with it. Second, the more stable a cleavage complex, the greater the likelihood that it will be converted to permanent DNA damage following a collision with a DNA tracking system.
As an initial step toward characterizing the persistence of cleavage complexes formed in the presence of genistein or etoposide, a neutral comet assay was utilized to monitor double-stranded DNA breaks. In this assay, cells are lysed by treatment with sarcosyl, which is the same denaturing detergent used to trap cleavage complexes in the ICE bioassay. Therefore, the majority of DNA breaks measured by this assay in the presence of a topoisomerase II poison should reflect the concentration of topoisomerase II-DNA cleavage complexes.
When cells were analyzed immediately following a 1 h exposure to 50 μM genistein or etoposide, significant levels of double-stranded DNA breaks (as determined by the length of the comet tail) were observed for both compounds (Figure 4, left panels). This finding confirms the results of the ICE bioassays shown in Figure 1. However, when cells were transferred to drug-free medium and allowed to recover for 30 min, a marked difference between genistein and etoposide was observed (Figure 4, right panels). While the levels of DNA damage in cells treated with the bioflavonoid were similar to control cultures grown in the absence of topoisomerase II poisons, the comet tail length in cells treated with the anticancer drug persisted. These results suggest that cleavage complexes formed in the presence of etoposide have a longer half-life in cells than those induced by genistein.
FIGURE 4.
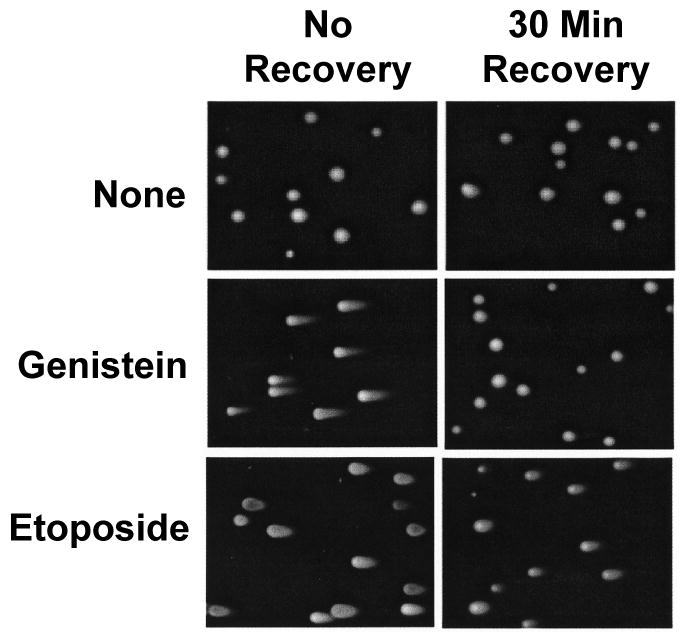
Effects of genistein and etoposide on the persistence of DNA cleavage complexes mediated by topoisomerase II in cultured human CEM cells. The neutral comet assay was utilized to monitor the levels of double-stranded DNA breaks in treated cells. Left panels show representative cells treated in the absence of compounds (None) or in the presence of 50 μM genistein or etoposide for 1 h and lysed immediately following treatment (i.e., No Recovery) Right panels show representative cells from parallel experiments in which cultures were transferred to drug-free medium for 30 min prior to lysis (i.e., 30 Min Recovery). The length of the “comet tail” of each cell is proportional to the level of double-stranded DNA breaks. Data are representative of at least three independent experiments. At least ten fields similar to those shown were observed for each set of experimental conditions.
To more thoroughly characterize the effects of genistein and etoposide on the persistence of topoisomerase II-DNA cleavage complexes, the ICE bioassay was utilized to monitor individual enzyme isoforms. Following a 1 h exposure of CEM cells to 50 μM genistein or etoposide, cells were transferred to culture medium that lacked either compound, and levels of cleavage complexes were monitored over time. Fifteen minutes after the removal of genistein from cell cultures, levels of topoisomerase IIα- and β-associated cleavage complexes dropped ∼10–fold (Figure 5, left panel). In marked contrast, etoposide-induced complexes were much more stable. Following removal of the anticancer drug, the half-lives of topoisomerase IIα- and β-associated cleavage complexes were ∼120 min and 25 min, respectively (Figure 5, right panel). These data provide strong evidence that etoposide-induced cleavage complexes formed with topoisomerase IIα and β persist considerably longer in CEM cells than do those induced by genistein.
FIGURE 5.
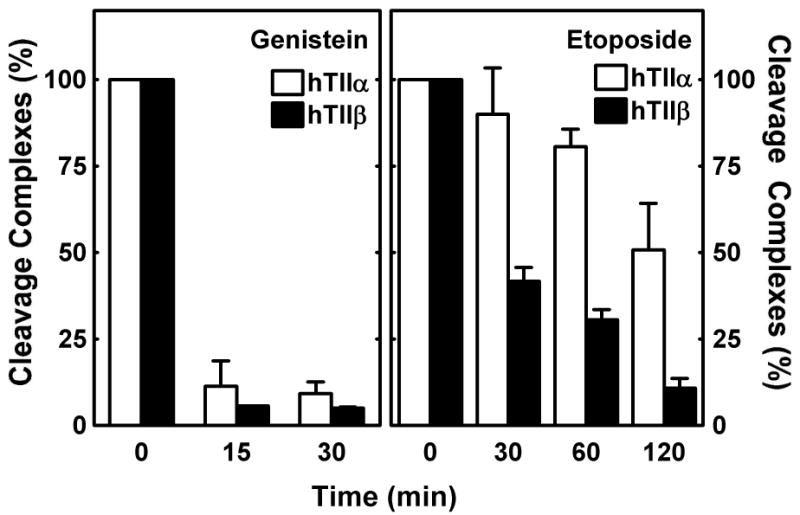
Effects of genistein and etoposide on the persistence of topoisomerase IIα- and β-DNA cleavage complexes in cultured human CEM cells. The ICE bioassay was utilized to monitor levels of isoform-specific DNA cleavage complexes induced by genistein (left panel) or etoposide (right panel). Cells were treated with 50 μM compounds for 1 h and transferred to drug-free medium for the indicated times. Data for topoisomerase IIα- (hTIIα; open bars) and topoisomerase IIβ-DNA cleavage complexes (hTIIβ; closed bars) are shown. Relative levels of complexes prior to recovery were set to 100%. Error bars represent the standard deviation for three independent experiments.
Two alternative explanations may account for the above findings. The first alternative is that genistein-induced complexes are processed more rapidly in CEM cells than those induced by etoposide. This explanation would appear to be improbable because faster processing would likely generate higher levels of permanent DNA damage. However, since previous studies indicate that topoisomerase II is processed from the termini of cleaved DNA in human cells (at least in part) by the 26S proteasome (47-49), levels of drug-induced cleavage complexes were monitored in the presence of 2 μM MG132, a potent proteasome inhibitor (50, 51). As seen in Figure 6, the addition of MG132 to cell cultures had virtually no effect on levels of genistein-induced DNA cleavage complexes. Consistent with the data in Figure 5, the concentration of topoisomerase IIα- and β-associated DNA strand breaks dropped precipitously following a 30 min recovery period in drug-free medium in the absence and presence of the proteasome inhibitor. A somewhat greater effect of MG132 was observed in etoposide-treated cells (Figure 6). Higher levels of both topoisomerase IIα- and β-associated cleavage complexes were seen in cells that were treated with the proteasome inhibitor. Taken together, these data demonstrate that the disappearance of genistein-induced cleavage complexes following a recovery period is not due to a more rapid processing by the 26S proteasome.
FIGURE 6.
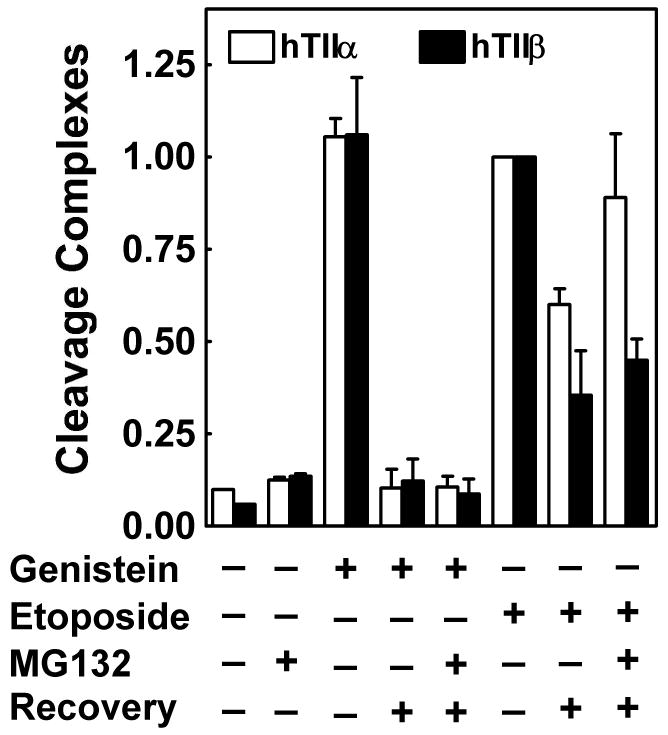
Role of the 26S proteasome in the degradation of topoisomerase IIα– and β–DNA cleavage complexes induced by genistein and etoposide. The ICE bioassay was utilized to monitor levels of isoform-specific DNA cleavage complexes. CEM cells were treated in the absence of compound or in the presence of 2 μM MG132 (a proteasome inhibitor), 50 μM genistein, or 50 μM etoposide for 1 h. In some cases, cells were transferred to drug-free medium for a 30 min recovery period following treatment. When experiments were carried out in presence of MG132 (2 μM), the proteasome inhibitor was included in cell cultures for 30 min prior to the addition of genistein or etoposide and also was present during the recovery period. Data for cleavage complexes mediated by topoisomerase IIα (hTIIα; open bars) or β (hTIIβ; closed bars) are shown. Levels of cleavage complexes prior to recovery were set to 100%. Error bars represent the standard deviation for three independent experiments.
The second alternative is that cleavage complexes formed in the presence of genistein are intrinsically less stable than those formed in the presence of etoposide. To examine this possibility, the stability of cleavage complexes formed in the presence of genistein or etoposide was examined in a cell-free system. A dilution experiment was designed to mimic the cellular recovery experiment performed with the ICE bioassay (see Figure 5). In this in vitro system, cleavage complexes were established for 6 min in the presence of the bioflavonoid or the anticancer drug, and reaction mixtures were diluted 10–fold with cleavage buffer. Levels of topoisomerase II-mediated DNA scission are dramatically reduced if cleavage reactions are initiated under these dilute conditions. Therefore, the rate at which DNA scission decreases after samples are diluted should reflect the intrinsic stability of cleavage complexes. Although the factors that control the decay of cleavage intermediates have not been addressed experimentally, the rate-determining step in this process is most likely the dissociation of the topoisomerase II poison from the ternary complex.
Results from this cell-free experiment paralleled those observed in cultured cells. The half-lives of cleavage complexes established in the presence of 50 μM genistein were less than 5 s for both topoisomerase II isoforms (Figure 7, left panel). In marked contrast, the half-lives of topoisomerase IIα- and β-associated DNA breaks formed in the presence of 50 μM etoposide were ∼30 min and 3 min, respectively. These findings demonstrate that topoisomerase II-associated DNA strand breaks formed in the presence of the anticancer drug are intrinsically more stable than those formed in the presence of the bioflavonoid. Furthermore, these in vitro data strongly suggest that the decreased persistence of genistein-induced cleavage complexes in CEM cells is due to a decreased stability of the cleavage intermediate, rather than enhanced cellular processing.
FIGURE 7.
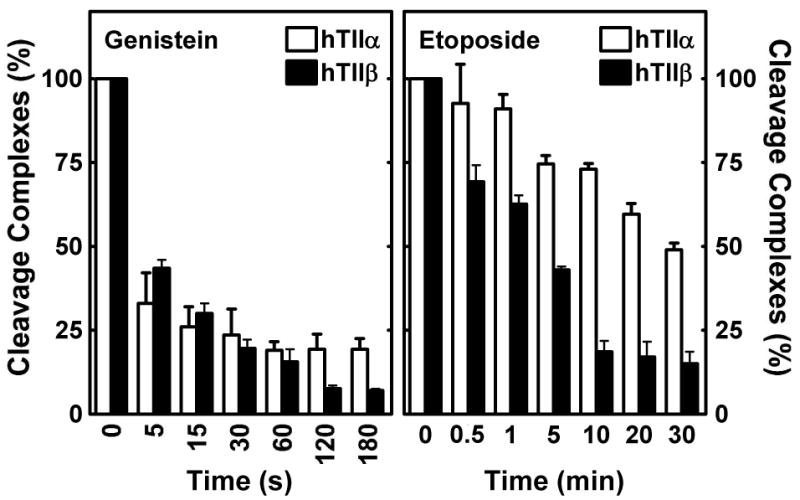
Effects of genistein and etoposide on the persistence of topoisomerase IIα- and β-DNA cleavage complexes in vitro. DNA cleavage assays were performed using purified human type II enzymes. Reactions were initiated in the presence of 50 μM genistein (left panel) or etoposide (right panel). After cleavage equilibrium was established (6 min), reaction mixtures were diluted 10–fold with DNA cleavage buffer, and levels of double-stranded DNA breaks were monitored over time. Data for DNA cleavage mediated by topoisomerase IIα (hTIIα; open bars) or β (hTIIβ; closed bars) are shown. Relative levels of DNA cleavage prior to dilution were set to 100%. Error bars represent the standard deviation for three independent experiments.
Preferential Targeting of Topoisomerase IIα-Mediated Cytotoxicity by Etoposide in Cultured Human Cells
All known anticancer drugs that act against topoisomerase II target both isoforms of the enzyme. However, several studies suggest that a drug that could preferentially target topoisomerase IIα might have beneficial ramifications for cancer patients. For example, while topoisomerase IIα is present only in rapidly proliferating tissues (including most cancer cells) (52-55), topoisomerase IIβ is present in all cell types (54, 56). Considerable evidence indicates that the action of topoisomerase II-targeted agents against the β isoform in differentiated tissues such as cardiac cells is responsible for much of the off-target toxicity of these drugs (30, 31). Recent studies also imply that topoisomerase IIα is the primary cytotoxic target of topoisomerase II poisons (30, 31), while topoisomerase IIβ may play a disproportionate role in generating the leukemogenic MLL gene 11q23 translocations observed in patients that are treated with these agents (30, 31).
The findings described in the previous section suggest that it may be possible to exploit the differential persistence of etoposide-induced DNA cleavage complexes in order to preferentially target topoisomerase IIα. Over the course of the 2 h recovery period following removal of etoposide from culture medium, levels of DNA scission mediated by topoisomerase IIα were ∼2.4–fold greater than those generated by the β isoform (Figure 8, inset). Therefore, it should be possible to preferentially target topoisomerase IIα using a “pulsed” schedule that includes recovery periods as opposed to “continuous” treatment with etoposide. However, it remains to be determined whether a pulsed schedule that induces lower levels of topoisomerase IIβ-DNA cleavage complexes will be as efficacious as a schedule that maintains high levels of cleavage complexes formed with both enzyme isoforms.
FIGURE 8.
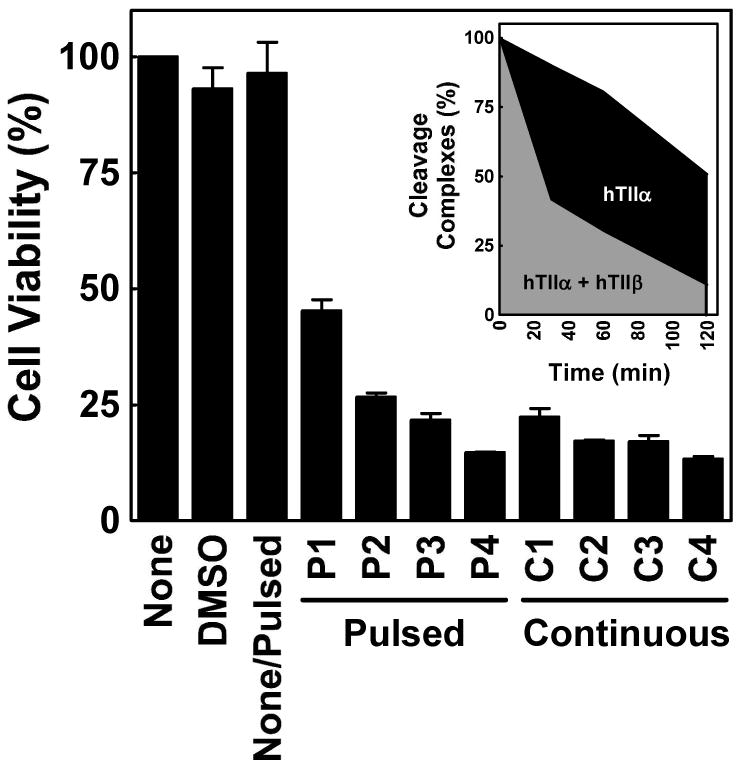
Cytotoxicity of etoposide using a pulsed treatment schedule in cultured human CEM cells. The CCK-8 proliferation assay was utilized to compare the viability of cells treated with a pulsed schedule (4 cycles of treatment with etoposide for 15 min followed by a 2 h recovery period in drug-free medium) vs. a continuous exposure to 50 μM etoposide for an equivalent period of time (9 h total). Control data are shown for cells treated in the absence of drug (None), in the continuous presence of drug diluent (DMSO), or in the absence of drug but subjected to the physical manipulation of the pulsed schedule (None/Pulsed). The viability of cells is shown following each cycle of the pulsed schedule (Cycle P1-4) and for samples that were removed at equivalent times (2.25 h intervals) for cultures that were continuously exposed to etoposide (Cycle C1-4). Cell viability was set to 100% in the absence of drug. Error bars represent the standard deviation for three independent experiments. The inset shows the area under the curve for levels of etoposide-induced DNA cleavage complexes mediated by topoisomerase IIα (black + gray filled areas) and β (gray filled area) for one pulse-recovery cycle, as monitored by the ICE bioassay.
To address this issue, cells were exposed to etoposide for 15 min followed by a 2 h recovery period in drug-free medium. Results with human CEM cells that were treated continuously with etoposide were compared to cultures that were pulsed for 4 treatment-recovery cycles (Figure 8). Lower cytotoxicity was observed following the first 2–3 cycles. However, after the completion of all four cycles, the survival of cells on the pulsed schedule was comparable to that of cultures treated with constant exposure to etoposide over an equivalent time course. The physical manipulation of cells required by the pulsed regimen had no effect on viability. Thus, it is concluded that, at least for the cell line and protocols employed, a schedule that is based on multiple cycles of etoposide exposure and recovery may be as efficacious as one that includes continuous drug treatment.
Discussion
Although etoposide and genistein display a similar ability to induce topoisomerase II-DNA cleavage complexes in vitro and in cultured human cells (28), their effects on the viability and generation of permanent DNA damage in cells differ considerably. Etoposide was ∼10 times more cytotoxic and genotoxic than the bioflavonoid in CEM cells. This difference was not due to the physiological effects of genistein on tyrosine kinases or other growth-related enzymes. Rather, it correlates with an intrinsic difference in the properties of topoisomerase II-DNA cleavage complexes generated in the presence of the two compounds. To this point, topoisomerase IIα- and β-DNA cleavage complexes formed in the presence of etoposide were dramatically more stable than those formed in the presence of genistein. Following dilution into drug-free medium (cellular experiments) or buffer (in vitro experiments), etoposide-induced cleavage complexes often persisted ten times longer than equivalent genistein-induced complexes.
Despite the difference in persistence, the actual levels of isoform-specific cleavage complexes generated during continual exposure of CEM cells to 50 μM genistein or etoposide were the same. This finding suggests that persistence is a property of cleavage complexes that is independent of the concentration of topoisomerase II-associated DNA cleavage intermediates. Rather, the intrinsic stability of any given cleavage complex may determine the likelihood that it can be converted to permanent DNA damage in the cell. For example, if a replication fork (or other tracking system) encounters a persistent topoisomerase II-DNA cleavage complex, there may be a high probability that the collision will render the enzyme unable to rejoin the cleaved DNA termini. Conversely, if the same replication fork encounters a less stable cleavage complex, the type II enzyme may still be able to ligate the DNA termini and allow the tracking system to traverse the site without generating permanent damage. Therefore, even if two drugs generate similar concentrations of topoisomerase II-DNA cleavage complexes in treated cells, the drug that remains associated with the ternary complex longer may induce higher levels of DNA strand breaks and subsequent toxicity. Similar conclusions have been proposed for topoisomerase I, based on the effects of mutant, self-poisoning enzymes (57, 58), and camptothecin derivatives on cell viability (59).
It should be noted that the DNA cleavage specificity of etoposide and genistein differ from one another, with the former preferring a cytosine at the -1 position relative to the scissile bond and the latter preferring a thymine (28, 60). At the present time, the role of DNA cleavage site selection in drug cytotoxicity is not understood. However, it is possible that it influences the relative abilities of etoposide and genistein to kill cultured human cells, despite the similar levels of topoisomerase II-DNA cleavage complexes induced by these two compounds.
An unexpected result from the present study was the finding that etoposide-induced DNA cleavage complexes formed with topoisomerase IIα persisted several–fold longer than those formed with topoisomerase IIβ. This observation may have potential ramifications for the scheduling of etoposide during cancer chemotherapy. As discussed earlier, all clinically relevant topoisomerase II-active anticancer drugs target both isoforms of the type II enzyme. However, a growing body of evidence indicates that the targeting of topoisomerase IIβ in cardiac cells is responsible for much of the off-target toxicity of these agents (30, 31). The β isoform also has been suggested to play a disproportionate role in generating leukemogenic 11q23 translocations involving the MLL gene (30, 31). Therefore, a drug that selectively targets topoisomerase IIα may be advantageous for patients.
Given the differential persistence of topoisomerase IIα- and β-DNA cleavage complexes, it may be possible to preferentially target the α isoform with etoposide by using a pulsed drug schedule as opposed to constant exposure. Results with cultured human CEM cells suggest that the two regimens yield similar levels of cytotoxicity after a few drug treatment-recovery cycles. Thus, the use of pulsed drug schedules in the clinical setting, at least in principle, might be feasible. Studies currently are underway to determine whether other topoisomerase II-targeted agents also display isoform specificity with regard to the stability of cleavage complexes.
In summary, the persistence of drug-induced topoisomerase II-DNA cleavage complexes may have a profound effect on both the therapeutic and leukemogenic actions of these anticancer agents. Consequently, it would be worthwhile to evaluate this property as part of the routine characterization of topoisomerase II-targeted drugs.
Acknowledgments
We are grateful to Joseph E. Deweese, Amanda C. Gentry, and Steven L. Pitts for critical reading of the manuscript. Comet assays were performed in part through the use of the VUMC Imaging Shared Resource.
Footnotes
This work was supported by National Institutes of Health research grant GM33944. O.J.B. was a trainee under grant 5 T32 CA09582 from the National Institutes of Health and was supported in part by Ruth L. Kirschstein National Research Service Award Predoctoral Fellowship F31 GM78744 from the National Institutes of Health.
References
- 1.Berger JM, Gamblin SJ, Harrison SC, Wang JC. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 2.Nitiss JL. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim Biophys Acta. 1998;1400:63–81. doi: 10.1016/s0167-4781(98)00128-6. [DOI] [PubMed] [Google Scholar]
- 3.Fortune JM, Osheroff N. Topoisomerase II as a target for anticancer drugs: When enzymes stop being nice. Prog Nucleic Acid Res Mol Biol. 2000;64:221–253. doi: 10.1016/s0079-6603(00)64006-0. [DOI] [PubMed] [Google Scholar]
- 4.Champoux JJ. DNA topisomerases: Structure, function, and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 5.Wang JC. Cellular roles of DNA topoisomerases: A molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 6.McClendon AK, Osheroff N. DNA topoisomerase II, genotoxicity, and cancer. Mutat Res. 2007;623:83–97. doi: 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu LF, Rowe TC, Yang L, Tewey KM, Chen GL. Cleavage of DNA by mammalian DNA topoisomerase II. J Biol Chem. 1983;258:15365–15370. [PubMed] [Google Scholar]
- 8.Sander M, Hsieh T. Double strand DNA cleavage by type II DNA topoisomerase from Drosophila melanogaster. J Biol Chem. 1983;258:8421–8428. [PubMed] [Google Scholar]
- 9.Zechiedrich EL, Christiansen K, Andersen AH, Westergaard O, Osheroff N. Double-stranded DNA cleavage/religation reaction of eukaryotic topoisomerase II: Evidence for a nicked DNA intermediate. Biochemistry. 1989;28:6229–6236. doi: 10.1021/bi00441a014. [DOI] [PubMed] [Google Scholar]
- 10.Hande KR. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur J Cancer. 1998;34:1514–1521. doi: 10.1016/s0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- 11.Li TK, Liu LF. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol. 2001;41:53–77. doi: 10.1146/annurev.pharmtox.41.1.53. [DOI] [PubMed] [Google Scholar]
- 12.Walker JV, Nitiss JL. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Invest. 2002;20:570–589. doi: 10.1081/cnv-120002156. [DOI] [PubMed] [Google Scholar]
- 13.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents. 2005;5:363–372. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 14.Felix CA. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta. 1998;1400:233–255. doi: 10.1016/s0167-4781(98)00139-0. [DOI] [PubMed] [Google Scholar]
- 15.Leone G, Voso MT, Sica S, Morosetti R, Pagano L. Therapy related leukemias: Susceptibility, prevention and treatment. Leuk Lymphoma. 2001;41:255–276. doi: 10.3109/10428190109057981. [DOI] [PubMed] [Google Scholar]
- 16.Felix CA. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med Pediatr Oncol. 2001;36:525–535. doi: 10.1002/mpo.1125. [DOI] [PubMed] [Google Scholar]
- 17.Felix CA, Kolaris CP, Osheroff N. Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amsterdam) 2006;5:1093–1108. doi: 10.1016/j.dnarep.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 18.Kurzer MS, Xu X. Dietary phytoestrogens. Annu Rev Nutr. 1997;17:353–381. doi: 10.1146/annurev.nutr.17.1.353. [DOI] [PubMed] [Google Scholar]
- 19.Galati G, O'Brien PJ. Potential toxicity of flavonoids and other dietary phenolics: Significance for their chemopreventive and anticancer properties. Free Radical Biol Med. 2004;37:287–303. doi: 10.1016/j.freeradbiomed.2004.04.034. [DOI] [PubMed] [Google Scholar]
- 20.Yao LH, Jiang YM, Shi J, Tomas-Barberan FA, Datta N, Singanusong R, Chen SS. Flavonoids in food and their health benefits. Plant Foods Hum Nutr. 2004;59:113–122. doi: 10.1007/s11130-004-0049-7. [DOI] [PubMed] [Google Scholar]
- 21.Sang S, Hou Z, Lambert JD, Yang CS. Redox properties of tea polyphenols and related biological activities. Antioxid Redox Signaling. 2005;7:1704–1714. doi: 10.1089/ars.2005.7.1704. [DOI] [PubMed] [Google Scholar]
- 22.Siddiqui IA, Adhami VM, Saleem M, Mukhtar H. Beneficial effects of tea and its polyphenols against prostate cancer. Mol Nutr Food Res. 2006;50:130–143. doi: 10.1002/mnfr.200500113. [DOI] [PubMed] [Google Scholar]
- 23.Ross JA, Potter JD, Reaman GH, Pendergrass TW, Robison LL. Maternal exposure to potential inhibitors of DNA topoisomerase II and infant leukemia (United States): A report from the Children's Cancer Group. Cancer Causes Control. 1996;7:581–590. doi: 10.1007/BF00051700. [DOI] [PubMed] [Google Scholar]
- 24.Strick R, Strissel PL, Borgers S, Smith SL, Rowley JD. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci USA. 2000;97:4790–4795. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, Felix CA, Davies SM, Slavin J, Potter JD, Blair CK, Reaman GH, Ross JA. Maternal diet and infant leukemia: The DNA topoisomerase II inhibitor hypothesis: A report from the children's oncology group. Cancer Epidemiol Biomarkers Prev. 2005;14:651–655. doi: 10.1158/1055-9965.EPI-04-0602. [DOI] [PubMed] [Google Scholar]
- 26.Austin CA, Patel S, Ono K, Nakane H, Fisher LM. Site-specific DNA cleavage by mammalian DNA topoisomerase II induced by novel flavone and catechin derivatives. Biochem J. 1992;282:883–889. doi: 10.1042/bj2820883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Waalwijk van Doorn-Khosrovani SB, Janssen J, Maas LM, Godschalk RW, Nijhuis JG, van Schooten FJ. Dietary flavonoids induce MLL translocations in primary human CD34+ cells. Carcinogenesis. 2007;28:1703–1709. doi: 10.1093/carcin/bgm102. [DOI] [PubMed] [Google Scholar]
- 28.Bandele OJ, Osheroff N. Bioflavonoids as poisons of human topoisomerase II< and II®. Biochemistry. 2007;46:6097–6108. doi: 10.1021/bi7000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandele OJ, Osheroff N. (–)-Epigallocatechin gallate, a major constituent of green tea, poisons human type II topoisomerases. Chem Res Toxicol. 2008;21:936–943. doi: 10.1021/tx700434v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JY, Wang JC, Liu LF. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci USA. 2007;104:11014–11019. doi: 10.1073/pnas.0704002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase IIβ mediated DNA double-strand breaks: Implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–8846. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 32.Drake FH, Hofmann GA, Bartus HF, Mattern MR, Crooke ST, Mirabelli CK. Biochemical and pharmacological properties of p170 and p180 forms of topoisomerase II. Biochemistry. 1989;28:8154–8160. doi: 10.1021/bi00446a029. [DOI] [PubMed] [Google Scholar]
- 33.Willmore E, Frank AJ, Padget K, Tilby MJ, Austin CA. Etoposide targets topoisomerase IIα and IIβ in leukemic cells: Isoform-specific cleavable comlexes visualized and quantified in situ by a novel immunofluorescence technique. Mol Pharmacol. 1998;53:78–85. doi: 10.1124/mol.54.1.78. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Lazaro M, Willmore E, Austin CA. Cells lacking DNA topoisomerase IIβ are resistant to genistein. J Nat Prod. 2007;70:763–767. doi: 10.1021/np060609z. [DOI] [PubMed] [Google Scholar]
- 35.Bender RP, Jablonksy MJ, Shadid M, Romaine I, Dunlap N, Anklin C, Graves DE, Osheroff N. Substituents on etoposide that interact with human topoisomerase II• in the binary enzyme-drug complex: contributions to etoposide binding and activity. Biochemistry. 2008;47:4501–4509. doi: 10.1021/bi702019z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worland ST, Wang JC. Inducible overexpression, purification, and active site mapping of DNA topoisomerase II from the yeast Saccharomyces cerevisiae. J Biol Chem. 1989;264:4412–4416. [PubMed] [Google Scholar]
- 37.Elsea SH, Hsiung Y, Nitiss JL, Osheroff N. A yeast type II topoisomerase selected for resistance to quinolones. Mutation of histidine 1012 to tyrosine confers resistance to nonintercalative drugs but hypersensitivity to ellipticine. J Biol Chem. 1995;270:1913–1920. doi: 10.1074/jbc.270.4.1913. [DOI] [PubMed] [Google Scholar]
- 38.Kingma PS, Greider CA, Osheroff N. Spontaneous DNA lesions poison human topoisomerase IIα and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry. 1997;36:5934–5939. doi: 10.1021/bi970507v. [DOI] [PubMed] [Google Scholar]
- 39.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 40.Fortune JM, Osheroff N. Merbarone inhibits the catalytic activity of human topoisomerase IIα by blocking DNA cleavage. J Biol Chem. 1998;273:17643–17650. doi: 10.1074/jbc.273.28.17643. [DOI] [PubMed] [Google Scholar]
- 41.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;276:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 42.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 43.Byl JA, Cline SD, Utsugi T, Kobunai T, Yamada Y, Osheroff N. DNA topoisomerase II as the target for the anticancer drug TOP-53: Mechanistic basis for drug action. Biochemistry. 2001;40:712–718. doi: 10.1021/bi0021838. [DOI] [PubMed] [Google Scholar]
- 44.Geahlen RL, Koonchanok NM, McLaughlin JL, Pratt DE. Inhibition of protein-tyrosine kinase activity by flavanoids and related compounds. J Nat Prod. 1989;52:982–986. doi: 10.1021/np50065a011. [DOI] [PubMed] [Google Scholar]
- 45.Meeran SM, Katiyar SK. Cell cycle control as a basis for cancer chemoprevention through dietary agents. Front Biosci. 2008;13:2191–2202. doi: 10.2741/2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banerjee S, Li Y, Wang Z, Sarkar FH. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008 doi: 10.1016/j.canlet.2008.03.052. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao Y, Desai SD, Ting CY, Hwang J, Liu LF. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J Biol Chem. 2001;276:40652–40658. doi: 10.1074/jbc.M104009200. [DOI] [PubMed] [Google Scholar]
- 48.Xiao H, Mao Y, Desai SD, Zhou N, Ting CY, Hwang J, Liu LF. The topoisomerase IIβ circular clamp arrests transcription and signals a 26S proteasome pathway. Proc Natl Acad Sci USA. 2003;100:3239–3244. doi: 10.1073/pnas.0736401100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang A, Lyu YL, Lin CP, Zhou N, Azarova AM, Wood LM, Liu LF. A protease pathway for the repair of topoisomerase II-DNA covalent complexes. J Biol Chem. 2006;281:35997–36003. doi: 10.1074/jbc.M604149200. [DOI] [PubMed] [Google Scholar]
- 50.Lee DH, Goldberg AL. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- 51.Lee DH, Goldberg AL. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 52.Heck MM, Hittelman WN, Earnshaw WC. Differential expression of DNA topoisomerases I and II during the eukaryotic cell cycle. Proc Natl Acad Sci USA. 1988;85:1086–1090. doi: 10.1073/pnas.85.4.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsiang YH, Wu HY, Liu LF. Proliferation-dependent regulation of DNA topoisomerase II in cultured human cells. Cancer Res. 1988;48:3230–3235. [PubMed] [Google Scholar]
- 54.Woessner RD, Mattern MR, Mirabelli CK, Johnson RK, Drake FH. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991;2:209–214. [PubMed] [Google Scholar]
- 55.Isaacs RJ, Davies SL, Sandri MI, Redwood C, Wells NJ, Hickson ID. Physiological regulation of eukaryotic topoisomerase II. Biochim Biophys Acta. 1998;1400:121–137. doi: 10.1016/s0167-4781(98)00131-6. [DOI] [PubMed] [Google Scholar]
- 56.Austin CA, Marsh KL. Eukaryotic DNA topoisomerase II®. BioEssays. 1998;20:215–226. doi: 10.1002/(SICI)1521-1878(199803)20:3<215::AID-BIES5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 57.Megonigal MD, Fertala J, Bjornsti MA. Alterations in the catalytic activity of yeast DNA topoisomerase I result in cell cycle arrest and cell death. J Biol Chem. 1997;272:12801–12808. doi: 10.1074/jbc.272.19.12801. [DOI] [PubMed] [Google Scholar]
- 58.Fiorani P, Bjornsti MA. Mechanisms of DNA topoisomerase I-induced cell killing in the yeast Saccharomyces cerevisiae. Ann N Y Acad Sci. 2000;922:65–75. doi: 10.1111/j.1749-6632.2000.tb07026.x. [DOI] [PubMed] [Google Scholar]
- 59.Tanizawa A, Kohn KW, Kohlhagen G, Leteurtre F, Pommier Y. Differential stabilization of eukaryotic DNA topoisomerase I cleavable complexes by camptothecin derivatives. Biochemistry. 1995;34:7200–7206. doi: 10.1021/bi00021a035. [DOI] [PubMed] [Google Scholar]
- 60.Capranico G, Binaschi M. DNA sequence selectivity of topoisomerases and topoisomerase poisons. Biochim Biophys Acta. 1998;1400:185–194. doi: 10.1016/s0167-4781(98)00135-3. [DOI] [PubMed] [Google Scholar]