

Abstract
An investigation of the use of Cp2Zr(H)Cl (Schwartz’s reagent) to reduce a variety of amides to the corresponding aldehydes under very mild reaction conditions and in high yields is reported. A range of tertiary amides, including Weinreb’s amide, can be converted directly to the corresponding aldehydes with remarkable chemoselectivity. Primary and secondary amides proved to be viable substrates for reduction as well, although the yields were somewhat diminished compared to the corresponding tertiary amides. Results from NMR experiments suggested the presence of a stable, 18-electron zirconacycle intermediate that presumably affords the aldehyde upon water or silica gel workup. A series of competition experiments revealed a preference of the reagent for substrates in which the lone pair of the nitrogen is electron releasing and thus more delocalized across the amide bond by resonance. This trend accounts for the observed excellent selectivity for tertiary amides versus esters. Experiments regarding the solvent dependence of the reaction suggested a kinetic profile similar to that postulated for the hydrozirconation of alkenes and alkynes. Addition of p-anisidine to the reaction intermediate resulted in the formation of the corresponding imine mimicking the addition of water that forms the aldehyde.
Introduction
The amide group is a robust functionality that is fairly inert to many oxidative and reductive conditions.1 While the simple reduction of amides with metal hydride reagents has been known since the late 1940s,2 many of these methods reduce this group to the corresponding alcohol and amine; several of which proceed in high yields under fairly mild conditions.3 However, controlling the reduction of an amide to the aldehyde oxidation state has proven more difficult, typically requiring substrate specific conditions to achieve reliable yields of the aldehyde products.4,5
In general, the reduction of amides to aldehydes using common commercially available metal hydrides provides the aldehydes in poor to moderate yields with isolation of the corresponding alcohols and amines as by-products in many of the cases.6 Early investigations demonstrated that the reaction of tertiary amides with lithium aluminum hydride generally furnished a mixture of the corresponding amine and the alcohol.2,7 It was then found that a reverse order of addition of the reagents (addition of the hydride to the amide) resulted in the formation of a significant amount of the corresponding aldehydes.7 It was further observed that an increase in the size of the substituents at the amide nitrogen provided a higher yield of the aldehyde. Monosubstituted amides and N,N-dimethylbenzamide did not react under these conditions or gave low yields of the aldehydes. Many other methods have been reported since and include reductions with LiAlH(OEt)3 and LiAlH2(OEt)2, 8,9, NaAlH2(OCH2CH2OMe)2 10 and other variously substituted aluminum hydride-based reagents,11 dissolving metals,12 L-Selectride and alkyl trifluoromethanesulphonate,13 substituted borohydride reagents,14 a SmI2-acid system,15 and a procedure based on a POCl3-mediated conversion to a Vilsmeier complex, followed by treatment with zinc dust and then water.16 Of these methods, the LiAlH(OEt)3 procedure has been the most successful in terms of generality and yield for the reduction of a variety of N,N-dimethyl and N,N-diethyl amides to the corresponding aldehydes. However, large N-substituents hinder this reaction as N,N-diisopropyl-n-butyric amide was unreactive under the reported conditions.8 Furthermore, very little information is available for this reaction with respect to functional group tolerance and selectivity. Brown has reported that the reduction of N,N-dimethyl-4-nitrobenzamide with LiAlH(OEt)3 afforded 75% of the desired aldehyde while observing partial reduction of the nitro group. This suggests that chemoselectivity could be problematic in amide reductions with this reagent in the presence of more electrophilic functionalities. Cha has reported the synthesis of aldehydes from primary and tertiary amides using lithium tris(dialkylamino)aluminum hydrides17 and dipyrrolidinoaluminum hydrides.18 These reactions furnished good yields of aldehydes but showed significant reactivity with accompanying nitro substituents. Borohydrides have been much less studied in this regard, however, Sia2BH (diisoamylborane) has been successful in the reduction of N,N-dimethyl and -diethyl amides.14 Unfortunately, there was no investigation into functional group compatibility with this reagent.
More recently there have been investigations into specialized amide derivatives to cleanly afford the aldehyde products. Generally, those in which the nitrogen lone pair competes for localization within the amide bond tend to give higher yields of the corresponding aldehydes. Specifically, N-acylcarbazoles,19 N-acylimidazoles,20 1-acylaziridines,21,22 1-acyl-3,5-dimethylpyrazoles,23 N-methylanilides,24 morpholine amides,25 3-acylthiazolidine-2-thiones,26 and an N-[2-(dimethylamino)ethyl]-N-methyl amide27 reacted with aluminum hydride reagents to give higher aldehyde yields. Of course the most notable specialized amide is the N,O-dimethylamide (Weinreb amide).4 It is presumed that the presence of the oxygen atom allows for selective association with the metal forming a stable five-membered chelate preventing further reduction. Hydrolysis then affords the aldehyde cleanly and in good yields.
Buchwald has reported a procedure that utilizes a presumed titanium hydride-like species that can efficiently reduce a variety of differentially N-substituted amides to the corresponding aldehyde.14b This method was successful on a variety of substrates including the usually problematic N,N-diisopropyl derived amides and those containing sensitive functionalities such as olefins, alkynes, nitriles, and epoxides. However, this particular transformation has been shown to proceed through an enamine intermediate and therefore is limited to α-enolizable amide substrates.
Thus there still remains a need for a mild method for the reduction of carboxamides to aldehydes that is generally free of alcohol and amine contaminants, is chemoselective, and works on a variety of alkyl and aromatic amide substrates without a dependence on the nature of the nitrogen substituents. We have found that Cp2Zr(H)Cl (1) is effective in this regard.
First reported by Wailes and Weigold, Schwartz’s reagent, Cp2Zr(H)Cl, is a 16-electron, d0 complex with the zirconium at the (+4) oxidation state.28 The one remaining coordination site renders the molecule Lewis-acidic while the absence of a valence-shell filled nonbonding orbital leaves it relatively non-nucleophilic. This empty orbital allows for complexation of the metal to a wide variety of functionalities that contain available nonbonding electron pairs, π-bonds/electrons or in some cases σ-bonds/electrons.29
Pioneering studies by Schwartz and collaborators have made the hydrozirconation of alkenyl and alkynyl substrates one of the most widely used zirconium-mediated reactions because hydrogen can be added stereo- and regioselectively across π-bonds.30 The resulting hydrozirconated products can then react with a variety of electrophiles including protons, halides, and certain carbon moieties or undergo transmetalation reactions.31,32 Hydrozirconation of heteroatomic functionalities are also known and include the reduction of nitriles,33 esters, ketones,34 thioketones,35 aldehydes,36 imines, nitro groups, phosphine oxides and sulfides,37 and secondary amides.38
We have previously communicated a novel procedure for the reduction of tertiary amides to aldehydes using Cp2Zr(H/D)Cl that operates under mild conditions.39,40 These reactions are generally high yielding and remarkably tolerant of many functionalities. Herein, we report an extended scope of this chemistry and the results of our studies aimed at elucidating the reaction mechanism.
Results and Discussion
Extended scope and utility
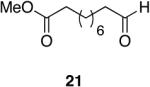
The mild reduction of tertiary amides to aldehydes was efficiently carried out on both aromatic and alkyl substrates with the Schwartz reagent41 as shown in equation 1.42 A variety of substrates that vary in electronic and steric properties were investigated for the reduction and are shown in Table 1.
 |
(1) |
Table 1.
Reduction of N,N-Dialkyl Amides39
| Entry | Amide | Product | Time (min) | Yield (%) |
|---|---|---|---|---|
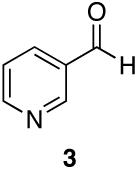
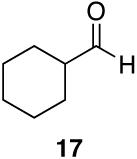
| 1 |  |
 |
15 | 99 |
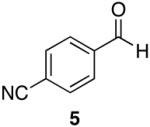
| 2 |  |
 |
30 | 90 |
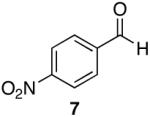
| 3 |  |
 |
30 | 81 |
| 4 |  |
 |
15 | 99 |
| 5 | 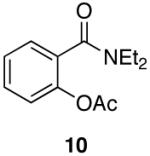 |
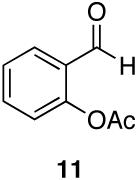 |
15 | 99 |
| 6 | 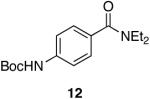 |
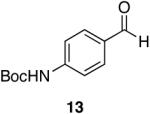 |
5 | 99 |
| 7 |  |
 |
15 | 90 |
| 8 | 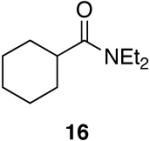 |
 |
15 | 82 |
| 9 | 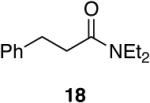 |
 |
15 | 96 |
| 10 |  |
 |
15 | 74 |
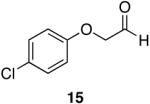
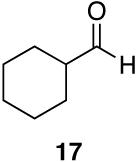
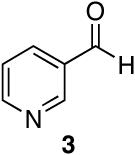
Aromatic, heteroaromatic, and aliphatic amides were efficiently reduced to the corresponding aldehyde in good to excellent yields (Table 1). Entry 1 highlights the ability of heteroaromatic compounds to participate in the reduction without a noticeable effect on the reaction rate or yield due to a potential interaction between zirconium and the pyridine nitrogen. Substitution of the aromatic substrates with electron donating or withdrawing groups did not significantly alter the reaction course (entries 2-7). Exceptional chemoselectivity was observed with fast reaction rates. Nitrile and nitro functionalities were maintained although they can be reduced with other hydride reagents (entries 2 and 3) including LiAlH(OEt)3.8 Remarkably, the tertiary amide was selectively reduced in the presence of an ester group (entries 5 and 10). This selectivity was observed on both aromatic and aliphatic substrates and are the only known general examples of such selectivity to the best of our knowledge.43 Tertiary amides are reduced exclusively in the presence of carbamate groups as is shown in entry 6. Aliphatic amides are viable substrates for this reaction (entries 7-10) highlighting the generality of this reduction.
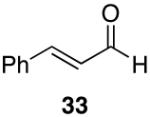
A variety of Weinreb amides were also examined (Table 2). For comparative reasons, many of the same substrates were used as in Table 1. In most cases similar yields, reaction times, and selectivities were observed. Entry 7 shows that aromatic ketones are reduced in addition to the amide functionality, when two equivalents of 1 are used. The cinnamic amide 32 (entry 10) afforded the aldehyde in good yield with the olefin remaining intact, but 12% of the corresponding alcohol was obtained.
Table 2.
Reduction of Weinreb Amides
| Entry | Amide | Product | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | 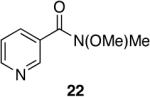 |
 |
15 | 85 |
| 2 |  |
 |
5 | 94 |
| 3 | 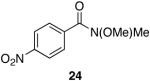 |
 |
5 | 89 |
| 4 | 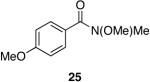 |
 |
5 | 93 |
| 5 |  |
 |
10 | 89 |
| 6 | 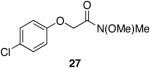 |
 |
10 | 82 |
| 7 | 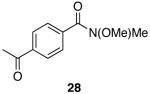 |
 |
20 | 91a |
| 8 |  |
 |
10 | 86 |
| 9 | 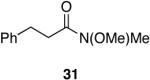 |
 |
20 | 93 |
| 10 | 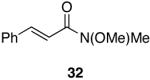 |
 |
20 | 87b |
Two equivalents of 1 were required.
The corresponding alcohol was also obtained (12%).
This protocol was also effective when applied to primary and secondary amides. Reaction times remained the same, but the yields were significantly reduced. It is worthwhile to note that only one equivalent of the reagent was employed for aldehyde formation from aliphatic and aromatic amides. Ganem has reported a procedure that requires two equivalents of Cp2Zr(H)Cl for the reduction of aliphatic secondary amides to the corresponding imines following a non-aqueous workup.38 Thus, this method gives an interesting alternative to the Ganem procedure on the same substrates. Again, functional group selectivity was maintained as shown in Table 3.
Table 3.
Reduction of Primary and Secondary Amides
| Entry | Amide | Product | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 |  |
 |
10 | 86 |
| 2 |  |
 |
10 | 60 |
| 3 |  |
 |
10 | 86 |
| 4 |  |
 |
30 | 60 |
| 5 | 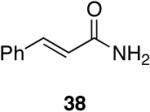 |
 |
5 | 56 |
| 6 |  |
 |
5 | 62 |
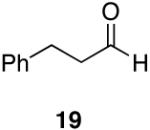
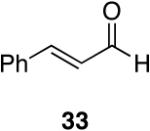
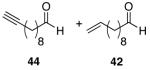
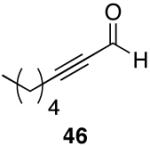
Alkenes and alkynes are excellent substrates for hydrozirconation and were investigated for their selectivity as compared to tertiary amides (Table 4). Cinnamic amides (entries 1 and 2) were selectively reduced without observed reduction of the double bond. Entry 1 afforded a 1:1 mixture of the aldehyde and alcohol each in 19% yield with no recovery of the starting amide. The problematic reduction of N,N-diethylcinnamamides has been previously reported with LiAlH4 in which only 29% of the cinnamyl alcohol was obtained.7 Interestingly, the Weinreb amide derivative 32 (Table 2, entry 10) yielded 87% of the aldehyde and 12% of the alcohol. The increase in yield for entry 2 (Table 4) suggests that the Weinreb amide may impart certain properties that increase intermediate stabilities or reactant selectivities; possibly by the same mechanism as proposed by Weinreb.4 In entry 3, the amide was reduced in the presence of the terminal olefin affording 63% of the corresponding aldehyde. On the contrary, the terminal alkyne furnished the alkynyl and alkenyl aldehydes in 13% and 9% yield respectively (entry 4). The conjugated internal alkyne (entry 5) afforded exclusively the corresponding aldehyde but in poor yields and mass balances. This selectivity is not unexpected as hydrozirconation of internal alkynes is typically kinetically disfavored when compared to terminal alkynes.33
Table 4.
Reduction of Alkenyl and Alkynyl Amides
| Entry | Substrate | Product(s) | Time (min) | Yield% (starting material) |
|---|---|---|---|---|
| 1 |  |
 |
20 | 19a |
| 2 | 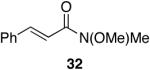 |
 |
20 | 87b |
| 3 |  |
 |
45 | 63(15) |
| 4 |  |
 |
20 | 13+9(67) |
| 5 |  |
 |
45 | 34 |
A mixture of aldehyde and alcohol (1:1) was obtained in 19% yield each.
The corresponding alcohol (12%) was also obtained
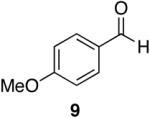
Cp2Zr(H)Cl is known to be sensitive to congestion at or near the reaction center of the substrate in olefin hydrozirconation and thus, we sought to examine the associated issues for amide reductions.33 Table 5 (entries 1-3) shows that steric encumbrance is tolerated on the amine portion of the carboxamide, although the yields are somewhat reduced when compared to the diethyl amide case (Table 1, entry 4). Diisopropyl amide 47 reacts in ten minutes providing 72% of the aldehyde, whereas its reaction with LiAlH4 or LiAlH(OEt)3 is extremely slow giving nearly all unreacted material after one hour.8,22 When 2,6-dichlorobenzamide (Table 5, entry 4) was subjected to hydrozirconation, only 17% of the aldehyde (with 63% of the starting material recovered) could be obtained even after prolonged reaction times and with the addition of excess reagent (three equivalents). This could be due to the presence of di-ortho substituents, the increased electron deficiency of this substrate, or both. Increased steric demand is most likely the major cause of inefficiency because the analogous Weinreb-derived 3,4-dichlorobenzamide reacts completely with the zirconium deuteride reagent affording the desired aldehyde in 93% yield after only five minutes (Table 7, entry 5).40 Overall, it appears that steric bulk is better tolerated on the amine side of the amide than on the carbonyl side.
Table 5.
Effects of Steric Bulk on the Reduction Reaction
| Entry | Amide | Product | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 |  |
 |
10 | 72 |
| 2 |  |
 |
5 | 88 |
| 3 |  |
 |
5 | 85 |
| 4 |  |
 |
18 hr | 17a |
63% yield of starting material recovered.
Table 7.
Amide Reduction Using Cp2Zr(D)Cl40
| Entry | Amide | Product | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 |  |
 |
10 | 92 |
| 2 |  |
 |
10 | 85 |
| 3 |  |
 |
15 | 70 |
| 4 | 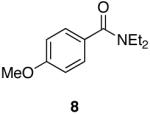 |
 |
15 | 99 |
| 5 | 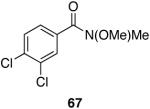 |
 |
5 | 93 |
| 6 |  |
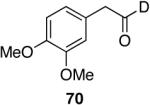 |
5 | 92 |
| 7 | 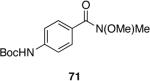 |
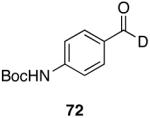 |
5 | 91 |
| 9 | 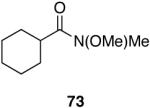 |
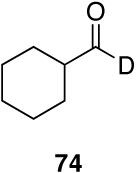 |
10 | 89 |
| 8 |  |
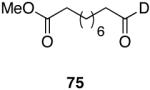 |
20 | 80 |
In addition to the reduction of a variety of amides discussed above, the Schwartz reagent was investigated for its ability to remove amide-based chiral auxiliaries. Initial attempts using the norephedrine-derived oxazolidinone developed by Evans44 proved successful on a model system (Table 6, entry 1), but provided only moderate yields when extended to other substrates (Table 6, entries 2-4). This is likely due to competitive reduction of the two carbonyls of the imide substrate with the more electron-rich endo carbonyl being preferentially reduced. As carbonyl conjugation is removed (Table 6, entries 2-4) the aldehyde yield decreases likely due to reduction of the endo carbonyl competing with the exo carbonyl group. It is also conceivable that enolization of the aliphatic imide substrates may account for the decreased yields for entries 2-4. The oxazolidinone auxiliary could not be recovered, presumably due to complexation of the norephedrine amino-alcohol with the zirconium byproduct (see the mechanistic discussion below).
Table 6.
Reduction of Amides Containing Chiral Auxiliaries
| Entry | Amide | Product | Time (min) | Yield (%) |
|---|---|---|---|---|
| 1 | 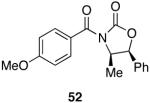 |
 |
15 | 92 |
| 2 | 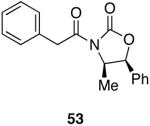 |
 |
90 | 51 |
| 3 |  |
 |
30 | 46 |
| 4 | 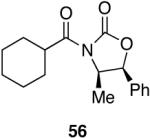 |
 |
90 | 57 |
| 5 | 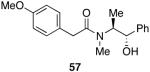 |
a | 10 | 0 |
| 6 |  |
 |
15 | 90 |
| 7 |  |
N.R. | 45 | 0 |
| 8 | 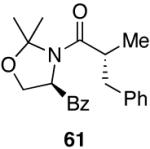 |
N.R. | 45 | 0 |
Complete loss of starting material without observation of aldehyde.
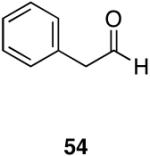
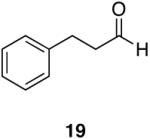
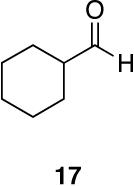
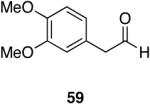
Myers’ pseudoephedrine derived auxiliary45 was examined due to its true tertiary amide nature versus the oxazolidinone-based systems. When a model system was subjected to the hydrozirconation conditions, a complete loss of starting material was observed without accompanying aldehyde formation (Table 6, entry 5). We believed this was due to deprotonation of the alcohol group on the auxiliary by the reagent.33 However, the addition of two or more equivalents was not able to elicit product formation. When the alcohol was protected as the tert-butyldimethylsilyl ether the desired aldehyde was isolated in a 90% yield as expected (Table 6, entry 6). This finding is notable as the pseudoephedrine auxiliary has recently been shown to impart asymmetric induction via alkylation using either an O-benyzlated “protected” auxiliary or a polymer-bound auxiliary (via a similar oxygen-polymer bond) with comparable enantioselectivity.46 Attempted hydrozirconation of α-methyl derivative 60 (Table 6, entry 7) did not result in product formation. Additionally, hydrozirconation of chiral compound 61 failed (Table 6, entry 8). Thus, it appears that α substitution, in these particular cases, confers a large enough steric environment to block hydrozirconation under these conditions. The discrepancies in yield between entries 6 and 7 (Table 6) strongly suggest that the inability of substrates 50, 60, and 61 to react is due to a steric interaction rather than an electronic bias. In support of this, it is important to note that the 4-nitrocarboxamides (compounds 6 and 24) and 3,4-dichlorobenzamide 67 react well.
Attempted hydrozirconation of various amide-based chiral auxiliaries have been reported since our initial communication and have been met with varying success.47 Mickel et al. were unsuccessful in the removal of an oxazolidinone-based chiral auxiliary containing an α-stereocenter with the Schwartz reagent in their process synthesis of (+)-discodermolide.47 Yamada et al. have reported the successful removal of 2,2-dimethyloxazolidine chiral auxiliaries using this methodology in the synthesis of 1,4-dihydropyridines affording the corresponding aldehyde.47 Apparently, the ability to remove such auxiliaries is substrate specific under these conditions.
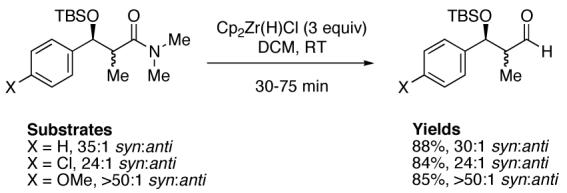
In a recent communication, Rawal et al. have reported the cleavage of α-substituted N,N-dimethyl amides utilizing a three-fold excess of 1 in methylene chloride that afforded the corresponding aldehydes in good yields (Scheme 1).48 Thus, it appears that certain α-substituted chiral aldehydes can be viable substrates for hydrozirconation under the appropriate conditions.
Scheme 1.
The use of the commercially available Cp2Zr(D)Cl was investigated for the ability to prepare deuterium labeled aldehydes. This proved to be an efficient reaction for the preparation of deuterio-aldehydes, which are often difficult to synthesize through other methods.40 Many of the existing methods involve expensive or difficult to synthesize reagents, multi-step procedures, or harsh reaction conditions en route to the specific labeling of the 1-aldehydic position.49 In addition to these concerns many of these procedures are substrate specific with unknown or low chemoselectivity.
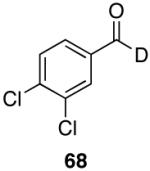
As is evident in Table 7, these labeled aldehydes were synthesized with results comparable to the hydrido reagent. Again, both aromatic and aliphatic substrates react well affording the desired deuterio-aldehydes in good yields. As expected, the chemoselectivity profile is retained in this reaction variant. In all cases deuterium incorporation was ≥95% as measured by 1H NMR spectroscopy. The ability to selectively label the aldehydic proton with a high deuterium incorporation in short reaction times with high yields from easily obtainable starting materials makes this method superior to existing procedures.
Mechanistic investigation
During the course of establishing the reaction scope, the presence of a seemingly stable intermediate became evident, in that over-reduction of the aldehyde to the alcohol was not observed in most cases, even when an excess of the zirconium hydride was employed. Aldehydes and ketones are known substrates for hydrozirconation34,36 and any aldehyde formed before the workup is expected to be reduced to the corresponding alcohol.
This observation led to the hypothesis of two intermediates. Hydrozirconation of the amide could result in an iminium ion species after hydride delivery and zirconium oxide expulsion (path a, intermediate II, Scheme 2) or by incorporation of the zirconium reagent to form an sp3 hybridized, 18-electron complex (path b, intermediate III, Scheme 2). Hydrolysis of either of these intermediates by the addition of water would then afford the aldehyde products. It is also feasible that the conversion of III to IV could occur by amine expulsion in which the amide oxygen in the carbonyl of the product aldehyde is retained. The proposed zirconacycle (III) should be stable as compounds related to intermediate III have been previously reported.50 Although iminium ions are generally highly reactive toward reducing agents, Buchwald has proposed them as intermediates in his reported reduction of amides.5 Thus, the iminium species II was initially considered a possible intermediate for this reaction.39
Scheme 2.

Extension of the reaction to generate deuterium labeled aldehydes using the commercially available Cp2Zr(D)Cl confirmed the obvious assumption that the hydride is transferred to the carbonyl carbon and is incorporated into the aldehyde. This gives direct evidence as to the hydride source and answers a preliminary question concerning the reaction mechanism.
Another question to the mechanistic details regarded the source of the aldehyde carbonyl oxygen. Did the addition of water (from silica gel employed in the workup)42 result in the hydrolysis of a reaction intermediate to form the aldehyde or was the amide carbonyl oxygen retained in the product?
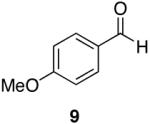
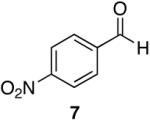
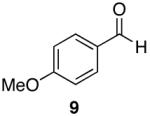
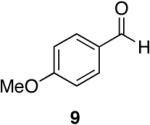
Towards this end, the addition of 18O labeled water (1:1 mixture of H2O16 and H2O18) to the reaction of 4-methoxy-N,N-diethylbenzamide (8) and 1 (equation 2) after 15 minutes demonstrated incorporation of the labeled oxygen into the aldehyde as observed by 13C NMR and MS [carbonyl peaks at δ 191.25 and 191.22 in a 1:1 ratio, and a 1:1 ratio for the pairs m/z 165, 163 (M+), and 164, 162 (M+-1)]. Control experiments with benzaldehyde and 18O-labeled water showed no isotopic incorporation after five minutes, one, and 24 hours. An additional control experiment with a catalytic amount of the Lewis acidic Cp2ZrCl2 was performed (1 is made from the dichloride and trace amounts may be present in the reaction mixture). Therefore, 8 and 1.1 equivalents of 18O labeled water in the presence of 10 mol% of Cp2ZrCl2 showed no isotopic enrichment after five minutes and a small peak was discernable after one hour as detected by 13C NMR (24:1 of 16O:18O). However, a 1.6:1 ratio of 16O:18O-labeled carbonyl peaks was apparent after 24 hours. Although this second control experiment confirms that organozirconium species can catalyze the oxygen exchange of water with p-anisaldehyde, we believe it unlikely that such a process is accounting for the observed 18O incorporation (1:1 ratio within 15 minutes) illustrated in equation 2 on the reaction time scale. This implies that the oxygen atom of the amide is sequestered by the zirconium reagent. This suggestion is supported by zirconium’s widely known oxophilicity and the strength of the zirconium-oxygen bond.51
 |
(2) |
In all cases the amine component of the amide is unrecoverable from the reaction mixture. In the particular case of lactams (both β-lactams and caprolactams were investigated), a complete loss of starting material was noted with no discernable amounts of the aldehyde observed by thin-layer chromatography. Thus, it seems that the residual amine component from the amide is strongly bound to the zirconium by-products.
Iminium ions react quite readily with a variety of nucleophiles, and the cyanide anion is well documented for its ability to derivatize such species.52 However, the addition of several cyanide anion sources failed to afford any of the aminonitrile after silica gel workup. Although these functional groups are reportedly stable to such chromatography,53 it was premature to rule out an iminium ion intermediate for various reasons: 1) cyanide retro-addition could occur, 2) the resulting amine product could be sequestered by the zirconium byproduct (as noted above), or 3) the nature of the counterion could change the reactivity of the iminium ion.54 It is worthwhile to note that the presence of the cyanohydrin from nitrile addition to the aldehyde was never observed.
Indirect evidence for complex III was gained from IR data as the differences in hybridization of the proposed intermediates would be distinct. The intermediate was prepared in an inert atmosphere from N,N-diethylbenzamide (77) and 1 and the corresponding spectrum was obtained as a solution in THF using an air free IR cell. The resulting spectrum did not show a peak definitively corresponding to an iminium ion.55 After the solution was exposed to water, a new peak formed at 1699 cm-1. Notably, there was no concomitant loss of an existing peak from the carbonyl region of the spectrum. If path a predominates, it would be expected that the addition of water would cause the loss of a peak corresponding to an iminium ion stretch and replace it with an aldehyde signal. The formation of a carbonyl stretch without concomitant loss of another signal from the same region supports intermediate III.
More direct evidence for intermediate III came from NMR spectroscopy. The intermediate was prepared in an NMR tube under anhydrous conditions with the addition of Cp2Zr(H)Cl to N,N-diethylbenzamide (77, in slight excess) in THF-d8. The resulting spectra are overlayed in Figure 1. Spectrum a is the N,N-diethylbenzamide substrate and spectrum b shows the intermediate after the addition of 1. As is evident, the initially rotameric diethyl peaks of the starting amide become defined multiplets and shift upfield (◆). This would be expected in the sp3 hybridized intermediate as shielding of the diethyl peaks by the cyclopentadienyl groups should occur. An iminium ion could show diastereotopicity of the ethyl peaks, but the chemical shifts would be expected downfield due to, formally, a quaternization of the nitrogen atom. Diastereotopicity of the alkyl amino portion suggests a closed cycle where the nitrogen coordinates to the zirconium atom resulting in a formal 18-electron intermediate. The formation of a new signal at 5.83 ppm (+) is clearly observed.
Figure 1.

1H-NMR of reaction intermediate III: (◆) denotes -CH2- and -CH3 peaks, (*) residual THF signals, (+) methine proton, (◇) minor product.
Most importantly, an expansion of the cyclopentadienyl region for intermediate III in Figure 2 shows diastereotopic splitting of the Cp groups with each integrating for five protons. This is only consistent with an sp3-hybridized intermediate. Iminium ion formation would require the elimination of the zirconium oxide from nitrogen lone pair donation. The zirconium oxide dimer has been well characterized and shows distinctly a singlet corresponding to the cyclopentadienyl ligands.56
Figure 2.
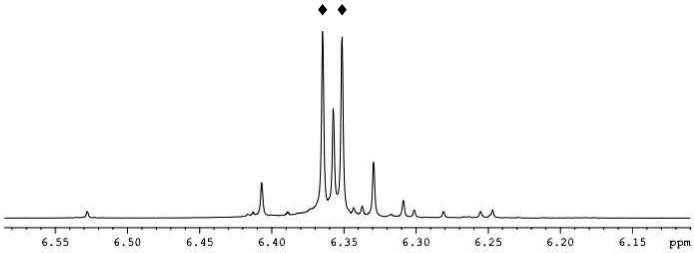
Expansion of Cp region for intermediate III: (◆) denotes diastereotopic Cp resonances.
A peak in the 13C NMR was observed at 97 ppm that correlates to the sp3 carbon based on DEPT data. The chemical shifts of the 1H (5.83 ppm) and 13C (97 ppm) peaks are very close to known N,O acetal shifts57 while the +N=CH(Ph) proton of N,N-dimethylbenzylideneammonium salts have been reported to be between 8.64-10.25 ppm, in the 1H NMR and 172.9-173.9 ppm in the 13C depending upon the counterion.58 Recently, Suginome and Murakami reported the proton and carbon chemical shifts for an N,N-diethylbenzylideammonium borate salt that displays a spectral profile different from the intermediate characterized from the hydrozirconation of tertiary amides unequivocally eliminating the iminium ion as a potential intermediate (Figure 3).59 The most striking difference lies in the upfield chemical shift of the methine proton in intermediate III as compared to the iminium ion reported by Sufinome and Murakami. The downfield shift of the methyl and methylene peaks in the ionic compound and their upfield shifts in intermediate III (as also compared to the starting amide) corroborates our hypothesis of zirconium complexation over iminium ion formation.
Figure 3.
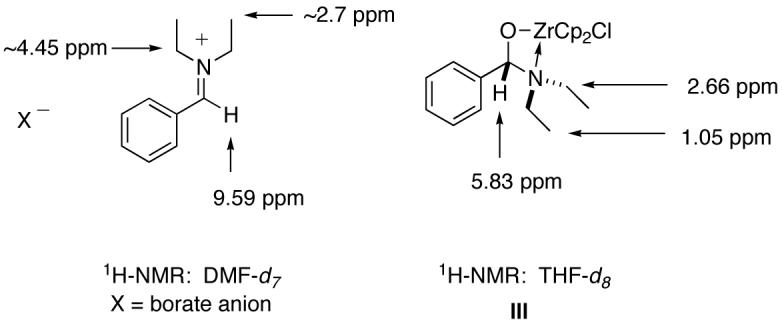
Comparison of 1H chemical shifts of iminium ion59 and intermediate III.
The reaction of 77 with Cp2Zr(D)Cl resulted in a complete loss of the methine peak in the 1H and 13C spectrum. This signal should exist as a triplet in the 13C spectrum due to splitting of the carbon from the deuterium atom but was not evident. It was anticipated that the carbon may not be observable due to the lack of an Overhauser effect and signal splitting. We therefore subjected N,N-diethylbenzamide 13C-enriched at the carbonyl carbon to Cp2Zr(D)Cl and observed a triplet at 97 ppm. Lastly, 2H-NMR of the deuterated intermediate showed a peak at 5.83 ppm confirming the origin and identity of the pre-aldehydic proton. This data collectively gives solid support to compound III as the intermediate in this reaction.
Careful examination of the 1H-NMR spectra revealed the existence of another compound in the reaction mixture (◇ - Figure 1). This minor compound is similar in all respects to the major product with the most pronounced change in the chemical shift for the methine proton at 5.07 ppm; a 0.76 ppm shift upfield from the major peak. Both signals were visible in the 2H spectrum, suggesting they are derived from the zirconium reagent.
Commercial and freshly prepared versions of 1 are usually contaminated with small to moderate amounts of Cp2ZrH2 (or Cp2ZrD2 for the deuterated Schwartz reagent).60 We hypothesized that it was quite likely that the minor compound could be the result of the reaction of this dihydride with one or two equivalents of the amide substrate. When 77 was reacted with the reagent containing an increased amount of the dihydride61 the relative intensity of the minor peak was increased along with the formation of three new peaks within close proximity to the original signal (along with corresponding signals for new -CH2- and -CH3 peaks upfield). Thus it appeared that the minor component was due to reaction of the substrate amide with amounts of a hydride contaminant, most likely Cp2ZrH2. Further evidence for the identity of this compound came when a substoichiometric amount of 1 was added to 77 followed by the addition of Cp2ZrH2. The resulting spectrum showed an increase in the relative intensity and integration area for the minor peak at 5.07 and those at 2.65. New peaks in the Cp region near 6.4 also accompanied this change. Additionally, the amount of this minor compound relative to the major methine peak seems to differ depending on the source (or lot) of 1.
As shown in Table 2, methoxymethyl (Weinreb) amides are excellent substrates for the reaction. NMR spectroscopy of the reaction of 1 and N-methyl-N-methoxy benzamide showed the presence of a single set of methyl groups upfield from the original amide peaks. The Weinreb-derived intermediate can possibly exist as one of two constitutional isomers depending upon which substrate heteroatom is bound to zirconium (V vs VI/VII, Figure 4). Should Zr-N coordination predominate, it is then possible for the existence of two diastereomers arising from the nitrogen substituents occupying two chemically distinct positions. 2D-NOESY experiments did not show a correlation between the N,O-acetal methine peak and any other protons in the molecule. This lack of an NOE interaction suggests that structure V may be the identity of the Weinreb-derived intermediate. This is not unexpected as the stability of the five-membered ring is expected to be greater than the four-membered ring.
Figure 4.

Possible products of hydrozirconation of Weinreb amides.
Attempted crystallization of the intermediate derived from 77 at ambient temperatures resulted in the unexpected formation of the μ-oxo zirconium dimer (80) after 24-48 hours, which was fully characterized by X-ray crystallography (equation 3).62 Temperature reduction to -35 °C again resulted in the μ-oxo zirconium species albeit after several weeks. This confirms that intermediate III is only transiently stable and it appears to degrade to the zirconium dimer over time. The intermediate exists as a pale-yellow oil when the solvent is removed in vacuo, which has been previously noted for the organozirconium products of the hydrozirconation of alkenes and alkynes.33 This property obviously limits their characterization to solution NMR spectroscopy.33 The addition of halogens to the substrate (79) or variation of the substituents on nitrogen (78) did not seem to confer solidity or crystallinity to the intermediate.
 |
(3) |
We next turned our attention to events that precede intermediate formation. Due to the heterogenicity of the reaction and very short reaction times, kinetic studies were not a viable investigational tool. We thus utilized simple competition studies to provide information pertaining to the initial pathway for formation of the intermediate.
A 1:1 mixture of each probe was subjected to 0.5 equivalents of 1 expecting this saturation to show a kinetic preference for the reaction (Table 8). Accordingly, N,N-diethylbenzamide substrates substituted at the para-position of the aryl ring with the methoxy and the nitro substituents showed no statistically significant preference by the reagent for either substrate (entry 1). The IR stretching frequencies for these amides are very close at 1625 cm-1 and 1622 cm-1. Thus it is apparent that the electronic substitution on the carbonyl side of these amides imparts little to no kinetic selectivity in this reaction.
Table 8.
Competition Experiments Between Amides with Different Electronic Properties
 | |||||
|---|---|---|---|---|---|
| Entry | X | Y | R | ratio of X:Ya | IR stretch of RCO cm-1 |
| 1 | -OMe | -NO2 | -NEt2 | 1:1 | 1622 |
| 2 | -OEt | -OMe | -NEt2 | 1:1.1 | 1625 |
| 3 | -OEt | -OMe | 22:1 | 1635 | |
| 4 | -OEt | -OMe | 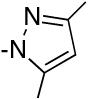 |
19:1 | 1683 |
determined by 1H NMR spectroscopy - average of two runs
Brown et al., has shown that aziridine amides are resistant to over-reduction to the amine with lithium aluminum hydride affording mostly the corresponding aldehyde even in the presence of 100% excess of the reagent.21,22 This is attributed to the developing strain by lone pair donation during formation of the iminium ion. The decrease in lone pair donation (resonance) is apparent in the amide itself as reflected by the higher IR carbonyl stretching frequency (1635 cm-1 versus 1625 cm-1 for 9, Table 2) and the pyramidalization of the nitrogen was observed in the solid state for the related 4-bromo-N-benzoylaziridine.63 Additionally, Brown has reported evidence for this amide distortion in which N-benzoylaziridine was shown to be ∼200,000-fold more susceptible to hydrolysis than N,N-dimethylbenzamide.64 We had anticipated that amides of this type would serve as useful probes to determine how the degree of lone pair donation would affect the kinetic selectivity for this reaction. Thus, a competitive reaction between an N-benzoyldiethylamide and an N-benzoylaziridine (Table 8, entry 3) resulted in a 22:1 selectivity for the diethyl amide showcasing a kinetic selectivity for the resonance-stabilized amide.
It was anticipated that the aromaticity requirement for the nitrogen lone pair for the dimethylpyrrazole probe in entry 4 would also bias the reagent against this substrate.23 This was again based on the IR carbonyl stretch observed for this amide probe (1683 cm-1). Accordingly, a 19:1 preference for the diethyl derivative was again observed for these compounds (entry 4).
As a control the 4-methoxy (1625 cm-1) and 4-ethoxy (1625 cm-1) diethylamide probes were compared under the same conditions resulting in a 1:1.1 mixture of the aldehyde products (entry 2). Thus, it is clear that the presence of the larger ethoxy group has little to no effect on the kinetic selectivity for this reaction.
These results collectively suggest that the reagent prefers substrates that do not decrease nitrogen lone pair availability. The participation for the nitrogen lone pair does not seem to be required for hydrozirconation to occur, but seems to kinetically activate the substrate.65 This “lone-pair activation” is also supported by the selective reduction of tertiary amides in the presence of esters as well as the reduced yields observed for primary and secondary amides, all of which have direct differences in the electron densities of the carbonyl oxygen. Based on these results, it is clear that an increase in the carbonyl electron densities of the substrate favors hydrozirconation and these results further imply that the degree of resonance in the amide linkage affects its kinetic reduction potential. This appears to be in contrast to the reduction of amides to aldehydes with LiAlH4 or LiAlH(OEt)3.8,21,22 In these case, it has been noted that substrates in which the unshared electron pair of the amide is involved in resonance with a heterocyclic ring (N-acylated pyrroles, indoles, and carbazoles) or in which the amide resonance develops strain (aziridine amides) give aldehydes in higher yields than N,N-alkyl substituted amides.8
It has been proposed that migratory insertion of zirconium hydride to alkenes and alkynes is not the rate-limiting step and that events prior to hydrozirconation may provide the energy maximum.66 Whether this holds true for hydrozirconation of tertiary amides is unconfirmed. Due to the experimental difficulty associated with the kinetics of heterogeneous reactions, we offer a qualitative explanation based on precedent and basic observations.
Wipf and Jahn have reported a comparison of the solvent dependence for the rate of hydrozirconation of 1-hexene.31 They disclosed a correlation between the coordinating ability of the solvent and the rate of hydrozirconation to which they ascribed solvent dissolution as the rate-determining step. Furthermore, it was observed that the reaction is first order in Cp2Zr(H)Cl in oxetane and zeroth order in reagent when carried out in THF. They conclude that in THF the rate-limiting step is dissolution of the oligomeric reagent and for oxetane the rate of dissolution is comparable to the rate of hydrozirconation.
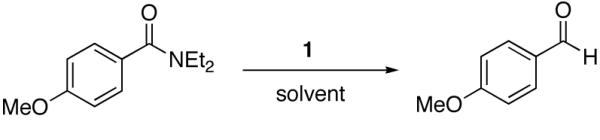
Not surprisingly, the hydrozirconation of tertiary amides displays a similar solvent dependency (Scheme 3 and Table 9). Strongly coordinating solvents favor hydrozirconation whereas weakly or non-coordinating solvents slow the reaction. Reactions in THF tend to be complete in ca. 10-15 minutes for most substrates while non-coordinating solvents such as chloroform afford no hydrozirconation products after extended reaction periods.67 Moderately to weakly coordinating solvents such as dioxane and toluene slow the rate of reaction and result in reduced reaction yields after an arbitrary time point. Table 8 outlines the yields after a reaction time point and gives the time of dissolution (tdiss) of the reagent for each solvent. For these cases, tdiss can be viewed as a crude indicator of the rate of the reaction as it has been generally noted that the time of reagent dissolution marks the completion of the reaction.33 We have noticed that the time it takes for the solution to clear corresponds to a complete loss of starting material by TLC (for reactions that run to completion in THF). Since Cp2Zr(H)Cl is an insoluble polymer, it is believed that the reaction rate is limited by the small amount of reagent in solution. Quite possibly, the more strongly coordinating solvents (THF, oxetane, and pyridine) are better able to dissociate these Zr-H polymers (e.g., k1THF > k1toluene, Scheme 3) and allow for reagent solvation as proposed by Wipf for alkene hydrozirconation. The coordinating solvent/ligand exchanges with the amide and hydrozirconation ensues. Although speculative, this solvent dependence seems to support reagent dissolution as the rate-determining step. With pyridine, dissolution (tdiss) occurs almost instantly resulting in a straw colored solution but gives poor chemical yields. We presume that pyridine is better able to dissolve the Schwartz reagent but forms a highly stable 18-electron complex that is resistant to exchange with the amide substrate.68 Tetrahydrofuran and oxetane are optimal solvents, in that reagent dissolution is favorable but does not bind too strongly. This is probably due to the availability of the non-bonding electron pairs from distortions of the tetrahedral geometry (of oxygen) caused by constraint in a four or five membered ring. Dioxane has a more tetrahedral geometry, likely impeding k1 and making reagent dissolution impractically slow.
Scheme 3.

Table 9.
Solvent Effects on Reaction
 | |||
|---|---|---|---|
| Entry | Solvent | Yield (%)a | tdissb |
| 1 | oxetane | 95 | 2-3 |
| 2 | tetrahydrofuran | 99 | 15 |
| 3 | 1,4 dioxane | 15 | - |
| 4 | pyridine | 15 | 1 |
| 5 | toluene | 15 | - |
| 6 | chloroform | 0 | - |
Aldehyde yield after 30 min period
Time of reagent dissolution in minutes
We anticipated that the migratory insertion event would be irreversible for this reaction based on the high oxophilicity of zirconium, the saturated coordination sphere of zirconium in the intermediate, and the polarized M-H bond nature of early transition metal hydrides.69 This would be in contrast to that of olefin hydrozirconation where the actual insertion event has been proposed to be reversible.31 To test this hypothesis we subjected a small excess of amide 2 to 1 (0.8 equiv) and allowed them to react completely (Scheme 4). Upon consumption of 1 an equimolar amount of amide 81 was added to the reaction mixture. An irreversible hydrozirconation would result in recovery of only aldehyde 9 while a reversible event should give a mixture of the aldehydes over time. Thus, a 1.5 hour incubation afforded exclusively aldehyde 9 as determined by 1H NMR and HPLC analysis, displaying the apparent irreversibility of the reaction.
Scheme 4.

The formation of aldehydes from the reaction intermediate by the addition of water raises the question if other nucleophiles can effectively react with the intermediate. The addition of a solution of p-anisidine in anhydrous THF to the reaction intermediate resulted in imine formation (82, equation 6) upon analysis of the crude reaction mixture after hexanes precipitation of the zirconium oxide byproducts and filtration.70 This addition was observed by NMR spectroscopy and initially the amount of imine product was small with large peaks corresponding to unreacted p-anisidine and the intermediate zirconacycle predominating (see supporting information). However, over time (ca. 40 minutes) the imine signals grew as the intermediate and nucleophile signals decreased. The diastereotopic ethyl groups on the amine slowly converged to defined multiplicities (a triplet for the -CH3 and a quartet for the -CH2- groups) as expected for freely rotating ethyl groups. These ethyl peaks retained their upfield chemical shifts suggesting that the amine component stays bound to the zirconium. This is supported by the inability to recover the amine portion of the carboxamide and the loss of lactam substrates (vide supra). Furthermore, the diastereotopic Cp signals resolved into a single Cp resonance as the zirconium byproduct formed. This was accompanied by a yellowish precipitation that was evident in the NMR tube. Some aldehyde was apparent in the initial experiment, most likely from adventitious water in the p-anisidine solution. The addition of aniline afforded imine 82b in the same manner as for p-anisidine but the reaction rate was significantly slower.
This observation is significant as Ganem has reported the conversion of secondary carboxamides to imines using the Schwartz reagent that apparently follows a mechanistically distinct pathway.38 This process could be considered complementary to Ganem’s method allowing tertiary amides to be cleanly converted to the corresponding imines. Thus the tertiary carboxamide can now be viewed as an accessible and chemically robust precursor or “protecting group” for the more labile aldehyde or imine derivatives giving it advantages over the generally more reactive ester functionality.
 |
(6) |
In summary, we have reported an investigation of the hydrozirconation of tertiary amides with the Schwartz reagent, outlining the scope and limitations for the transformation. We have demonstrated that hydrozirconation is the mildest and most general method for the formation of aldehydes from amides with the highest functional group tolerance reported to date. This reaction proceeds with very short reaction times and gives excellent to good yields of aldehyde with an unprecedented chemoselectivity profile. Importantly, the reaction can be performed using standard inert-atmosphere benchtop techniques and employs a convenient workup procedure. Also presented is the utility of the reagent to effectively reduce secondary and primary amides albeit with reduced yields. We have characterized the intermediate for the reduction as well as provided insight into the possible pathways for formation of the intermediate and the aldehyde product. The high chemoselectivity of this reaction and the robust nature of the amide functional group could allow one to consider this usually inert moiety as a protected form of the more reactive aldehyde and imine oxidation states.
Acknowledgment
The authors would like to thank Profs. Jon Tunge and Apurba Dutta for useful discussions, Dr. Doug Powell for X-ray determination, and Dr. David Vander Velde and Sarah Neuenswander for NMR assistance. J.T.S. is a recipient of an American Foundation for Pharmaceutical Education predoctoral fellowship and a Department of Defense predoctoral fellowship (DAMD17-99-1-9243). J.M.W. would like to thank the National Institutes of Health for a predoctoral training grant (GM-08545). This work was supported by the NIH (CA-084173).
Footnotes
Supporting Information Available: General methods, experimental procedures, a complete listing of authors for reference 47a, and spectral data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. John Wiley & Sons; New York: 2000. [Google Scholar]
- (2).Nystrom RF, Brown WG. J. Am. Chem. Soc. 1948;70:3738–3740. doi: 10.1021/ja01191a057. [DOI] [PubMed] [Google Scholar]
- (3).Larock RC. Comprehensive Organic Transformations: A Guide to Functional Group Preparation. VCH; New York: 1989. [Google Scholar]
- (4).Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- (5).Bower S, Kreutzer K, Buchwald SL. Angew. Chem., Int. Ed. Engl. 1996;35:1515–1516. [Google Scholar]
- (6).Hudlický M. Reductions in Organic Chemistry. Ellis Horwood Limited; Chichester, England: 1984. pp. 164–171. [Google Scholar]
- (7).Mićović VM, Mihailović ML. J. Org. Chem. 1953;18:1190–1200. [Google Scholar]
- (8).Brown HC, Tsukamoto A. J. Am. Chem. Soc. 1964;86:1089–1095. [Google Scholar]
- (9).Brown HC, Tsukamoto A. J. Am. Chem. Soc. 1959;81:502–503. [Google Scholar]
- (10).Ramegowda NS, Modi MN, Koul AK, Bora JM, Narang CK, Mathur NK. Tetrahedron. 1973;29:3985–3986. [Google Scholar]
- (11)(a).Cha JS, Lee JC, Lee HS, Lee SE, Kim JM, Kwon OO, Min SJ. Tetrahedron Lett. 1991;47:6903–6904. [Google Scholar]; (b) Muraki M, Mukaiyama T. Chem. Lett. 1975:875–878. [Google Scholar]; (c) Yoon NM, Ahn JH, An DK, Shon YS. J. Org. Chem. 1993;58:1941–1944. [Google Scholar]; (d) Zakharkin LI, Maslin DN, Gavrilenko VV. Tetrahedron. 1969;25:5555–5569. [Google Scholar]
- (12).Bedenbaugh AO, Payton AL, Bedenbaugh JH. J. Org. Chem. 1979;25:4703–4705. [Google Scholar]
- (13).Tsay S-C, Robl JA, Hwu JR. J. Chem. Soc., Perkin Trans. 1. 1990:757–759. [Google Scholar]
- (14)(a).Brown HC, Bigley DB, Arora SK, Yoon NM. J. Am. Chem. Soc. 1970;92:7161–7167. [Google Scholar]; (b) Godjoian G, Singaram B. Tetrahedron Lett. 1997;38:1717–1720. [Google Scholar]
- (15).Kamochi Y, Kudo t. Tetrahedron. 1992;48:4301–4312. [Google Scholar]
- (16).Rahman A, Basha A. J. Chem. Soc., Chem. Commun. 1976:594–595. [Google Scholar]
- (17).Cha JS. Bull. Korean Chem. Soc. 1992;13:670–676. [Google Scholar]
- (18).Cha JS, Kwon OO, Kim JM, Lee JC. Bull. Korean Chem. Soc. 1994;15:644–649. [Google Scholar]
- (19).Wittig G, Hornberger P. Ann. Chim. (Paris) 1952;577:11–15. [Google Scholar]
- (20).Staab HA, Bräunling H. Ann. Chim. (Paris) 1962;654:119–130. [Google Scholar]
- (21).Brown HC, Tsukamoto A. J. Am. Chem. Soc. 1961;83:2016–2017. [Google Scholar]
- (22).Brown HC, Tsukamoto A. J. Am. Chem. Soc. 1961;83:4549–4552. [Google Scholar]
- (23).Ried W, Königstein FJ. Angew. Chem. 1958;70:165. [Google Scholar]
- (24)(a).Weygand R, Eberhardt G. Angew. Chem. 1952;64:458. [Google Scholar]; (b) Weygand R, Eberhardt G, Linden H, Schäfer F, Eigen I. Angew. Chem. 1953;65:525–531. [Google Scholar]; (c) Weygand R, Linden H. Angew. Chem. 1954;66:174–175. [Google Scholar]; (d) Weygand R, Mitgau R. Chem. Ber. 1955;88:301–308. [Google Scholar]; (e) Weygand R, Bestmann HJ. Chem. Ber. 1959;92:528–529. [Google Scholar]
- (25).Douat C, Heitz A, Martinez J, Fehrentz J-A. Tetrahedron Lett. 2000;41:37–40. [Google Scholar]
- (26)(a).Nagao Y, Kawabata K, Seno K, Fujita E. J. Chem. Soc., Perkin Trans. 1. 1980:2470–2473. [Google Scholar]; (b) Izawa T, Mukaiyama T. Chem. Lett. 1977:1443–1446. [Google Scholar]; (c) Izawa T, Mukaiyama T. Bull. Chem. Soc. Jpn. 1979;52:555–558. [Google Scholar]
- (27).Comins DL, Brown JD. Journal of Organic Chemistry. 1986;51:3566–3572. [Google Scholar]
- (28)(a).Wailes PC, Weigold H, Bell AP. J. Organomet. Chem. 1971;27:373–378. [Google Scholar]; (b) Wailes PC, Weigold H. J. Organomet. Chem. 1970;24:405–411. [Google Scholar]
- (29).Marek I, editor. Titanium and Zirconium in Organic Synthesis. Wiley-VCH; Weinheim: 2002. [Google Scholar]
- (30)(a).Hart DW, Schwartz J. J. Am. Chem. Soc. 1974;96:8115–8116. [Google Scholar]; (b) Schwartz J, Labinger JA. Angew. Chem., Int. Ed. Engl. 1976;15:333–340. [Google Scholar]
- (31).For a review up to ca. 1995 see:Wipf P, Jahn W. Tetrahedron. 1996;52:12853–12910.
- (32).For a review from ca. 1995-2001 see:Lipshutz BH, Pfeiffer SS, Noson K, Tomioka T. In: Titanium and Zirconium in Organic Synthesis. Marek I, editor. Wiley-VCH; Weinheim: 2002. pp. 110–148.
- (33).Labinger JA. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 8. Pergamon; New York: 1991. pp. 667–702. [Google Scholar]
- (34).Cesarotti E, Chiesa A, Maffi S, Ugo R. Inorg. Chim. Acta. 1982;64:L207–L208. [Google Scholar]
- (35).Laycock DE, Alper H. J. Org. Chem. 1981;26:289–293. [Google Scholar]
- (36).Majoral JP, Zablocka M, Igau A, Cenac N. Chem. Ber. 1996;129:879–886. doi: 10.1021/jo951222w. [DOI] [PubMed] [Google Scholar]
- (37).Zablocka M, Delest B, Igau A, Skowronska A, Majoral JM. Tetrahedron Lett. 1997;38:5997–6000. [Google Scholar]
- (38)(a).Schedler DJA, Godfrey AG, Ganem B. Tetrahedron Lett. 1993;34:5035–5038. [Google Scholar]; (b) Schedler DJA, Li J, Ganem B. J. Org. Chem. 1996;61:4115–4119. doi: 10.1021/jo960286j. [DOI] [PubMed] [Google Scholar]
- (39).White JM, Tunoori AR, Georg GI. J. Am. Chem. Soc. 2000;122:11995–11996. [Google Scholar]
- (40).Spletstoser JT, White JM, Georg GI. Tetrahedron Lett. 2004;45:2787–2789. [Google Scholar]
- (41).The commercially available reagent is stable for several months if the bottle is purged with argon following each use, the cap wrapped with parafilm, and stored in a desiccator at room temperature. The reagent should exist as a white powder and discoloration to an off-white or pinkish hue is usually indicative of impure reagent. Lipshutz has noted that the reagent can last ca. 3 months with careful handling - see reference 5. After this time, the reagent loses its efficiency even in the absence of appearance changes. We have noticed a similar trend. In addition to its commercial availability, Cp2Zr(H)Cl may also be synthesized from the dichloride in high purity following the procedure of Buchwald (seeBuchwald SL, LaMaire SJ, Nielsen RB, Watson BT, King SM. Tetrahedron Lett. 1987;28:3895–3898.andBuchwald SL, LaMaire SJ, Nielson RB, Watson BT, King SM. Org. Synth. 1993;71:77–82.). The reagent can be assayed using 1H- NMR following the procedure of Schwartz (Gell KI, Posin B, Schwartz J, Williams GM. J. Am. Chem. Soc. 1982;104:1846–1855.andBuchwald Org. Synth. above.
- (42).A representative procedure for the hydrozirconation of tertiary amides is as follows: Cp2Zr(H)Cl (0.29 mmol) is suspended in THF (3 mL) under argon at room temperature in a flame-dried round bottom flask. To this suspension, is added the substrate (0.24 mmol) in THF (2 mL) also at room temperature. The mixture is stirred until the reaction turns clear usually within 15-30 minutes. Workup by the addition of silica gel (ca. 3 g) and removal of the solvent in vacuo, followed by elution through a short path silica gel column affords the aldehyde.
- (43).Hudlický M. Reductions in Organic Chemistry. 2nd ed. American Chemical Society; Washington, DC: 1996. [Google Scholar]
- (44).Evans DA. In: Top. Stereochem. Nelson JV, Taber T, editors. Vol. 13. John Wiley & Sons; 1982. pp. 1–115. [Google Scholar]
- (45)(a).Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496–6511. [Google Scholar]; (b) Myers AG, Gleason JL, Yoon T. J. Am. Chem. Soc. 1995;117:8488–8489. [Google Scholar]
- (46)(a).Hutchison PC, Heightman TD, Procter DJ. J. Org. Chem. 2004;69:790–801. doi: 10.1021/jo0354950. [DOI] [PubMed] [Google Scholar]; (b) Hutchison PC, Heightman TD, Procter DJ. Org. Lett. 2002;4:4583–4585. doi: 10.1021/ol0268788. [DOI] [PubMed] [Google Scholar]
- (47)(a).Mickel SJ, et al. Org. Process Res. Dev. 2004;8:107–112. [Google Scholar]; (b) Yamada S, Morita C. J. Am. Chem. Soc. 124:8184–8185. doi: 10.1021/ja0203317. 124. [DOI] [PubMed] [Google Scholar]
- (48).This information was published while this manuscript was under review:McGilvra JD, Unni AK, Modi K, Rawal VH. Angew. Chem., Int. Ed. Engl. 2006;118:6276–6279. doi: 10.1002/anie.200601638.
- (49)(a).Axenrod TA, Loew L, Pregosin PS. J. Org. Chem. 1968;33:1274.Bennett DJ, Kirby GW, Moss VA. J. Chem. Soc., Chem. Commun. 1967:218–219.(c) Craig, J. C.; Kray,L R. J. Org. Chem. 1968;33:871–872.Degani I, Fochi R. Synthesis. 1976:759–761.Franzen VV. Liebigs Ann. Chem. 1956;600:109–114.Meyers AI, Nabeya A, Adickes HW, Fitzpatrick JM, Malone GR, Politzer IR. J. Am. Chem. Soc. 1969;91:764–765.Meyers AI, Nabeya A, Adickes HW, Politzer IR. J. Am. Chem. Soc. 1969;91:763–764.Olofson RA, Zimmerman DM. J. Am. Chem. Soc. 1967;89:5057–5059.Schlosser M. Chem. Ber. 1964;97:3219–3233.Schowen RL, Burgstahler AW, Walker DE, Kuebrich JP. J. Org. Chem. 1972;37:1272–1273.Scott CA, Smith DG, Smith DJH. Synth. Commun. 1976;6:135–139.Seebach D, Erickson BW, Singh G. J. Org. Chem. 1966;31:4303–4304.Thompson AF, Cromwell NH. J. Am. Chem. Soc. 1939;61:1374–1376.Yamashita M, Miyoshi K, Nakazono Y, Suemitsu R. Bull. Chem. Soc. Jpn. 1982;55:1663–1664.
- (50)(a).For selected examples see:Gately DA, Norton JR, Goodson PA. J. Am. Chem. Soc. 1995;117:986–996.Plössl K, Norton JR, Davidson JG, Barefield EK. Organometallics. 1992;11:534–539.
- (51).Ferreri C, Palumbo G, Caputo R. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 1. Pergamon; New York: 1991. pp. 139–172. [Google Scholar]
- (52).For a recent example of iminium ion trapping with cyanide, see:Myers AG, Kung DW, Zhong B, Movassaghi M, Kwon S. J. Am. Chem. Soc. 1999;121:8401–8402.
- (53).The authors report the flash chromatography purification of various α-aminonitriles:Heydari A, Fatemi P, Alizadeh A-A. Tetrahedron Lett. 1998;39:3049–3050.
- (54).For an example of different iminium counterions affording different reaction products see:Ofial AR, Mayr H. Angew. Chem., Int. Ed. Engl. 1997;36:143–145.
- (55).Imine salt stretching vibrations (-C=N-) are found approximately at ν 1680 cm-1:Conley RT. Infrared Spectroscopy. 2nd ed. Allyn and Bacon; Boston: 1972.
- (56).Reid AF, Shannon JS, Swan JM, Wailes PC. Aust. J. Chem. 1965;18:173–181. [Google Scholar]
- (57).The authors report the -NCHO- proton at 5.64 ppm for an indole-derived cyclic N,O-acetal:Austin JF, Kim S-G, Sinz CJ, Xiao W-J, MacMillan DWC. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5482–5487. doi: 10.1073/pnas.0308177101.
- (58)(a).For NMR data of iminium salts see:Mayr H, Ofial AR, Würthwein EU, Aust NC. J. Am. Chem. Soc. Vol. 119. 1997. pp. 12727–12733.Merényi R. In: Iminium Salts in Organic Chemistry. Böhme H, Viehe HG, editors. Vol. 9. Interscience; New York: 1976. pp. 23–105. Part 1.
- (59).1H and 13C spectra are available in the Supporting Information of:Suginome M, Uehlin L, Murakami M. J. Am. Chem. Soc. 2004;126:13196–13197. doi: 10.1021/ja045827y.
- (60).Buchwald SL, LaMaire SJ, Nielsen RB, Watson BT, King SM. Tetrahedron Lett. 1987;28:3895–3898. [Google Scholar]
- (61).Gell KI, Posin B, Schwartz J, Williams GM. J. Am. Chem. Soc. 1982;104:1846–1855. [Google Scholar]
- (62).The crystal structure for compound 80 can be found in the Supporting Information. It was first reported in:Clarke JF, Drew MGB. Acta Cryst. 1974;B30:2267–2269.
- (63).Shibaeva RP, Atovmyan LO, Kostyanovskii RG. Dokl. Bulg. Akad. Nauk. 1968;12:669. [Google Scholar]
- (64).Ślebocka-Tilk H, Brown RS. J. Org. Chem. 1987;52:805–808. [Google Scholar]
- (65).Compound 81 was treated independently with approx. 1.5 equiv. of Cp2Zr(H)Cl and the aldehyde was obtained as a 2:1 mixture with the starting material (as judged by 1H-NMR) along with 30% of p-methoxybenzyl alcohol. This shows that 81 is a viable substrate for hydrozirconation. The formation of the alcohol is most likely due to instability of the intermediate to the reagent as the nitrogen lone pair should be less available for coordination to zirconium.
- (66).Doherty NM, Bercaw JE. J. Am. Chem. Soc. 1985;107:2670–2682. [Google Scholar]
- (67).This observation could also be due to H-Cl exchange between the reagent and solvent.
- (68).The addition of pyridine to 1 before the addition of substrate results in a deep red color that changes upon the addition of substrate in pyridine.
- (69).Fachinetti G, Floriani C, Roselli A, Pucci S. J. Chem. Soc., Chem. Commun. 1978:269–270. [Google Scholar]
- (70).The crude proton spectrum clearly shows the imine product with excess p-anisidine. See the Supporting Information.