

Abstract
Maternal overweight and obesity in pregnancy often result in fetal overgrowth, which increases the risk for the baby to develop metabolic syndrome later in life. However, the mechanisms underlying fetal overgrowth are not established. We developed a mouse model and hypothesized that a maternal high-fat (HF) diet causes up-regulation of placental nutrient transport, resulting in fetal overgrowth. C57BL/6J female mice were fed a control (11% energy from fat) or HF (32% energy from fat) diet for 8 wk before mating and throughout gestation and were studied at embryonic day 18.5. The HF diet increased maternal adiposity, as assessed by fat pad weight, and circulating maternal leptin, decreased serum adiponectin concentrations, and caused a marked increase in fetal growth (+43%). The HF diet also increased transplacental transport of glucose (5-fold) and neutral amino acids (10-fold) in vivo. In microvillous plasma membranes (MVMs) isolated from placentas of HF-fed animals, protein expression of glucose transporter 1 (GLUT1) was increased 5-fold, and protein expression of sodium-coupled neutral amino acid transporter (SNAT) 2 was elevated 9-fold. In contrast, MVM protein expression of GLUT 3 or SNAT4 was unaltered. These data suggest that up-regulation of specific placental nutrient transporter isoforms constitute a mechanism linking maternal high-fat diet and obesity to fetal overgrowth.—Jones, H. N., Woollett, L. A., Barbour, N., Prasad, P. D., Powell, T. L., Jansson, T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice.
Keywords: maternal obesity, macrosomia
Fetal overgrowth results in the birth of a large-for-gestational age (LGA) baby, which is typically defined as birth weight > 90th centile and occurs in ∼10% of all pregnancies (1). LGA babies have an increased risk of developing obesity and metabolic syndrome in childhood and adolescence (2, 3), thereby contributing to the increased incidence of diabetes (4) currently seen worldwide. Although an association between obesity in pregnancy and intrauterine growth restriction has been reported in a few studies (5), the typical outcome with respect to fetal growth in pregnancies of overweight and obese women is fetal overgrowth (6,7,8) Indeed, women with increased body mass index (BMI) are 2–3 times more likely to give birth to a large baby (9), which represents a significant medical problem, considering that >50% of all American women enter pregnancy overweight (BMI 25–29.9 kg/m2) or obese (BMI≥30 kg/m2) (9).
Maternal dietary energy intake, including total fat intake, is elevated in overweight and obese pregnant women in both the first and third trimesters (10). Thus, a “vicious cycle” is created, with overweight and obese women consuming a diet rich in energy, fat, and carbohydrates and giving birth to large babies. These babies are more likely to develop obesity and diabetes later in life and, in the case of females, are prone to deliver a large baby when they become pregnant (7, 8, 11). Measures to alleviate fetal overgrowth in these women represent an early intervention strategy that could contribute to a decrease in the prevalence of obesity and diabetes in future generations. However, a lack of appropriate animal models to study the mechanisms underlying fetal overgrowth in these pregnancies has hampered advances in this area.
Fetal growth is strongly influenced by nutrient supply, which is dependent on placental transport functions. Placental nutrient transporter activity is known to be altered in cases of abnormal fetal growth, and these changes may contribute directly to the changes in fetal growth rate (12, 13). Fetal overgrowth in pregnancies complicated by type 1 diabetes (14) but not by gestational diabetes mellitus (GDM) (15) is associated with increased placental glucose transporter activity and protein expression. The capacity of the placenta to transport the essential amino acid leucine is increased in GDM complicated by fetal overgrowth (16) and placental system A amino acid transporter activity was reported to be increased in both type 1 diabetes and GDM (16). In contrast to these findings, a previous study indicated that system A activity is reduced and the activity of system L is unaltered in microvillous plasma membrane (MVM) vesicles isolated from type 1 diabetic pregnancies with LGA babies (17). However, no data are currently available with respect to placental transport function in pregnancies associated with increased maternal BMI.
Compared with women with normal BMI, overweight and obese women have increased serum concentrations of insulin, interleukin-6 (IL-6), and leptin and lower adiponectin levels in the third trimester (10, 18, 19), and many of these metabolic alterations are already present in the first trimester (10). Previous studies have demonstrated that leptin and insulin stimulate the activity of the system A amino acid transporter in primary villous fragments and cultured trophoblast cells (20, 21). Similarly, placental glucose transporters in primary villous fragments obtained in early pregnancy have been shown to be up-regulated in vitro by insulin (22). Therefore, the alterations in the maternal levels of these regulatory factors in overweight and obese pregnant women may influence the placental nutrient transport pathways and increase nutrient supply to the developing fetus.
There are few, if any, published animal models of obesity in pregnancy that display the key features of the human condition, i.e., increased maternal adiposity without diabetes, increased levels of maternal metabolic hormones, low adiponectin, and fetal overgrowth. Therefore, the aim of this study was to develop a mouse model fed a moderately high-fat diet before and throughout gestation, resulting in fetal overgrowth and associated with a maternal environment reflective of that found in the pregnant woman with increased BMI. Once established, we used the model to test the hypothesis that the mechanisms underlying fetal overgrowth involved increased placental nutrient transport.
MATERIALS AND METHODS
Animals and diets
All protocols were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati. Eight-week-old female C57/BL6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were housed 4/cage in PIV barrier housing and fed ad libitum with a high-fat (HF) diet (32% energy from fat, 52% from carbohydrate, and 16% from protein; catalog no. D12266B) or control (C) diet (11% energy from fat, 73% from carbohydrate, and 16% from protein; catalog no. D12489B) for 8 wk. Diets were purchased from Research Diets (New Brunswick, NJ, USA). Daily food intake was estimated by weighing remaining food at the end of each week and was used to calculate daily caloric intake. After 8 wk of feeding with these diets, mice were weighed and mated. Females were checked daily for postcopulatory plugs. The presence of a plug represented embryonic day (E) 0.5. At E18.5, dams were weighed and then used either for collection of blood and tissue samples, glucose tolerance tests, or in vivo placental transport studies.
Collection of blood and tissue samples
Dams were sacrificed at E18.5, and maternal blood was collected by cardiac puncture, allowed to clot, and spun at 4000 rpm to collect serum and red blood cell pellets. After laparotomy, fetuses and placentas were removed and weighed, and placentas in each litter were pooled. Approximately 0.5 g of pooled placental tissue was washed in saline and transferred to 3 ml of buffer D [250 mM sucrose, 10 mM Hepes-Tris, and 1 mM EDTA (pH 7.4) at 4°C], protease inhibitor cocktail (Sigma-Aldrich Corp., St. Louis, MO, USA) was added at a dilution of 1:1000, and the mixture was homogenized using a Polytron homogenizer (Kinematica, Bohemia, NY, USA). Homogenates were used for isolation of trophoblast MVMs, as described below, or snap-frozen for subsequent analysis. In a subset of dams, the inguinal, parametrial, mesometrial, and retroperitoneal fad pads were dissected and weighed to assess maternal adiposity. Maternal serum analyses of adiponectin (Alpco Diagnostics, Salem, NH, USA), IL-6, and tumor necrosis factor-α (TNF-α) (R&D Systems Inc., Minneapolis, MN, USA) were performed using commercially available ELISA kits. Glycated hemoglobin (HbA1c) levels were determined in lysed red blood cell samples using the Helena Glyco-Tek Affinity column standard method (Helena Laboratories, Beaumont, TX, USA). Maternal serum was analyzed for leptin, insulin, and lipid profile, and packed red blood cell samples were analyzed for lipid composition by the Mouse Metabolic Phenotyping Center at the University of Cincinnati.
Isolation of trophoblast MVMs
The protocol for isolating trophoblast MVMs from mouse placenta was adapted from that used previously in human placenta (16). All procedures were performed on ice and centrifugation steps were performed at 4°C. Homogenates were centrifuged at 10,000 g for 15 min; pellets were resuspended, rehomogenized in 1 ml of buffer D, and centrifuged at 10,000 g for 10 min. The resulting supernatants were combined and spun at 125,000 g for 30 min. The pelleted crude membrane fraction was resuspended in 2 ml of buffer D, and 12 mM MgCl2 was added. The suspension was stirred for 20 min on ice. After centrifugation at 2500 g for 10 min, the supernatant was centrifuged at 125,000 g for 30 min. The final pellet was resuspended in buffer D to give a protein concentration of ∼10 mg/ml. MVM purity was assessed by MVM/homogenate enrichments of alkaline phosphatase (23) and was found to be 13 ± 1.49-fold (n=16).
Western blot
Protein expression of the system A transporter isoforms sodium-coupled neutral amino acid transporter (SNAT) 2 and SNAT4 and the glucose transporter isoform GLUT1 was analyzed in placental MVMs using Western blotting. Expression of GLUT3 was analyzed in placental homogenates. A polyclonal SNAT2 antibody was generated in rabbits (24) and validated for immunoblotting in rat placental homogenate (13). Affinity-purified polyclonal anti-SNAT4 antibodies were generated against the peptide sequence YGEVEDELLHAYSKV in rabbits (Eurogentec, Seraing, Belgium). GLUT1 and GLUT 3 antibodies were purchased from Millipore (Temecula, CA, USA), and anti-β-actin was from Sigma-Aldrich Corp. Protein concentrations were determined by Bradford assay, and Western blotting was performed. In brief, 15 μg of total protein was loaded onto a 10% SDS-PAGE gel, and electrophoresis was performed at a constant 100 V for 2 h. Proteins were transferred onto nitrocellulose membranes overnight at a constant 30 V. Membranes were incubated with primary antibodies overnight (SNAT2, 1:1000) or for 1 h at room temperature (GLUT1, GLUT3, and β-actin, 1:5000; SNAT4, 1:2000), washed, and incubated with the appropriate secondary peroxidase-labeled immunoglobulin G (IgG; 1:1000–1:5000) for 1 h. After washing, bands were visualized using enhanced chemiluminescence (ECL) detection reagents (GE Healthcare, Chalfont St. Giles, UK). Blots were stripped and reprobed for β-actin as a loading control. Analysis of the blots was performed by densitometry using an Alpha Imager (Alpha Innotech Corporation, San Leandro, CA, USA).
Glucose tolerance test
Glucose tolerance tests were performed in a subset of mice on E18.5 after a 6-h fast. Conscious unrestrained mice were injected i.p. with glucose (2 g/kg body weight), and blood was sampled in triplicate from the tail vein at 0, 15, 30, 60, and 90 min after the glucose load. Blood was obtained by amputation of the tip of the tail at time zero and by removal of wound crust at subsequent time points. Blood glucose was measured immediately using a Freestyle glucometer (Abbott Laboratories, Abbott Park, IL, USA).
In vivo placental transport studies
At E18.5 dams were anesthetized using Ketaset (Fort Dodge Animal Health, Fort Dodge, IA, USA) plus inactin (Sigma-Aldrich Corp.), a neck incision was made, and the jugular vein was identified and catheterized. A bolus of two radiolabeled tracers (50 μCi/kg [14C]methylaminoisobutyric acid (MeAIB) and 250 μCi/kg [3H]methylglucose; Perkin Elmer, Waltham, MA, USA) was injected into the jugular vein. After 3 min, the dam was sacrificed using an overdose of inactin, a sample of maternal arterial blood was obtained by cardiac puncture, laparotomy was performed, and fetuses were killed by decapitation. Fetuses and placentas were removed, weighed, and solubilized for 4 h in Biosol (National Diagnostics, Atlanta, GA, USA). Radioactivity in maternal plasma and solubilized placentas and fetuses was determined by beta counting. The maternofetal unidirectional clearance (Kmf; μl·min−1·g−1) for each tracer was calculated as follows:
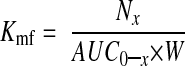 |
where Nx is counts in the fetus taken at time x when the mother was killed (dpm), AUC0–x is the area under the maternal isotope concentration curve from time 0 to time of sacrificing the mother (dpm·min·μl−1) (25), and W is the wet weight of the placenta (g).
Data presentation and statistics
Data are presented as means ± se. For fetal and placental data, a mean for each litter was calculated, and this litter mean was used in subsequent statistical analysis. Thus, n represents the number of litters. Statistical significance of differences between the C and HF groups was assessed using an unpaired Student’s t test. Pearson correlations were used for multivariate analysis. P < 0.05 was considered significant.
RESULTS
There were no statistically significant differences in maternal weight at the time of mating (data not shown). Mice fed the HF diet had a higher daily calorie intake than those fed the C diet (Table 1). At E18.5, HF-fed dams were 12% heavier than dams fed the C diet; however, there was no difference in maternal weight after removal of fetuses by cesarean section (Table 1). The total weight of the maternal fat pads was increased by 7% in animals fed the HF diet (Table 1). At E18.5 maternal adiponectin levels were reduced by 35% in HF-fed dams, and leptin levels were significantly increased by >80% in the animals fed the HF diet (Table 1). No differences were seen in maternal levels of IL-6 (Table 1), and TNF-α levels were below the level of detection in all animals. Maternal HbA1c (Table 1) and fasting blood glucose (Fig. 1) were similar in the two groups, indicating that HF-fed animals did not develop diabetes. Maternal nonfasting insulin levels on E18.5 were similar in the two diet groups (Table 1). The blood glucose response to an intraperitoneal glucose load was similar in the control and HF-fed dams (Fig. 1).
TABLE 1.
Maternal weights and metabolic parameters at E18.5
| Parameter | C diet | HF diet | Significancea |
|---|---|---|---|
| Daily calorie intake (kcal) | 14.82 ± 0.25 | 16.76 ± 0.21 | P < 0.01 (9) |
| Body weight (g) | 28.7 ± 0.22 | 32.1 ± 0.33 | P < 0.05 (21) |
| Post-cesarean section weight (g) | 23.1 ± 0.19 | 23.7 ± 0.23 | NS (21) |
| Fat pad weight (g) | 0.87 ± 0.01 | 0.93 ± 0.02 | P < 0.05 (10) |
| Serum adiponectin (μg/ml) | 69.7 ± 5.3 | 45.0 ± 2.4 | P < 0.01 (11) |
| Serum leptin (ng/ml) | 2.45 ± 0.29 | 4.78 ± 0.27 | P < 0.05 (10) |
| Serum IL-6 (pg/ml) | 14.7 ± 4.2 | 13.6 ± 3.4 | NS (6) |
| Serum insulin (ng/ml) | 0.73 ± 0.22 | 0.71 ± 0.08 | NS (10) |
| HbA1c (%) | 5.94 ± 0.28 | 5.76 ± 0.37 | NS (8) |
Values are means ± se. C and HF diets were fed to female mice 8 wk before and throughout gestation. All serum measurements were nonfasting. aStatistical significance was determined using an unpaired Student’s t test; P < 0.05 was considered significant. Values in parentheses indicate n/diet group.
Figure 1.

Maternal glucose tolerance test at E18.5 after a 6-hour fast. Values are means ± se; n = 5/diet. C (○) and HF diets (▪) were given to female mice 8 wk before and throughout gestation. An i.p. injection of 2 g/kg of a 30% glucose solution was given, and blood glucose measurements from the tail vein at time points are shown. No significant difference was seen between the two diet groups.
Maternal lipid status was studied in both serum and red blood cells. HF-fed dams had 44% higher serum triglyceride levels and 19% lower serum concentrations of nonesterified fatty acids than dams fed the C diet but similar cholesterol levels (Table 2). Despite no change in total serum lipid levels, the relative percentages of stearic, oleic, and linoleic acids in red blood cells were altered in HF-fed dams (Table 2). Total n-6 content was also significantly reduced (P<0.05, n=6) in the animals fed the HF diet (33.8±0.48%) compared with those fed the C diet (37.3±0.42%).
TABLE 2.
Maternal serum and red blood cell membrane lipids at E18.5
| Parameter | C diet | HF diet | Significancea |
|---|---|---|---|
| Serum lipids (mg/dl) | 137 ± 8.3 | 142 ± 7.9 | NS (8) |
| Serum triglycerides (mg/dl) | 45.8 ± 4.21 | 65.8 ± 4.6 | P < 0.05 (8) |
| Serum nonesterified fatty acids (mEq/L) | 0.59 ± 0.03 | 0.48 ± 0.03 | P < 0.05 (8) |
| Serum cholesterol (mg/dl) | 85.4 ± 8.48 | 81.0 ± 4.3 | NS (8) |
| Red blood cell stearic acid (%) | 14.8 ± 0.21 | 16.9 ± 0.32 | P < 0.01 (6) |
| Red blood cell oleic acid (%) | 14.0 ± 0.42 | 11.8 ± 0.23 | P < 0.01 (6) |
| Red blood cell linoleic acid (%) | 6.23 ± 0.09 | 10.8 ± 0.16 | P < 0.01 (6) |
Values are means ± se. C and HF diets were fed to female mice 8 wk before and throughout gestation. All serum measurements were nonfasting. aStatistical significance was determined using an unpaired Student’s t test; P < 0.05 was considered significant. Values in parentheses indicate n/diet group.
Fetal growth was markedly stimulated in response to the HF diet, resulting in 43% higher fetal weights at E18.5 in the HF-fed group than in the control group (Fig. 2A). There was no significant difference in average litter size between the HF (8.2±0.7) and C groups (8.3±1.1), resulting in an increase in total litter weight in the HF-fed dams (Fig. 2B). Placental growth was not affected by the HF diet (Fig. 2C). There was no significant correlation between maternal weight after cesarean section and fetal weight in either the C or HF groups (data not shown).
Figure 2.

Fetal, placental, and total litter weights at E18.5. Values are means ± se. C and HF diets were given to female mice 8 wk before and throughout gestation. P < 0.05 was considered significant; unpaired Student’s t test; n ≥ 18 litters/diet.
In vivo transport studies demonstrated marked increases in placental clearances of both [3H]methylglucose and [14C]MeAIB in response to the HF diet (Fig. 3). The unidirectional maternofetal clearance of [3H]methylglucose was increased by 5-fold and the clearance of [14C]MeAIB was increased by 10-fold in the HF group compared with the C group. The increase in placental clearance rates was associated with a significant increase in GLUT1 expression (5-fold) in isolated placental MVMs from animals fed the HF diet (Figs. 4 and 5). In addition, SNAT2 protein expression was significantly increased (9-fold) in MVMs from animals fed the HF diet (Figs. 4 and 5). In contrast, MVM protein expression of SNAT4 was unaffected by the HF diet (Fig. 4). GLUT3 expression was measured in total placental homogenate; however, no difference was found between the two diet groups (Fig. 5).
Figure 3.

Placental nutrient transport capacity in vivo at E18.5. Unidirectional maternal-fetal clearances (Kmf) for methylglucose and MeAIB were measured in anesthetized dams (n=5/diet). Values are means± sem. C and HF diets were fed to female mice 8 wk before and throughout gestation. *P < 0.05, **P < 0.001; unpaired Student’s t test.
Figure 4.

Protein expression of placental nutrient transporters at E18.5. C and HF diets were given to female mice 8 wk before and throughout gestation. Representative Western blots are shown for GLUT1, SNAT2, SNAT4, and β-actin in isolated MVM and GLUT3 and β-actin in homogenates of pooled placentas from animals fed either the C or HF diet.
Figure 5.

Protein expression of placental nutrient transporters at E18.5. Summary of densitometry of SNAT 2 and GLUT 1 band (normalized for β-actin). Values are means ± sem; n = 7 (pooled placentas from 7 litters for each diet). No difference was seen in SNAT4 or GLUT3 expression. *P < 0.05, **P < 0.001; unpaired Student’s t test.
DISCUSSION
Maternal overweight and obesity in pregnancy often results in fetal overgrowth, which increases the risk of the baby developing metabolic syndrome later in life. The mechanisms underlying fetal overgrowth in overweight or obese pregnant women without diabetes are unknown, and the lack of relevant animal models has hampered progress in this area. We describe a novel mouse model in which a moderately HF diet results in increased maternal adiposity, normal glucose tolerance, elevated maternal serum leptin and triglycerides, decreased serum adiponectin concentrations, and a marked increase in fetal growth. The most significant novel finding in this study is that a HF diet resulted in markedly increased placental transport of glucose and neutral amino acids, mediated by an up-regulation of the protein expression of specific nutrient transporter isoforms in the placental barrier. We propose that the increase in placental transfer of nutrients constitutes one of the mechanisms underlying fetal overgrowth in pregnancies complicated by increased dietary fat intake and/or overweight or obesity.
There are a number of reports in the literature describing animal models of overnutrition in pregnancy, but in most cases these interventions have not resulted in fetal overgrowth. In the sheep, increased maternal nutrition in late gestation has been shown to alter the appetite-regulating network in the fetal brain (26) and the gene expression of adiponectin and leptin in fetal adipose tissue (27). However, this overnutrition model does not result in fetal overgrowth (26), and maternal metabolism and adiposity in response to overnutrition were not investigated (26, 27). Offspring of pregnant rats fed a diet with a HF or increased cholesterol content developed abnormal glucose metabolism (28, 29) and serum lipid profiles (28, 30) and increased leptin (29) and adiposity (28, 31). Pregnant rats fed a cafeteria-style diet have increased adiposity and develop GDM (32). In the study by Srinivasan et al. (33) pregnant rats fed a HF diet were obese and developed diabetes and their offspring developed metabolic syndrome after birth; however, fetal weights did not differ between diet groups.
Mouse models with moderate to pronounced obesity have been developed and include leptin (34) and leptin receptor (35, 36) knockouts and those with adipose tissue-specific overexpression of 11β-hydroxysteroid dehydrogenase type 1 (37). These models are of limited use for the study of obesity in pregnancy as they are associated with infertility and/or diabetes. Female C57BL/6J mice fed a HF diet (60% of caloric intake as fat) before pregnancy developed obesity, and their offspring developed obesity and type 2 diabetes (38). However, no information was provided on fetal growth, and mothers were fed a control diet throughout the periconceptual and gestational periods, resulting in significant weight loss. Samuelsson et al. (39) demonstrated that the offspring of mice fed a high-fat and high-sugar diet developed cardiovascular and metabolic dysfunction and adult adiposity. Although the maternal metabolic environment in this model resembled the human condition, including increased leptin and insulin levels, fetal overgrowth was not reported (39). Han et al. (40) demonstrated that dams with the agouti yellow modification were moderately obese in pregnancy but did not develop overt diabetes and gave birth to larger pups, but the study did not address placental involvement.
In the current study, mice fed a moderately high-fat diet had increased adiposity but did not develop diabetes as evidenced by normal HbA1c values, fasting blood glucose, and glucose tolerance test results. Maternal serum concentrations of leptin and triglycerides were increased, and adiponectin levels were decreased in response to a HF diet, resembling the metabolic profile in overweight and obese subjects in both the nonpregnant and pregnant states (3, 41). Furthermore, in response to the HF diet, fetal growth was increased in our mouse model similar to the fetal overgrowth typically associated with overweight and obesity in pregnant women (9). However, dams fed the HF diet in our study were not obese and nonfasting insulin was not elevated, indicating that any insulin resistance present in the HF-fed animals was unlikely to be pronounced. Furthermore, HF-fed mice had an increased intake of fat but not of carbohydrates or total energy. This result is in contrast to findings in overweight and obese pregnant women, who typically have elevated fasting insulin (3) and increased intake of fat, carbohydrates, and total energy (10). The mouse model described in this report therefore provides the opportunity to explore the effect of a HF diet per se on maternal hormones, placental nutrient transport, and fetal growth, without the influence of overt obesity, insulin resistance, or increased intake of total energy and carbohydrates. We found that a diet rich in fat had profound effects on maternal serum triglyceride, leptin, and adiponectin levels and caused a marked up-regulation of placental nutrient transporters, resulting in fetal overgrowth. It remains to be established whether the fat content in the diet is also of particular importance for fetal overgrowth in overweight and obese pregnant women. If this is the case, it may be speculated that a change in dietary composition in these women may be effective in alleviating fetal overgrowth.
As expected for animals fed a HF diet, maternal serum triglyceride levels were significantly increased. Increases in erythrocyte membrane linoleic acid content is reflective of the increase in linoleic acid content of the HF compared with the C diet, and this measure has been previously used as a marker of dietary intake of linoleic acid (42). Studies suggested that the fatty acid composition of the erythrocyte, specifically the arachidonic component and the total n-6 acid component, is inversely related to fasting serum insulin levels and therefore indicative of insulin resistance (43). In our study, total n-6 fatty acid erythrocyte levels were slightly reduced in HF-fed dams. This finding is compatible with relatively normal insulin sensitivity in our model. Despite increased dietary intake of oleic acid in HF-fed dams, erythrocyte membrane oleic acid content was decreased. The underlying mechanisms for this change and the decreased serum nonesterified fatty acid levels in our HF diet animals remain to be established; however, it may suggest increased utilization of maternal stores to supply the fetus.
Babies of women with increased BMI are often large at birth (3, 4, 8). The mechanisms underlying fetal overgrowth in response to increased maternal adiposity and high dietary fat intake have not previously been investigated, and this is the first study to identify increased placental nutrient transport as one such mechanism. In addition, we show that these changes in our animal model are likely to be due to increased protein expression of system A amino acid and glucose transporter isoforms in the placental barrier. Our data do not unequivocally demonstrate that the up-regulated placental nutrient transport is the cause of fetal overgrowth, because the possibility that these changes are due to the increased fetal growth rate cannot be excluded. Further studies are required to address this question definitely. However, we recently provided evidence that in maternal protein deprivation in the rat, a model of intrauterine growth restriction and fetal programming, placental transport changes occurred before reductions in fetal size (13). These findings are compatible with the hypothesis that placental transport changes are a cause, rather than a consequence, of altered fetal growth.
The increase in placental nutrient transport and fetal growth in response to a HF diet in our mouse model is reminiscent of our previous reports in pregnant women in which fetal overgrowth in women with type 1 diabetes was associated with increased glucose transporter activity and GLUT 1 expression (13) and increased system A amino acid transporter activity in isolated syncytiotrophoblast plasma membranes (16). To identify transporter isoforms responsible for the increased placental nutrient transport we studied the protein expression of transporters for glucose and neutral amino acids. In the mouse GLUT1 (SLC2A1) and GLUT3 (SLC2A3) are the primary glucose transporter isoforms expressed in the labyrinthine zone, which represents the part of the placenta where maternal-fetal nutrient transfer occurs (44), providing the rationale for studying GLUT1 and GLUT3 protein expression. System A amino acid transport is mediated by the three isoforms, SNAT1 (SLC38A1), SNAT2 (SLC38A2), and SNAT4 (SLC38A4), which are all expressed in human (45) and rodent placenta (46). However, we limited our protein expression studies to SNAT2 and SNAT4, as our SNAT1 antibody failed to identify a specific band in mouse placental MVMs.
We found a striking specificity in the up-regulation of placental nutrient transporters in response to a HF diet when the protein expression of placental GLUT1 and SNAT2 was markedly increased whereas the expression of GLUT3 and SNAT4 was unaltered. Differential regulation of placental GLUT1 and GLUT3 has been reported previously in response to various perturbations in the mouse. For example, in thioredoxin-1-overexpressing mice placental oxidative stress is reduced, which was shown to be associated with increased protein expression of placental GLUT1 but not GLUT3 (47). The effect of experimental diabetes in pregnant mice on placental glucose transporter expression is variable in that increased (44), unaltered (48), and decreased (49) placental GLUT3 or GLUT1 protein expression has been reported. On the other hand, maternal undernutrition in pregnant mice causes a decrease in the protein expression of GLUT3 but not of GLUT1 in the placenta (50). Collectively, these data in the mouse suggest that perturbations in the maternal compartment have distinct effects on the regulation of placental glucose transporter isoform expression. With respect to the regulation of placental SNAT isoforms no comparable data are available in the mouse or in the human. Interestingly, preliminary data from our laboratory show that the system A activity and protein expression of SNAT2,but not of SNAT1 or SNAT4 are up-regulated in MVMs isolated from placentas obtained from pregnancies of obese women giving birth to large babies (N. Jansson, personal communication, January 18, 2008). Thus, with respect to the effect on placental nutrient transporter isoforms, there appear to be distinct similarities between HF-fed mice and obese pregnant women.
The factors linking a maternal HF diet and increased adiposity to up-regulation of SNAT 2 and GLUT 1 protein expression in the placental barrier remain to be elucidated; however, maternal metabolic hormones are likely candidates (51, 52). For example, leptin has been shown to stimulate system A amino acid transport activity in isolated term placental villous fragments (21), and it is possible that the increased circulating maternal leptin levels in the HF-fed dams represent one mechanism for increasing placental nutrient transport. Adiponectin is thought to be anti-inflammatory and increases the sensitivity of tissues to insulin, and lower levels may lead to insulin resistance (53). However, in studies of placental tissue, adiponectin appears to be proinflammatory (54), stimulating the release of proinflammatory cytokines. Very little is known regarding the regulation of placental nutrient transport by adiponectin. However, infusion of adiponectin to pregnant rats has been reported to decrease the mRNA expression of glucose transporter GLUT3 and lipoprotein lipase in the placenta (55). Therefore, the reduced levels of maternal serum adiponectin in our model may lead to increased placental nutrient transport.
It is of interest to compare our model of overnutrition in pregnant mice with a recently published elegant study in which the effect of an obesogenic diet (16% fat and 33% sugar), given to female mice before mating and throughout pregnancy and lactation, on offspring metabolism and cardiovascular function was studied at 3 and 6 months of age (39). In this model, the caloric intake was significantly greater, abdominal fat mass was 4-fold higher, and leptin and insulin levels were increased at E18 in dams fed the obesogenic diet compared with control dams. The composition of the diets providing the overnutrition in these two studies is likely to explain the differences in metabolic outcome in the dams. Of note, however, fetal overgrowth was not reported in the mouse model of Samuelsson et al. (39) but led to increased offspring weight at 5 wk of age.
Our novel mouse model offers the potential for further studies into the effects of maternal HF diet and increased adiposity on placental function, fetal growth, and metabolic and cardiovascular function in the offspring. We suggest that this model will be particularly valuable to further elucidate the mechanisms underlying fetal overgrowth, which is the characteristic outcome in overweight and obese women, and test interventions targeting these mechanisms. Future studies of mice fed an obesogenic diet, which results in increased intake of fat, carbohydrates, and total energy as well as maternal obesity (39), will provide additional information on the maternal factors that determine placental nutrient transport function and fetal growth. Furthermore, to determine whether the marked effects of the HF diet on placental transport functions and fetal growth in the current study are specific to the inbred strain used, it will be interesting to perform similar studies in an outbred strain of mice. Successful strategies to alleviate fetal overgrowth in women with increased BMI have the potential to reduce the numbers of children susceptible to the development of obesity and the metabolic syndrome in childhood and later life.
Acknowledgments
We are indebted to Dr. Jo Glazier for sharing her expertise in isolating MVM vesicles from mouse placenta. We thank the staff at the Mouse Metabolic Phenotyping Center (DK59630), University of Cincinnati, for their assistance and the University of Cincinnati Millennium Scholars Program for funding this project. This project was also supported by award R03HD058032 and in part by HD058032 (T.J.) and HD34089 (L.A.W.) from the National Institute of Child Health and Human Development. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Child Health and Human Development or the National Institutes of Health. We are indebted to Tamara Hagen for technical assistance. Abstracts including work from this study were presented at the Society for Gynecological Investigation Meeting 2008 and the Aspen Perinatal Symposium 2007.
References
- Ananth C V, Wen S W. Trends in fetal growth among singleton gestations in the United States and Canada, 1985 through 1998. Semin Perinatol. 2002;26:260–267. doi: 10.1053/sper.2002.34772. [DOI] [PubMed] [Google Scholar]
- Boney C M, Verma A, Tucker R, Vohr B S. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes. Pediatrics. 2005;115:e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- Catalano P M, Ehrenberg H M. The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG. 2006;113:1126–1133. doi: 10.1111/j.1471-0528.2006.00989.x. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association Type 2 diabetes in children and adolescents. Diabetes Care. 2000;23:381–389. doi: 10.2337/diacare.23.3.381. [DOI] [PubMed] [Google Scholar]
- Perlow J H, Morgan M A, Montgomery D, Towers C V, Porto M. Perinatal outcome in pregnancy complicated by massive obesity. Am J Obstet Gynecol. 1992;167:958–962. doi: 10.1016/s0002-9378(12)80019-6. [DOI] [PubMed] [Google Scholar]
- Sebire N J, Jolly M, Harris J P, Wadsworth J, Joffe M, Beard R W, Regan L, Robinson S. Maternal obesity and pregnancy outcome: a study of 287,213 pregnancies in London. Int J Obes Relat Metab Disord. 2001;25:1175–1182. doi: 10.1038/sj.ijo.0801670. [DOI] [PubMed] [Google Scholar]
- Jansson N, Nilsfelt A, Wennergren M, Rossander-Hulthen L, Powell T L, Jansson T. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am J Clin Nutr. 2008;87:1743–1749. doi: 10.1093/ajcn/87.6.1743. [DOI] [PubMed] [Google Scholar]
- Cnattingius S, Bergstrom R, Lipworth L, Kramer M S. Prepregnancy weight and the risk of adverse pregnancy outcomes. N Engl J Med. 1998;338:147–152. doi: 10.1056/NEJM199801153380302. [DOI] [PubMed] [Google Scholar]
- Ogden C L, Caroll M D, Curtin L R, McDowell M A, Tabak C J, Flegal K M. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- Ehrenberg H M, Mercer B M, Catalano P M. The influence of obesity and diabetes on the prevalence of macrosomia. Am J Obstet Gynecol. 2004;191:964–968. doi: 10.1016/j.ajog.2004.05.052. [DOI] [PubMed] [Google Scholar]
- Catalano P M. Obesity and pregnancy—the propagation of a viscous cycle? J Clin Endocrinol Metab. 2003;88:3505–3506. doi: 10.1210/jc.2003-031046. [DOI] [PubMed] [Google Scholar]
- Jansson T, Powell T L. Human placental transport in altered fetal growth: does the placenta function as a nutrient sensor?—a review. Placenta. 2006;27:91–97. doi: 10.1016/j.placenta.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Jansson N, Pettersson J, Haafiz A, Ericsson A, Palmberg I, Tranberg M, Ganapathy V, Powell T L, Jansson T. Down-regulation of placental transport of amino acids precedes the development of intrauterine growth restriction in rats fed a low protein diet. J Physiol. 2006;576:935–946. doi: 10.1113/jphysiol.2006.116509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson T, Wennergren M, Powell T L. Placental glucose transport and GLUT 1 expression in insulin dependent diabetes. Am J Obstet Gynecol. 1999;180:163–181. doi: 10.1016/s0002-9378(99)70169-9. [DOI] [PubMed] [Google Scholar]
- Jansson T, Ekstrand Y, Wennergren M, Powell T L. Placental glucose transport in gestational diabetes. Am J Obstet Gynecol. 2001;184:111–116. doi: 10.1067/mob.2001.108075. [DOI] [PubMed] [Google Scholar]
- Jansson T, Ekstrand Y, Björn C, Wennergren M, Powell T L. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes. 2002;51:2214–2219. doi: 10.2337/diabetes.51.7.2214. [DOI] [PubMed] [Google Scholar]
- Kuruvilla A G, D'Souza S W, Glazier J D, Mahendran D, Maresh M J, Sibley C. Altered activity of the system A amino acid transporter in microvillous membrane vesicles from placentas of macrosomic babies born to diabetic women. J Clin Invest. 1994;94:689–695. doi: 10.1172/JCI117386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay J E, Ferrell W R, Crawford L, Wallace A M, Greer I A, Sattar N. Maternal obesity is associated with dysregulation of metabolic. vascular and inflammatory pathways. J Clin Endocrinol Metab. 2006;87:4231–4237. doi: 10.1210/jc.2002-020311. [DOI] [PubMed] [Google Scholar]
- Hendler I, Blackwell S C, Metha S H, Whitty J E, Russell E, Sorokin Y, Cotton D B. The levels of leptin, adiponectin, and resistin in normal weight, overweight, and obese pregnant women with and without preeclampsia. Am J Obstet Gynecol. 2005;193:979–983. doi: 10.1016/j.ajog.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Karl P I, Alpy K L, Fischer S E. Amino acid transport by the cultured human placental trophoblast: effect of insulin on AIB transport. Am J Physiol. 1992;262:C834–C839. doi: 10.1152/ajpcell.1992.262.4.C834. [DOI] [PubMed] [Google Scholar]
- Jansson N, Greenwood S, Johansson B R, Powell T L, Jansson T. Leptin stimulates system A activity in human placental villous fragments. J Clin Endocrinol Metab. 2003;88:1205–1211. doi: 10.1210/jc.2002-021332. [DOI] [PubMed] [Google Scholar]
- Ericsson A, Hamark B, Powell T L, Jansson T. Glucose transporter isoform 4 is expressed in the syncytiotrophoblast of first trimester human placenta. Hum Reprod. 2005;20:521–530. doi: 10.1093/humrep/deh596. [DOI] [PubMed] [Google Scholar]
- Bowers G N, Jr, McComb R B. A continuous spectrophotometric method for measuring the activity of serum alkaline phosphatase. Clin Chem. 1966;12:70–89. [PubMed] [Google Scholar]
- Ling R, Bridges C C, Sugawara M, Fujita T, Leibach F H, Prasad P D, Ganapathy V. Involvement of transporter recruitment as well as gene expression in the substrate-induced adaptive regulation of amino acid transport system A. Biochim Biophys Acta. 2001;1512:15–20. doi: 10.1016/s0005-2736(01)00310-8. [DOI] [PubMed] [Google Scholar]
- Constancia M, Hemberger M, Hughes J, Dean W, Ferguson-Smith A C, Fundele R, Stewart F, Kelsley G, Fowden A, Sibley C, Reik W. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417:945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- Muhlhausler B S, Adam C L, Findlay P A, Duffield J A, McMillen I C. Increased maternal nutrition alters development of the appetite-regulating network in the brain. FASEB J. 2006;20:E556–E565. doi: 10.1096/fj.05-5241fje. [DOI] [PubMed] [Google Scholar]
- Muhlhausler B S, Duffield J A, McMillen I C. Increased maternal nutrition stimulates peroxisome proliferator activated receptor-γ, and leptin messenger ribonucleic acid expression in adipose tissue before birth. Endocrinology. 2007;148:878–885. doi: 10.1210/en.2006-1115. [DOI] [PubMed] [Google Scholar]
- Guo F, Jen K L. High-fat feeding during pregnancy and lactation affects offspring metabolism in rats. Physiol Behav. 1995;57:681–686. doi: 10.1016/0031-9384(94)00342-4. [DOI] [PubMed] [Google Scholar]
- Taylor P D, McConnell J, Khan I Y, Holemans K, Lawrence K M, Asare-Anane H, Persaud S J, Jones PM, Petrie L, Hanson M A, Poston L. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol. 2005;288:R234–R239. doi: 10.1152/ajpregu.00355.2004. [DOI] [PubMed] [Google Scholar]
- Karnik H B, Sonawane B R, Adkins J S, Mohla S. High dietary fat feeding during perinatal development of rats alters hepatic drug metabolism of progeny. Dev Pharmacol Ther. 1989;14:135–140. [PubMed] [Google Scholar]
- Khan I Y, Dekou V, Douglas G, Jensen R, Hanson M A, Poston L, Taylor PD. A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am J Physiol Regul Integr Comp Physiol. 2004;288:R127–R133. doi: 10.1152/ajpregu.00354.2004. [DOI] [PubMed] [Google Scholar]
- Holemans K, Caluwaerts S, Poston L, Van Assche F A. Diet-induced obesity in the rat: a model for gestational diabetes mellitus. Am J Obstet Gynecol. 2004;190:858–865. doi: 10.1016/j.ajog.2003.09.025. [DOI] [PubMed] [Google Scholar]
- Srinivasan M, Katewa S D, Palaniyappan A, Pandya J D, Patel M S. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab. 2006;291:E792–E799. doi: 10.1152/ajpendo.00078.2006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman J M. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;479:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Chen H, Charlat O, Tartaglia L A, Woolf E A, Weng X, Ellis S J, Lakey N D, Culpepper J, Moore K J, Breitbart R E, Duyk G M, Tepper R I, Morgenstern J P. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- Tartaglia L A, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards G J, Campfield L A, Clark F T, Deeds J, Muir C, Sanker S, Moriarty A, Moore K J, Smutko J S, Mays G G, Wool E A, Monroe C A, Tepper R I. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:12163–12171. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Paterson J, Shinyama H, Morton N M, Mullins J J, Seckl J R, Flier J S. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- Gallou-Kabani C, Vige A, Gross M S, Boileau C, Rabes J P, Fruchart-Najib J, Jais J P, Junien C. Resistance to high-fat diet in the female progeny of obese mice fed a control diet during the periconceptual, gestation, and lactation periods. Am J Physiol Endocrinol Metab. 2007;292:E1095–E1000. doi: 10.1152/ajpendo.00390.2006. [DOI] [PubMed] [Google Scholar]
- Samuelsson A-M, Matthews P A, Argenton M, Christie M R, McConnell J M, Jansen E H J M, Piersma A H, Ozanne S E, Fernandez Twinn D, Remacle C, Rowlerson A, Poston L, Taylor P D. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension and insulin resistance. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- Han J, Xu J, Epstein P N, Lui Y Q. Long-term effect on maternal obesity on pancreatic β cells of offspring: reduced β cell adaptation to high glucose and high-fat diet challengers in adult female mouse offspring. Diabetologia. 2005;48:1810–1818. doi: 10.1007/s00125-005-1854-8. [DOI] [PubMed] [Google Scholar]
- Sattar N, Greer I A, Pirwani I, Gibson J, Wallace A M. Leptin levels in pregnancy: marker for fat accumulation and mobilization? Acta Obstet Gynecol Scand. 1998;77:278–283. [PubMed] [Google Scholar]
- Sarkkinen E S, Agren J J, Ahola I, Ovaskainen M L, Uusitupa M I. Fatty acid composition of serum cholesterol esters, and erythrocyte and platelet membranes as indicators of long term adherence to fat modified diets. Am J Clin Nutr. 1994;59:364–370. doi: 10.1093/ajcn/59.2.364. [DOI] [PubMed] [Google Scholar]
- Clifton P M, Nestel P J. Relationship between plasma insulin and erythrocyte fatty acid composition. Prostaglandins Leukot Essent Fatty Acids. 1998;59:191–194. doi: 10.1016/s0952-3278(98)90062-x. [DOI] [PubMed] [Google Scholar]
- Boileau P, Mrejen C, Girard J, Hauguel-de Mouzon S. Overexpression of GLUT3 placental glucose transporter in diabetic rats. J Clin Invest. 1995;96:309–317. doi: 10.1172/JCI118036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desforges M, Lacey H A, Glazier J D, Greenwood S L, Mynett K J, Speake P F, Sibley C P. SNAT4 isoform of system A amino acid transporter is expressed in human placenta. Am J Physiol Cell Physiol. 2006;290:C305–C312. doi: 10.1152/ajpcell.00258.2005. [DOI] [PubMed] [Google Scholar]
- Novak D, Lehman M, Bernstein H, Beveridge M, Cramer S. SNAT expression in rat placenta. Placenta. 2006;27:510–516. doi: 10.1016/j.placenta.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Umekawa T, Sugiyama T, Kihira T, Murabayashi N, Zhang L, Nagao K, Kamimoto Y, Ma N, Yodoi J, Sagawa N. Overexpression of thioredoxin-1 reduces oxidative stress in the placenta of transgenic mice and promotes fetal growth via glucose metabolism. Endocrinology. 2008;149:3980–3988. doi: 10.1210/en.2007-1682. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaskar S U, Devaskar U P, Schroeder R E, deMello D, Fiedorek F T, Jr, Mueckler M. Expression of genes involved in placental glucose uptake and transport in the nonobese diabetic mouse pregnancy. Am J Obstet Gynecol. 1994;171:1316–1323. doi: 10.1016/0002-9378(94)90154-6. [DOI] [PubMed] [Google Scholar]
- Ogura K, Sakata M, Yamaguchi M, Kurachi H, Murata Y. High concentration of glucose decreases glucose transporter-1 expression in mouse placenta in vitro and in vivo. J Endocrinol. 160:443–452. doi: 10.1677/joe.0.1600443. [DOI] [PubMed] [Google Scholar]
- Lesage J, Hahn D, Leonhardt M, Blondeau B, Breant B, Dupouy J P. Maternal undernutrition during late gestation-induced intrauterine growth restriction in the rat is associated with impaired placental GLUT3 expression, but does not correlate with endogenous corticosterone levels. J Endocrinol. 2002;174:37–43. doi: 10.1677/joe.0.1740037. [DOI] [PubMed] [Google Scholar]
- Jones H N, Powell T L, Jansson T. Regulation of placental transport—a review. Placenta. 2007;28:763–774. doi: 10.1016/j.placenta.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Ericsson A, Hamark B, Jansson N, Johansson B R, Powell T L, Jansson T. Hormonal regulation of glucose and system A amino acid transport in first trimester placental villous fragments. Am J Physiol. 2005;288:R656–R662. doi: 10.1152/ajpregu.00407.2004. [DOI] [PubMed] [Google Scholar]
- Fang X, Palanivel R, Zhou X, Liu Y, Xu A, Wang Y, Sweeney G. Hyperglycemia- and hyperinsulinemia-induced alteration of adiponectin receptor expression and adiponectin effects in L6 myoblasts. J Mol Endocrinol. 2005;35:465–476. doi: 10.1677/jme.1.01877. [DOI] [PubMed] [Google Scholar]
- Lappas M, Yee K, Permezel M, Rice G E. Leptin and adiponectin stimulate the release of proinflammatory cytokines and prostaglandins from human placenta and maternal adipose tissue via nuclear factor-κB, peroxisomal proliferator-activated receptor-γ and extracellularly regulated kinase 1/2. Endocrinology. 2005;146:3334–3342. doi: 10.1210/en.2005-0406. [DOI] [PubMed] [Google Scholar]
- Caminos J E, Nogueiras R, Gallego R, Bravo S, Tovar S, García-Caballero T, Casanueva F F, Diéguez C. Expression and regulation of adiponectin and receptor in human and rat placenta. J Clin Endocrinol Metab. 2005;90:4276–4286. doi: 10.1210/jc.2004-0930. [DOI] [PubMed] [Google Scholar]