

Abstract
Antigen with dual costimulation through CD137 and CD134 induces powerful CD8 T cell responses. These effector T cells are endowed with an intrinsic survival program resulting in their accumulation in vivo, but the signaling components required for survival are unknown. We tested a cadre of pathway inhibitors and found one preclinical compound, Bay11-7082 (Bay11), which prevented survival. Even the γc cytokine family members IL-2, -4, -7, and -15 could not block death, nor could pretreatment with IL-7. We found that dual costimulation caused loading of phosphorylated IκBα (p-IκBα) and high basal levels of NF-κB activity in the effector CD8 T cells. Bay11 trumped both events by reducing the presence of p-IκBα and ensuing NF-κB activity. Not all pathways were impacted to this degree, however, as mitogen-mediated ERK phosphorylation was evident during NF-κB inhibition. Nonetheless, Bay11 blocked TCR-stimulated cytokine synthesis by rapidly accentuating activation-induced cell death through elicitation of a caspase-independent pathway. Thus, in effector CD8 T cells, Bay11 forces a dominant caspase-independent death signal that cannot be overcome by an intrinsic survival program nor by survival-inducing cytokines. Therefore, Bay11 may be a useful tool to deliberately kill death-resistant effector T cells for therapeutic benefit.
Keywords: apoptosis, T cell survival, costimulatory molecules, cytokines
INTRODUCTION
The combination of CD137 and CD134 costimulation generates high-quality supereffector CD8 T cells [1, 2]. Both costimulators use comparable signaling pathways such as NF-κB as well as TNFR-associated factor (TRAF)1–3 and -5 [3, 4]. TRAF2 and TRAF5 are crucial for activation of NF-κB and JNK, and TRAF1 and TRAF3 inhibit the classical and alternative NF-κB pathways [5, 6]. The NF-κB family consists of five different components: RelA (p65), RelB, c-Rel, NF-κB1 (p50, precursor is p105), and NF-κB2 (p52, precursor is p100). These members share an N-terminal Rel homology domain for DNA binding, dimerization with other NF-κB members, and association with the inhibitory protein, IκB. In resting cells, the NF-κB dimers are sequestered in the cytoplasm by IκB proteins such as IκBα (sequestrating p65-p50) or p100 (masking RelB). The central transcription factors p65, p50, and cRel can be activated by the classical pathway through the binding of microbial products to TLRs, proinflammatory cytokines, TCR stimulation, and costimulatory signals that lead to activation of the IκB kinase (IKK) complex, which consists of the catalytic kinase subunits IKKα and IKKβ and the essential regulatory subunit IKKγ or NF-κB essential modulator (NEMO). Activated IKK phosphorylates IκBα, which subsequently becomes ubiquitinated and degraded by the proteosome, thus releasing the p65-p50 dimer. These NF-κB dimers translocate into the nucleus, where they bind specific DNA regulatory elements to promote transcription of various genes. On the other hand, the noncanonical or alternative NF-κB pathway has been shown to be stimulated by members of the TNFR family such as CD40, lymphotoxin β receptor, and B cell activation factor [7,8,9,10]. Engagement of these costimulatory receptors promotes activation of NF-κB-inducing kinase, leading to phosphorylation of the IKKα-IKKα complex and subsequently, p100 cleavage into p52 in a NEMO- and IKKβ-independent manner [11, 12]. Eventually, p52-RelB heterodimers translocate into the nucleus and stimulate gene transcription. As the NF-κB pathway is involved in many cellular processes, deviant or constitutively active NF-κB may be considered as a token of malignancy [13,14,15,16,17,18]. Thus, the NF-κB pathway is a major therapeutic target for cancer therapy [19,20,21].
The complete details of these downstream signaling pathways driving CD137 and CD134 dual costimulation are unclear. In this report, some of these concepts were tested using primary CD8 T cells that were primed with peptide and agonist anti-CD137 and -CD134 mAb. We show that dual costimulation-mediated, intrinsic cell survival was sensitive to specific inhibitors of the NF-κB pathway. This included Bay11-7082 (Bay11), a compound that specifically uncouples phosphorylation of IκBα [22,23,24] and is a potential treatment for colorectal cancer [25], breast cancer [26], leukemia [27], and chronic lymphocyte leukemia [28]. Although IL-7 was able to promote cell survival substantially, NF-κB inhibitor-induced cell death dominated over cytokine-induced cell survival. Further, Bay11 even rerouted activation-induced cell death (AICD) from a caspase-dependent process to a caspase-independent pathway.
Thus, the combination of CD137 and CD134 endows antigen-specific CD8 T cells with the ability to use the γc cytokine family members for optimal cell survival [2], but here, we show that these supereffector CD8 T cells are also exquisitely sensitive to the NF-κB inhibitor Bay11. Therefore, the TNFR superfamily members can be used to eradicate tumors and enhance vaccine development, but now, we show a counter-regulatory means of purging any deleterious or renegade T cells by controlling cell fate with Bay11.
MATERIALS AND METHODS
Mice and reagents
C57BL/6 mice were purchased from the National Cancer Institute (NCI; Frederick, MD, USA) and the Jackson Laboratory (Bar Harbor, ME, USA). The OT-I Rag−/− transgenic mice were bred by our laboratory. All mice were maintained in the animal facility at the University of Connecticut Health Center (Farmington, CT, USA) in accordance with National Institutes of Health (NIH) guidelines.
SIINFEKL peptide was purchased from Invitrogen Life Technologies (Grand Island, NY, USA). PD98059, U0126, SB203580, LY294002, Bay11, NF-κB activation inhibitor, and caspase 8 Inhibitor II [benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone (Z-IETD-FMK)] were purchased from Calbiochem (San Diego, CA, USA). PMA and ionomycin were purchased from Sigma-Aldrich (St. Louis, MO, USA).
The anti-CD137 from the 3H3 rat hybridoma [29] and the anti-CD134 from the OX86 rat hybridoma [30] were purified using protein G (Invitrogen Life Technologies). Bath-to-batch variation was titered for maximal effect using minimal antibody. Injection schedules and immunization procedure were as follows: For the adoptive transfer model, 2 × 106 splenocytes from OT-I Rag−/− transgenic mice were transferred into C57BL/6 mice by the i.v. route. The following day, 100 μg SIINFEKL peptide diluted into PBS was injected i.p., and at the same time, enforced costimulation was induced with anti-CD137 (25 μg) or -CD134 (10 μg) or both (dual costimulation) by the i.p. route, as we have done in the past [1]. All data in the figures and tables used this immunization approach.
Cell processing, purification, and culturing
Spleens from Day 2.5 mice were removed and treated with collagenase (Roche Applied Science, Indianapolis, IN, USA) in MEM-HEPES solution (2% FCS, 10 mM HEPES in MEM) at 37°C for 30 min. An EDTA solution (100 μl of 0.5 M) was added to inactivate the activity of collagenase. In the case of Days 3 and 4 spleens, this step was unnecessary and was bypassed directly by crushing spleens through cell strainers (Falcon, BD Biosciences, San Jose, CA, USA). Splenocytes were crushed through cell strainers, resuspended with balanced salt solution (BSS), and treated with ammonium chloride solution to lyse RBC.
To remove APCs, splenocytes were run through a nylon wool column (PerkinElmer Life and Analytical Sciences, Boston, MA, USA) as described previously [31]. As CD45.1+ OT-I T cells were adoptively transferred into CD45.2+ C57BL/6 mice, activated OT-I CD8 T cells were positively selected using the CD45.1 congenic marker. Briefly, cells were labeled with biotinylated or fluorochrome-conjugated anti-CD45.1 mAb (BD Biosciences) and placed on ice for 30 min. Magnetic labeling was performed by using antibiotin or antifluorochrome MicroBeads (Miltenyi Biotec, Auburn, CA, USA), according to the manufacturer’s recommendation. These magnetically labeled CD45.1+ OT-I CD8 T cells were positively separated in a MACS column using a MACS magnet separator (Miltenyi Biotec). After several washes, the purity of the CD45.1+ OT-I CD8 T cells was typically at 80–99% CD45.1+.
The purified OT-I T cells were resuspended in MEM containing amino acids, salts, antibiotics, and FBS (complete tumor media). The cells were counted using a Z1 particle counter (Beckman Coulter, Miami, FL, USA), and 1 × 105 cells were placed into a well of a 96-well plate and cultured at 37°C. In some experiments, recombinant mouse (rm)IL-2, rmIL-4, rmIL-7, or rmIL-15 (Pepro Tech, Rocky Hill, NJ, USA) or human (h)IL-7 (Biological Resources Branch, NCI/NIH, Bethesda, MD, USA) was added at 10 ng/ml (mouse ILs) or 600 ng/ml (hIL-7) at the beginning of the culture.
Cell staining and flow cytometry
The following mAb were purchased from eBioscience (San Diego, CA, USA): anti-CD45.1, anti-CD25, anti-IFN-γ, anti-TNF, and control rat IgG1; and the following from BD Biosciences: anti-CD127, control rat IgG2a, anti-STAT5a (pY694), control mouse IgG1, and antiactive caspase 3, which recognizes the active form of caspase 3, not the proenzyme form. Cells were stained with primary antibodies in the presence of a blocking solution containing 5% normal mouse serum (Sigma-Aldrich), 10 μg/ml human γ-globulin (Sigma-Aldrich), and 0.1% sodium azide in culture supernatant from the 2.4.G.2 hybridoma {anti-FcR [32] for 30 min on ice and then washed in BSS wash buffer (3% FBS and 0.1% sodium azide in BSS)}.
For Mito Flow staining, at the end of in vitro culture, the cells were treated with Mito Flow reagent (Cell Technology, Mountain View, CA, USA) and anti-CD45.1 for 30 min at 37°C, according to the manufacturer’s recommendation.
For phosphorylated (p)-STAT5 and active caspase 3 staining, cells were fixed with 2% paraformaldehyde in BSS for 10 min at 37°C. After a couple of washes with BSS wash buffer, cells were resuspended with 100% methanol and incubated at room temperature for 30 min. Cells were then washed with BSS wash buffer and stained with either of the following combinations for 30 min at room temperature: p-STAT5 staining with anti-CD45.1, anti-CD25, anti-p-STAT5 (pY694), or isotype control antibodies; active caspase 3 staining with anti-CD45.1 and antiactive caspase 3.
For intracellular cytokine staining, 1 × 106 splenocytes were cultured with 1 μg Brefeldin A (Calbiochem) in the presence or absence of SIINFEKL peptide at 37°C for 4 h. The cells were stained with anti-CD45.1 mAb on ice for 30 min, and after a couple of washes, the cells were fixed with 2% paraformaldehyde in BSS. The cells were placed in permeabilization buffer (0.25% saponin in wash buffer) and then incubated with anti-IFN-γ, anti-TNF, or an isotype control rat IgG1 for 20 min at room temperature. Cells were washed twice with permeabilization buffer. All stained cells were assayed on a FACSCalibur (BD Biosciences), and the data were analyzed with CellQuest (BD Biosciences) or FlowJo software (Tree Star, San Carlos, CA, USA).
NF-κB ELISA and Western blot analysis
To examine NF-κB activity, Days 2.5–4 OT-I CD8 T cells were purified from splenocytes of immunized mice as described earlier. Five million cells were washed with PBS containing phosphatase inhibitors (10 mM NaF, 1 mM β-glycerophosphate, and 1 mM Na3VO4; all chemicals from Sigma-Aldrich), and the cell pellets were frozen at –80°C. A nuclear extract was prepared from the cell pellets using a nuclear extract kit according to the manufacturer’s recommendation (Active Motif, Carlsbad, CA, USA). Briefly, the pelleted cells were resuspended with hypotonic buffer and incubated for 15 min on ice. Detergent was added, and cell lysates were pelleted and then resuspended in Complete lysis buffer, incubated for 30 min on ice, and finally spun down. Supernatants were stored at –80°C, and protein concentration of the extract was determined using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL, USA).
The nuclear extracts were tested using a TransAM NF-κB family ELISA kit, according to the manufacturer’s protocol (Active Motif). Briefly, the nuclear extract was diluted to a concentration of 0.1 μg/μl, and 20 μl each sample was added into each well of a 96-well plate, which was coated with immobilized oligonucleotides containing the NF-κB consensus site sequence. The plate was incubated for 1 h at room temperature and then washed with washing buffer. NF-κB antibodies (p65 and p50) were diluted 1:1000 and added to each well. The wells were incubated for 1 h at room temperature and then washed three times. HRP antibody was added and incubated for 1 h at room temperature. After several washes with washing buffer, developing solution was added to all wells, and the plate was incubated for 5 min at room temperature. The reaction was halted by adding stop solution, and a microplate model 680 reader from Bio-Rad (Hercules, CA, USA) was used to read the sample absorbance at 450 nm (A450) with a reference wavelength of 655 nm.
For immunoblot (Western blot) analysis, cell pellets consisting of 5 × 106 cells were lysed in 40 μl lysis buffer A (50 mM Tris, 0.1% SDS, 40 μg/ml DNase I, 20 μg/ml RNase A, and 2 mM MgCl2) for 5 min at room temperature. Lysis buffer B [25.5 μl; 0.5 M Tris, 150 mM NaCl, 0.5% Triton X-100, 0.5% octyl-β-D-glucopyranoside, 10 mM NaF, 5 mM EDTA (pH 6.8)] was added into the homogenized lysates and incubated for 5 min at room temperature, and cell lysates were centrifuged at 12,000 rpm at 4°C for 10 min. Supernatants were removed, boiled for 5 min under reducing conditions [SDS sample buffer (40 mM Tris-HCl, pH 6.8, 8% SDS, 40% glycerol) with 20% β-ME], and then run on 4–15% SDS-PAGE (Bio-Rad). Gels were transferred onto a 0.2-μm nitrocellulose membrane (Bio-Rad) and typically incubated overnight with 1:1000 dilution of primary antibody for p-IκBα (Cell Signaling Technology, Danvers, MA, USA), IκBα, p-ERK1/2, ERK1, or β-actin (Sigma-Aldrich). The next day, blots were incubated with 1:1000 diluted chicken anti-rabbit IgG-HRP secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunoblot detection was performed using the ECL plus Western blotting detection kit (Amersham Biosciences, Piscataway, NJ, USA). The image was developed on an Amersham Storm 860 imaging system and analyzed with Adobe Photoshop CS2 software. Briefly, raw images were opened in Photoshop and adjusted in auto level. Based on molecular weight, a window area around each protein (i.e., IκBα, 41 kDa; ERK1, 44 kDa; ERK2, 42 kDa; actin, 45 kDa) was cropped, and the image was optimized and adjusted using brightness/contrast.
RESULTS
To characterize how dual-costimulated CD8 T cells accumulate, we examined mitochondrial membrane potential during the process of intrinsic cell survival. OT-I CD8 T cells were adoptively transferred into C57BL/6 mice and immunized with antigen and costimulation as described in Materials and Methods, and on Day 2.5, OT-I CD8 T cells were purified from spleen. To assess the intrinsic survivability of these cells, we cultured them overnight without any added stimulus besides media. The next day, using Mito Flow, the mitochondrial membrane potential was quantitated by flow cytometry (Fig. 1A). In healthy cells, mitochondria maintain an electrochemical gradient and a sufficient membrane potential to generate ATP. Mito Flow is a cell-permeable, cationic dye that accumulates in healthy mitochondria but not in mitochondria of apoptotic or dead cells [33]. Before culture, all of the costimulated cells showed maximal Mito Flow staining with comparable mean fluorescence intensity (MFI) levels (Fig. 1A, upper set). After overnight culture, however, the dual costimulation treatment group not only had a higher percentage of viable cells (20% live cells and at least threefold greater than the other groups) but also contained more cells that accumulated Mito Flow compared with the single costimulation groups (Fig. 1A, lower set). This was also true for purified Day 4 OT-I CD8 T cells from each group (data not shown). Thus, CD137 and CD134 dual costimulation conditions primary CD8 T cells to acquire an intrinsic survival program that maintains mitochondrial health.
Fig. 1.

Dual-costimulated CD8 T cell intrinsic survival is NF-κB-dependent. (A) OT-I Rag−/− splenocytes were adoptively transferred into C57BL/6 mice and then treated the next day with SIINFEKL peptide plus rat IgG control antibody or anti-CD137, anti-CD134, or both mAb. On Day 2.5, OT-I CD8 T cells were isolated from splenocytes using MACS column purification. Purified OT-I CD8 T cells (1×105) were cultured, and the following day, the mitochondrial membrane potential was measured using Mito Flow. Before (upper set) or after (lower set) culture, histograms were gated from forward-scatter (FSC)/side-scatter (SSC) plots and show representative Mito Flow staining from total cells or viable cells. The gray-tinted, dotted line indicates Mito Flow staining from gated dead cells, and the black line represents the mitochondrial membrane potential level from each sample. Numbers indicate the percentage of cells positive for Mito Flow dye. The culturing experiment was performed at two other time-points, and our analysis before culture was completed once. (B) OT-I Rag−/− recipient C57BL/6 mice were given SIINKFEKL peptide plus dual costimulation (Fig. 1B). On Day 2.5 or 4, OT-I CD8 T cells were purified from splenocytes using MACS columns. Fifty thousand cells were treated overnight with Bay11 (left panel) or NF-κB activation inhibitor (right panel) at the indicated concentrations. The line graphs show mean ± sem of the percentage of viable cells as measured by FSC/SSC from triplicate cultures in each group. The data are from one experiment but repeated at least three times. (C) Fifty thousand purified cells were cultured overnight with Bay11 (left panel) or NF-κB activation inhibitor (right panel) at the indicated concentrations in the presence of 20 ng/ml rmIL-2 (□), rmIL-4 (⋄), rmIL-7 (○), or rmIL-15 (▵). The next day, cells were analyzed for viability. The line graphs represent the percentage of live cells in each treatment. These are data from one experiment and were repeated at least two times with IL-2 or IL-15 and at least three times with IL-7 or IL-4.
To study the intracellular mechanism of this intrinsic survival effect by dual costimulation, we examined its requirements of various signaling components. Thus, dual-costimulated OT-I CD8 T cells were cultured with various inhibitors. PD98059 (MEK inhibitor [34]), U0126 (MEK1/2 inhibitor [35]), and SB203580 (p38 MAPK inhibitor [36]) had no significant effect on survival of dual-costimulated CD8 T cells after overnight culture compared with vehicle alone (data not shown). We also tested other inhibitors such as wortmanin (PI-3K [37]) and LY294002 (PI-3K [38]), and no significant difference in survival was observed compared with vehicle alone (data not shown). SP600125, an inhibitor of JNK [39], had a minimal effect inhibiting cell survival (data not shown). Nevertheless, when the NF-κB inhibitor Bay11 was tested, we did not detect viable cells, even after the cells were pulsed with this inhibitor for just 3 h (data not shown). Taken together, these data suggest that the NF-κB pathway, but neither the MEK nor p38 MAPK pathway, plays a crucial role in dual costimulation-induced intrinsic cell survival.
To explore the role of NF-κB signaling in dual costimulation-mediated cell survival, Bay11 and NF-κB activation inhibitor, another NF-κB-specific inhibitor, were compared. When purified, Day 2.5 OT-I CD8 T cells were treated separately with pharmacological and noncytotoxic doses of these NF-κB-specific inhibitors, and the viability of these cells was reduced profoundly in a titrable manner after overnight culture (Fig. 1B). Overall, our results show that cell death by Bay11 was consistently robust at 10 μM. However, as the primary effector T cells that we generated may be intrinsically different from other cell types or even T cells primed differently, it will be crucial for others to titer Bay11 against graded cell numbers and assay a variety of time-points.
It was hypothesized that IL-7 or other survival-inducing cytokines [40] may rescue the T cells from Bay11 (or NF-κB activation inhibitor)-induced cell death. As IL-7 uses different signaling components such as JAK-STAT signaling [41], we asked whether survival cytokines were able to uncouple the effect of Bay11 on cell death. The OT-I CD8 T cells were treated overnight with a titration of Bay11 (Fig. 1C, left panel) or the NF-κB activation inhibitor (Fig. 1C, right panel) in the presence of rIL-2, rIL-4, rIL-7, or rIL-15. Although these cytokines substantially enhanced cell survival compared with cultures not containing cytokines (data not shown, and compare the maximal percentage of viable cells from Fig. 1, B with C), Bay11 and the NF-κB activation inhibitor blocked cytokine-induced cell survival profoundly in a consistent and titratable manner.
To delve further into the idea that the death pathway induced by interfering with NF-κB signaling overrides a survival signal, we tested whether IL-7-pre-established cell survival was able to block Bay11-driven death. We chose IL-7 because of its documented role in keeping these effector CD8 T cells alive [41]. OT-I-purified Day 4 CD8 T cells were cultured for 4 h with rIL-7, and at the same time, 10 μM Bay11 was added at Time 0, 1, or 3 h (Fig. 2). After 4 h, all cells were assayed for mitochondrial membrane potential by flow cytometry (Fig. 2A). There was no significant difference in the percentage of viable cells or the Mito Flow-positive population between media alone and the IL-7 treatment group. However, when cells were cultured with Bay11 plus IL-7 for 4 h, the percentage of live cells was reduced (Fig. 2A). Also, this demonstrated that cell death was not a cytotoxic effect, as the dead cells from the Bay11 cultures were the same size and granularity as the dead cells in the absence of Bay11. Even a 3-h preculture with IL-7 could not prevent the death-inducing effects of Bay11 (Fig. 2A, fifth column). To address whether IL-7-driven STAT5 signaling was affected in the presence of Bay11, we examined p-STAT5 and CD25 expression (Fig. 2B). As expected, cells cultured with IL-7 showed higher p-STAT5 and CD25 expression compared with those with nothing (Fig. 2B). When Bay11 and IL-7 were added at the beginning of culture, p-STAT5 and CD25 expression was inhibited. On average, we observed less inhibition when cells were cultured with IL-7 for 3 h first and then with Bay11 for the last hour. For example, 46% of the viable cells in the IL-7 treatment group were p-STAT5- and CD25-double-positive, whereas cells from the IL-7-first, Bay11-later group were 41% double-positive (Fig. 2B, compare second and fifth columns). The inhibition was greater when MFI for p-STAT5 was calculated (Supplemental Table 1). Thus, inhibiting NF-κB did not completely block all aspects of IL-7-induced signaling but was nevertheless able to dominate over IL-7-induced survival. Together, these data imply that an active NF-κB pathway is loaded in the dual-costimulated T cells and that Bay11 (or NF-κB activation inhibitor) abrogates this signal and dominates over cytokine-induced cell survival.
Fig. 2.

Cell survival established by IL-7 cannot prevent Bay11-induced cell death. Day 4 OT-I CD8 T cells were purified from splenocytes, and 1 × 105 cells were cultured for 4 h under the following conditions: The first column indicates cells cultured with nothing; second through fifth columns represent cells treated with 0.6 μg/ml hIL-7; the third column cells were incubated with 10 μM Bay11 during the entire 4-h culture; in the fourth column, Bay11 was added into a well at t = 1 h; in the fifth column, Bay11 was added during the last hour of culture. (A) The mitochondrial membrane potential was measured using the Mito Flow reagent. Histograms show representative staining of Mito Flow from total cells (middle row) or the viable cell population (bottom row). The gray-tinted line indicates staining of Mito Flow from gated dead cells, and the black line represents the mitochondrial membrane potential level from each sample. Numbers indicate mean ± sem of the percentage of cells that accumulated Mito Flow from two cultures derived from two separate experiments. We also performed this experiment three times using AnnexinV staining, resulting in comparable data (data not shown). (B) Cells were analyzed for CD25 expression and p-STAT5. Dot plots are representative staining from all cells (upper row) or from the live cell population (lower row). These data are representative of three experiments.
As Bay11 can inhibit the action of the IKK complex [22], we examined the effect of Bay11 on IκBα phosphorylation in our experimental system. Day 4 CD8 T cells contained p-IκBα (data not shown), and after 1 h, these cells managed to maintain p-IκBα (Fig. 3A, upper, top left panel). However, after 5 h, p-IκBα was not detected, which was probably a result of degradation (Fig. 3A, upper, top right panel) [42]. In the presence of Bay11, we did not detect p-IκBα at 1 or 5 h of culture. Thus, Bay11 completely inhibited the maintenance of p-IκBα. Furthermore, the level of IκBα expression was lower in the Bay11 treatment groups compared with that of the vehicle-alone-treated cells, implying that Bay11 might block NF-κB induction of IκBα gene transcription [43]. Next, we examined the effects of Bay11 and IL-7 on IκBα phosphorylation. IL-7-treated cells maintained phosphorylation of IκBα, but in the presence of Bay11, whether IL-7 was provided or not, p-IκBα declined rapidly (Fig. 3A, lower panels). We also observed similar data from a 1-h culture (data not shown). Taken together, these results show that the NF-κB inhibitor, Bay11, dismantled IκBα phosphorylation rapidly, and IL-7 was unable to block this effect.
Fig. 3.

Bay11 blocks phosphorylation of IκBα and NF-κB activity even in the presence of IL-7. Day 4 OT-I CD8 T cells were purified from spleen cells, and 5 × 106 OT-I T cells were cultured with vehicle alone, 10 μM Bay11, 0.6 μg/ml rhIL-7, or IL-7 plus Bay11 for the indicated times. (A) Cell lysates were prepared and immunoblotted with antibodies specific for p-IκBα, IκBα, or β-actin. P+I, Positive control lysates for p-IκBα derived from naïve splenocytes that were stimulated with 125 ng/ml PMA plus 2.5 μg/ml ionomycin for 1 h. β-Actin was used as protein-loading controls. The tabulated values represent the relative ratios of p-IκBα or IκBα to β-actin (relative ratio with β-actin was set at 1.0). (B) A nuclear extract was prepared, and NF-κB DNA-binding activity was examined. Data show p65 and p50 NF-κB activity, and the black bar indicates activity at t = 0 h (before culture), the darker gray bar for the vehicle-treated group, the dark gray for IL-7 treatment, the light gray for Bay11, the lighter gray for IL-7 plus Bay11, and the open bar is a negative control. NF-κB activity data from a 0.5-h culture was performed once, and the experiment for a 2.5-h culture was repeated twice. The summary of data from four other experiments is shown in Table 1. (C) Five million OT-I CD8 T cells were culture for 1 h with 10 μM Bay11 or 50 μM PD98059 and then pulsed or not with 125 ng/ml PMA plus 2.5 μg/ml ionomycin for 10 min. Cell lysates were immunoblotted for p-ERK1/2 (ExpI). The tabulated values show the relative ratios of p-ERK1 to p-ERK2 (relative ratio of p-ERK2 was set as 1.0). In experiment 2 (ExpII), lysates were blotted simultaneously for p-ERK1/2 and p-IκBα and ERK1 and IκBα, the latter two used as protein-loading controls. The tabulated values indicate the relative ratios of p-ERK1 or p-ERK2 to ERK1 and p-IκBα to IκBα.
These data suggested that the dual-costimulated CD8 T cells possessed intrinsic NF-κB activity, and we asked whether Bay11 or IL-7 would impact this activity. First, purified CD8 T cells were cultured with vehicle alone or Bay11, nuclear extracts were prepared, and DNA-binding activity to a NF-κB consensus sequence was measured as described in Materials and Methods. We found that the cells possessed a high p65 and p50 basal activity at the time of purification, but by 0.5 or 2.5 h in culture, this activity declined gradually (Fig. 3B, compare t=0 with vehicle). For example, p65 activity of vehicle-treated cells for 0.5 h was ∼91% of t = 0 h cells, whereas those from a 2.5-h culture were ∼24% (Fig. 3B). Second, Bay11 treatment massively reduced the activity of p65 and p50. Third, IL-7 was not fully able to block this decline, nor was IL-4 (data not shown), although these cytokines are potent inducers of survival [40]. For example, even 0.5 h exposure of Bay11 substantially reduced the capacity of IL-7 to maintain the level of NF-κB activity by at least threefold. A summary of our experiments examining NF-κB activity in this system is given in Table 1.
TABLE 1.
Bay11 Reduces NF-κB Activity Rapidly and Profoundly
| Exp. I | Exp. II | Exp. III | Exp. IV | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 h with Bay 11, wash-out, 3 h culture
|
1 h
|
1 h
|
1 h
|
||||||
| Vehicle | Bay11 5 μM | Vehicle | Bay11 10 μM | Vehicle | Bay11 5 μM | Bay11 1 μM | Vehicle | Bay11 1 μM | |
| p65 | 100 | 6 | N.D.a | N.D.a | 100 | –4 | 27 | 100 | 14 |
| p50 | 100 | –0.9 | 100 | 15 | 100 | –7 | 37 | 100 | 26 |
Five million purified Day 2.5 OT-I CD8 T cells were cultured with vehicle alone or Bay11. In Experiment I, cells were incubated with 5 μM Bay11 for 1 h, washed twice with media, and then cultured for another 3 h without Bay11. In Experiments II–IV, cells were treated for 1 h with 10 μM (Exp. II), 5 μM or 1 μM (Exp. III), or 1 μM (Exp. IV) Bay11 or vehicle alone. A nuclear extract was prepared from each sample, and NF-κB DNA-binding activity (p65 and p50) was examined as described in Materials and Methods. Data show the percentage of remaining NF-κB activity after Bay11 treatment in each experiment, which was determined by setting activity of vehicle alone after a culture as 100 and that of negative control (without lysates) as 0.
N.D., Not done.
Based on these data, we asked whether other signaling pathways were turned off by Bay11 treatment. Thus, dual-costimulated OT-I T cells were incubated for 1 h with 10 μM Bay11 or with 50 μM PD98059 as a comparison, and then cells were pulsed for 10 min in the absence or presence with the strong T cell mitogen PMA plus ionomycin (P+I). p-ERK1/2 was determined by immunoblotting (Fig. 3C, ExpI). Even with a 1-h exposure to Bay11, the T cells retained the ability to phosphorylate ERK1/2 after mitogen stimulation, which was inhibited by the PD98059 (MEK inhibitor) treatment. Moreover, when p-ERK1/2 and p-IκBα were examined in Bay11-treated cells, p-IκBα was not detected, whereas ERK1/2 was phosphorylated after PMA plus ionomycin stimulation (Fig. 3C, ExpII). Thus, Bay11-treated cells were able to respond to mitogen stimulation, although NF-κB signaling was turned off completely and specifically.
As a mitogen could signal into the Bay11-treated T cells, we tested whether effector cytokines after TCR or mitogen stimulation would be affected by Bay11. Splenocytes from peptide and dual-costimulated mice were rechallenged in vitro with SIINFEKL peptide or PMA plus ionomycin and were then assayed for effector cytokine production using intracellular cytokine staining (Fig. 4A). Consistent with the effect of Bay11 on NF-κB activity, Bay11 inhibited CD8 T cell effector cytokine production profoundly in a concentration-dependent manner. For example, after TCR triggering, ∼45% IFN-γ/TNF double-positive cells were detected with 0.5 μM Bay11, whereas 17% were seen with 10 μM Bay11 treatment (Fig. 4A). Similar data were also obtained for PMA plus ionomycin stimulation in the presence of Bay11 (Fig. 4A, middle panel), and our data are summarized in Table 2.
Fig. 4.

Bay11 reduces CD8 T cell effector cytokine production by enhancing AICD. (A) Day 3 spleen cells were stimulated for 5 h with 1 μg SIINFEKL peptide (top panel), 125 ng/ml PMA plus 2.5 μg/ml ionomycin (middle panel), or nothing (bottom panel) in the presence of vehicle alone or Bay11 (0.5 μM, 5 μM, or 10 μM) and then assayed for intracellular cytokine production. Representative zebra plots are shown from each group of gated OT-I CD8 T cells. The numbers in the upper right quadrant indicate the percentage of OT-I CD8 T cells producing IFN-γ and TNF. This experiment was repeated at least three times, and the data are summarized in Table 2. (B) Day 4 dual-costimulated OT-I CD8 T cells (1×105 cells) were cultured for 4 h with media alone (left panel), 5 μg/ml SIINFEKL peptide (middle panel), or SIINFEKL peptide plus 10 μM Bay11 (right panel). All cells were assayed for mitochondrial membrane potential, and histograms show representative Mito Flow staining of total cells. Comparable data were obtained from at least three other experiments.
TABLE 2.
Bay11 Inhibits Effector Cytokine Production by Antigen-Specific CD8 T Cells
| Percent of OT-I T cells producing IFN-γ and TNF
|
||||||||
|---|---|---|---|---|---|---|---|---|
| with SIINFEKL peptide
|
with PMA + ionomycin
|
|||||||
| No Bay11 | 0.5–1.1 μM | 3.3–5 μM | 10 μM | No Bay11 | 0.5–1.1 μM | 3.3–5 μM | 10 μM | |
| Exp. I Day 6 | 58 ± 2.0 | 42 ± 1.6 | 20 ± 0.1 | 4.6 ± 1.2 | 70 ± 1.8 | 72 ± 0.5 | 39 ± 0.9 | 13 ± 2.7 |
| Exp. II Day 3 | 51 ± 1.4 | 43 ± 1.8 | 31 ± 1.5 | 14 ± 2.0 | 55 ± 3.8 | 50 ± 3.8 | 39 ± 4.8 | 21 ± 3.1 |
| Exp. III Day 3 | 38 ± 4.4 | 35 ± 4.7 | 14 ± 2.1 | 2.5 ± 0.3 | 60 ± 4.9 | 58 ± 4.0 | 30 ± 6.6 | 6.7 ± 3.3 |
| Exp. IV Day 3 | 32 | 27 | 14 | 0.5 | 47 | 45 | 34 | 2.3 |
| Exp. V Day 3 | 30 | 29 | 4.3 | N.D. | 26 | 31 | 14 | N.D. |
Day 3 or 6 spleen cells were restimulated with SIINFEKL peptide or 125 ng/ml PMA plus 2.5 μg/ml ionomycin for 4 h in the absence or presence of Bay11 and then assayed by intracellular cytokine production. Data are the mean ± sem percentage of IFN-γ and TNF double-positive cells within the OT-I T cell population. Experiments IV and V have one mice, and the others have three mice per experiment.
One possibility for the reduced cytokine production was enhanced AICD. As expected, stimulation with the TCR-specific peptide (SIINFEKL) began to induce AICD within 5 h, but inclusion of Bay11 inhibited all cell survival as measured by Mito Flow staining (Fig. 4B). Thus, Bay11 accentuated AICD to a much greater extent than TCR stimulation alone, possibly pre-empting the initiation of cytokine synthesis.
Because of the rapidity of the Bay11 cell death response, the mechanism involved was examined. Thus, dual-costimulated OT-I CD8 T cells were cultured for 4 h with SIINFEKL peptide alone or SIINFEKL peptide plus 10 μM Bay11 and then assayed for the active form of caspase 3 (Fig. 5A), an executor of apoptosis [44, 45]. At Time 0 h, there was a low level of active caspase 3 (data not shown); however, after a 4 h-culture with peptide, 57% of the cells were positive for active caspase 3 and underwent AICD. Nonetheless, when cells were cultured with SIINFEKL peptide and Bay11 only, 5.5% expressed the active form of caspase 3 (Fig. 5A, right panel). We also examined this idea extensively in live- and dead-gated cells and in the presence of IL-7 (Table 3). The active form of caspase 3 within the Day 4 OT-I CD8 T cell population was evaluated, and as expected, cells treated with SIINFEKL peptide alone showed increases of active caspase 3 after a 3-h culture, whether cells were from the live or dead gate (Table 3, see Exp. I). Interestingly, IL-7 treatment had no significant effect on active caspase 3 compared with those cells with vehicle alone, suggesting that AICD dominated over cytokine-induced survival. Cells cultured with Bay11 again showed a much lower percentage of active caspase 3 compared with those with vehicle alone, which was also true for those with Bay11 treatment in the presence of IL-7. Therefore, these data suggest that the Bay11-mediated death of CD8 effector T cells is caspase-independent (Fig. 5A and Table 3).
Fig. 5.
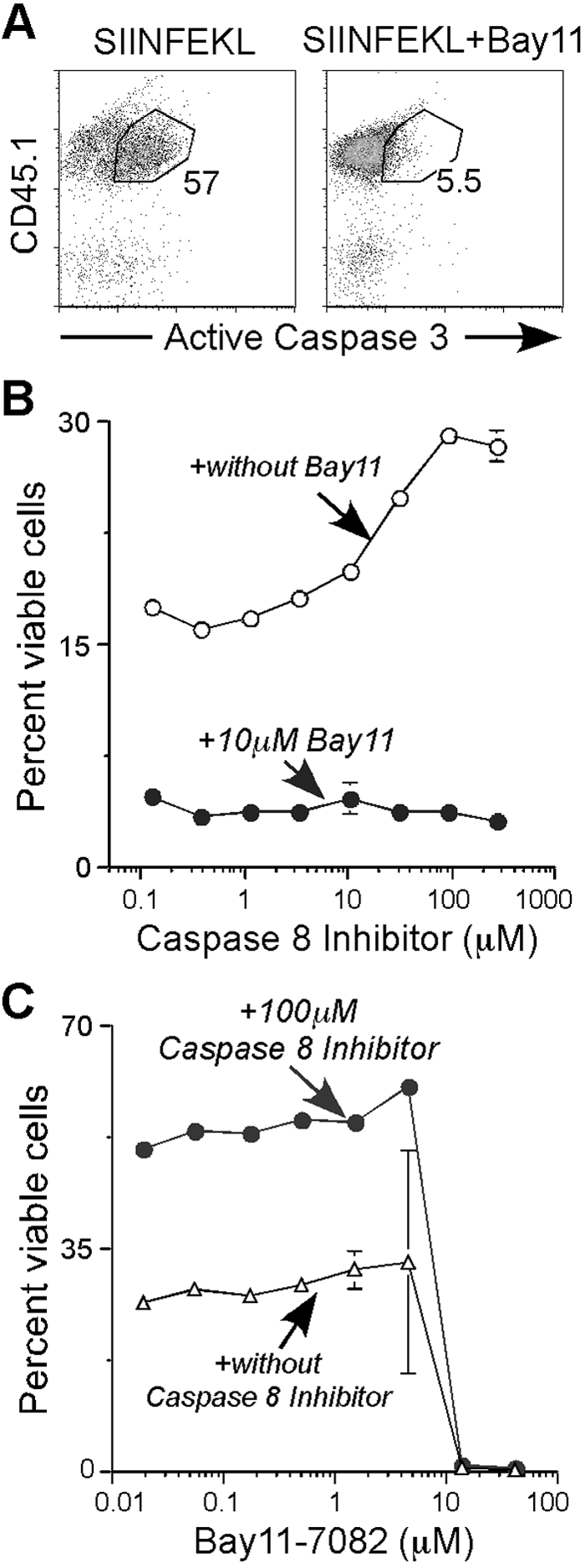
Bay11 reroutes CD8 effector T cell AICD from a caspase-dependent to caspase-independent process. On Day 4, purified OT-I CD8 spleen T cells were cultured under the following conditions: (A) Cells (1×105) were incubated with 5 μg/ml SIINFEKL peptide (left panel) or SIINFEKL peptide plus 10 μM Bay11 (right panel), and after 4 h, cells were stained for CD45.1 and the active form of caspase 3. Dot blots represent CD45.1 and active caspase 3 of each sample gated from total cells. This experiment was repeated at least three times and also at different time-points (Table 3). (B) Fifty thousand SIINFEKL-stimulated cells were incubated with caspase 8 Inhibitor II at the indicated concentrations in the absence (○) or presence (•) of 10 μM Bay11. The mean ± sem of the percentage of viable cells from triplicate cultures in each treatment is given. This experiment was done twice. (C) Cells were stimulated with SIINFEKL and cultured in duplicate with 10 μM Bay11 in the absence of inhibitor (▵) or in single cultures in the presence of 100 μM caspase 8 Inhibitor II (•). Line graph represents the percentage of live cells in each group.
TABLE 3.
Bay11 Treatment Prevents Active Action of Caspase 3
| Time of culture | Percent of OT-I cells positive for active caspase 3
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Vehicle
|
Bay11
|
IL-7
|
Bay11 + IL-7
|
||||||
| Live | Dead | Live | Dead | Live | Dead | Live | Dead | ||
| Exp. I | 1 h | 6.8 | 8.8 | 1.1 | 5.2 | 8.8 | 9.4 | 1.1 | 2.5 |
| 3 h | 36 | 31 | 1.0 | 3.0 | 38 | 35 | 2.1 | 2.3 | |
| O/N | 62 | 82 | 4.6 | 2.6 | 57 | 80 | 6.1 | 3.2 | |
| Exp. II | 4.5 h | 3.4 | 4.5 | 1.2 | 1.2 | 4.6 | 7.8 | 1.4 | 1.8 |
| O/N | 42 | 45 | 18 | 3.1 | 47 | 34 | 14 | 2.6 | |
| 2 days | 37 | 47 | 12 | 3.7 | 59 | 57 | 10 | 3.4 | |
| Exp. III | 4 h | 26 | 29 | 3.9 | 1.0 | 25 | 27 | 4.6 | 1.0 |
| Exp. IV | 4.5 h | 66 | 79 | 13 | 11 | 70 | 81 | 16 | 14 |
| Exp. V | 4 h | 38 | 39 | 9.9 | 10 | 34 | 36 | 13 | 12 |
| 4 h (w/ APC) | 40 | 35 | 14 | 14 | 37 | 37 | 14 | 13 | |
Purified Day 4 OT-I CD8 T cells (105) were placed into a well of a 96-well plate and cultured with 5 μg/ml SIINFEKL peptide for the indicated times. At the same time, cells were treated with vehicle alone, 10 μM Bay 11, 0.6 μg/ml rh IL-7, or Bay11 plus IL-7. At the end of culture, the percentage of live or dead OT-I cells positive for the active form of caspase 3 is given. O/N, Overnight; (w/ APC), OT-I CD8 T cells were purified and mixed with naive spleen cells (105) for the duration of the culture.
To further explore the role of caspases, purified Day 4 OT-I CD8 T cells were treated with SIINFEKL peptide to induce AICD in the presence of caspase 8 Inhibitor II (Z-IETD-FMK), with or without Bay11 (Fig. 5B). The caspase 8 inhibitor blocked AICD, as there was approximately a twofold increase of viable cells when the cells were treated with 90 μM inhibitor compared with those exposed to a lower concentration. Nevertheless, the survival-inducing effect of the caspase 8 inhibitor was blocked completely in the presence of Bay11. Consistent with these data, when cells were cultured with a titration of Bay11, with or without 100 μM caspase 8 inhibitor, Bay11 significantly blocked the survival effect by the inhibitor in a titrable manner (Fig. 5C). We also tested other inhibitors such as caspase 3 Inhibitor II [Z-Asp-Glu-Val-Asp-FMK (Z-DEVD-FMK)], caspase 3 Inhibitor III (acetyl-DEVD-chloromethylketone), and a pan-caspase inhibitor (Z-Val-Ala-Asp-FMK), all of which showed similar results as those with the caspase 8 inhibitor (data not shown). Thus, Bay11-mediated CD8 effector T cell death is caspase-independent and overrides caspase-dependent AICD, and both pathways dominate over IL-7 cytokine-induced survival.
DISCUSSION
Previously, it was demonstrated that simultaneous administration of agonist anti-CD137 and -CD134 mAb synergistically enhanced CD8 T cell effector function leading to rapid rejection of murine tumors [1]. IL-7 signaling was required for accumulation of these supereffector CD8 T cells [2], and in this study, we set out to uncover pathways that were important for this process. After testing many pathways, we found that CD137 and CD134 programmed intrinsic cell survival in a NF-κB-sensitive manner. The specific NF-κB inhibitor, Bay11, overrode costimulation- and cytokine-induced T cell survival. Moreover, interfering with NF-κB signaling through the action of Bay11 induced caspase-independent death even when cells were undergoing caspase-dependent AICD. These data suggest that basal NF-κB activity is a switch-point to control effector CD8 T cells and provide a means to purge them on demand.
This latter issue of removing undesirable effector T cells is naturally pertinent to treating autoimmune disease, and recently, Bay11 was shown to suppress inflammation and erosion in knee joints of arthritic mice in an antigen-dependent manner [46]. A different scenario, however, where it might be useful to prime and subsequently terminate an effector T cell response would be with T cell-based therapies that target tumor differentiation antigens that are expressed on tumors as well as the normal tissues from which they derive. Thus, permitting the effector T cell response to proceed for a limited time might allow for significant tumor destruction while minimizing the autoimmune destruction of healthy tissues that can often occur [47, 48]. Bay11 may by useful under these circumstances by rapidly impacting the life and death of stimulated T cells, even when they are induced with powerful costimulation, as might be necessary for cancer immunotherapy.
CD137 and CD134 receptors are excellent examples of powerful costimulators. In combination, they induce robust CD8 T cell responses, and although the signaling pathways are not resolved completely, there is evidence for NF-κB, JNK, and p38 MAPK activity in this process, possibly through recruitment of various TRAFs [49]. Consistent with these data, CD137 and CD134 dual costimulation-mediated cell survival was dependent on the NF-κB pathway. This included intrinsic survival, where there were no added factors besides media, and cytokine-mediated survival (Fig. 1). The NF-κB inhibitor, Bay11, blocked both processes and even overrode IL-7-pre-establisied cell survival (Fig. 2). As cell viability is also dependent on the balance between reactive oxygen species generation and inactivation by mitochondria [50, 51], we used a synthetic superoxide dismutase mimetic compound, manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), which has been shown to decrease staurosporin-induced cell death [52]. MnTBAP did not block Bay11-induced cell death, although this chemical increased survival of the vehicle-alone group (data not shown). Therefore, besides blocking cytokine-induced survival, Bay11 inhibited survival induced by the antioxidant MnTBAP. Ultimately, these data suggest a hierarchical power of NF-κB activity over antioxidant- and cytokine-induced cell survival, which is dependent on engagement of the classical NF-κB pathway.
To better understand the molecular process of survival, we examined the lasting and downstream effect of CD137 and CD134 signaling. We found that the dual-costimulated cells were loaded with p-IκBα at the time of purification, which correlated with a high basal level of NF-κB. Bay11 rapidly inhibited both processes, which may help to explain its potent, antitumorigenic potential. These data suggest that Bay11 inhibited the survival effect of IL-7 by blocking phosphorylation of IκBα, even in the presence of p-STAT-5 (Fig. 2B). Therefore, this shows the potency of Bay11, as a constitutively expressing STAT5b transgenic mouse, was shown to contain death-resistant CD8 T cells [53]. Interestingly, IL-7 did not induce NF-κB, suggesting that the IL-7 pathway is distinct from the CD137 and CD134 pathways that promoted NF-κB activation in vivo (Fig. 3B). Perhaps IL-7 promotes cell survival by maintaining residual NF-κB activity rather than enhancing it. Taken together, our data show that once the classical NF-κB pathway is uncoupled, cells are prone to die, and even the presence of a survival signal cannot block this effect.
Unexpectedly, Bay11 minimally affected molecules in the TCR signaling pathway (Fig. 3C), but in contrast, effector cytokine production was impaired substantially in a concentration-dependent manner (Fig. 4A). A likely explanation was the rapid induction of cell death (Fig. 4B), and consistent with these data are reports showing that Bay11 promotes apoptosis or inhibits growth of various cancer cells [25]. In particular, one report [54] demonstrated that Bay11 sensitized high-risk myelodysplastic syndrome patient-derived cell lines to starvation-induced cell death, occurring with the complete characteristics of apoptosis, such as release of cytochrome c, apoptosis-inducing factor, and endonuclease G from mitochondria and a loss of mitochondria membrane potential. However, they and another study [28] have observed that Bay11 promoted activation of caspase 3, which differed from our study (Fig. 5A). In our model system, we used primary T cells and the SIINFEKL peptide as the apoptosis trigger, as AICD can be induced in activated T cells by stimulating with the TCR-specific peptide [55]. To study caspase-independent death by Bay11, we tested the effects of a caspase 8 inhibitor and demonstrated that at 10 μM Bay11, the caspase 8 inhibitor was ineffective (Fig. 5B), but at lower Bay11 concentrations, the caspase 8 inhibitor prevented Bay11-induced death and enhanced survival (Fig. 5C). Thus, after TCR triggering, Bay11 rerouted AICD from a caspase-dependent death process to a caspase-independent pathway.
Although we showed that Bay11-induced death overrode cytokine-mediated cell survival in a caspase-independent manner, the details about the molecular process of how dual costimulation programs intrinsic cell survival and how Bay11 promotes cell death are unknown. Perhaps the combination of CD137 and CD134 may redistribute TRAFs, leading to activation of the classical NF-κB pathway, consistent with results from Arch et al. [56]. Second, the combination of CD137 and CD134 costimulation may be able to recruit TRAF6, which predominantly activates the classical NF-κB pathway [57]. We also cannot rule out a role for the alternative NF-κB pathway in our model system. Lastly, Bay11 may induce death downstream to all of these effects by rapidly modulating mitochondrial activity, thereby trumping the role of these pathways. One interesting possibility is that Bay11 recruits the autophagic pathway to induce T cell death [58]. In particular, this may involve a physical process of mitochondrial collapse, which may or may not be dependent on the changes to the NF-κB pathway.
In summary, our results demonstrated how CD8 T cells intrinsically survive better after dual costimulation. This was based on concomitant CD137 and CD134 direct signaling leading to the augmentation of the NF-κB pathway in the CD8 T cell population and ultimately enhancing intrinsic survival of these cells. This study may reveal approaches for inducing and then rapidly dismantling powerful CD8 effector T cell responses in vivo.
Acknowledgments
This work was supported by NIH grants AI 142858 an AI 52108 (A. T. V.). The authors thank Dr. Menoret (Department of Immunology, The University of Connecticut Health Center) for providing expertise and assistance with immunoblotting.
References
- Lee S J, Myers L, Muralimohan G, Dai J, Qiao Y, Li Z, Mittler R S, Vella A T. 4-1BB and OX40 dual costimulation synergistically stimulate primary specific CD8 T cells for robust effector function. J Immunol. 2004;173:3002–3012. doi: 10.4049/jimmunol.173.5.3002. [DOI] [PubMed] [Google Scholar]
- Lee S J, Rossi R J, Lee S K, Croft M, Kwon B S, Mittler R S, Vella A T. CD134 costimulation couples the CD137 pathway to induce production of supereffector CD8 T cells that become IL-7 dependent. J Immunol. 2007;179:2203–2214. doi: 10.4049/jimmunol.179.4.2203. [DOI] [PubMed] [Google Scholar]
- Dempsey P W, Doyle S E, He J Q, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- Arch R H, Thompson C B. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor κB. Mol Cell Biol. 1998;18:558–565. doi: 10.1128/mcb.18.1.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsikov E N, Laouini D, Dunn I F, Sannikova T Y, Davidson L, Alt F W, Geha R S. TRAF1 is a negative regulator of TNF signaling. Enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–657. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- Liao G, Zhang M, Harhaj E W, Sun S C. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- Coope H J, Atkinson P G, Huhse B, Belich M, Janzen J, Holman M J, Klaus G G, Johnston L H, Ley S C. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Wu L, Wesche H, Arthur C D, White J M, Goeddel D V, Schreiber R D. Defective lymphotoxin-β receptor-induced NF-κB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- Thompson J S, Bixler S A, Qian F, Vora K, Scott M L, Cachero T G, Hession C, Schneider P, Sizing I D, Mullen C, Strauch K, Zafari M, Benjamin C D, Tschopp J, Browning J L, Ambrose C. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-κ B2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- Xiao G, Harhaj E W, Sun S C. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Xiao G, Fong A, Sun S C. Induction of p100 processing by NF-κB-inducing kinase involves docking IκB kinase α (IKKα) to p100 and IKKα-mediated phosphorylation. J Biol Chem. 2004;279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay T, Roth J A, Maxwell S A. Altered expression of the p50 subunit of the NF-κ B transcription factor complex in non-small cell lung carcinoma. Oncogene. 1995;11:999–1003. [PubMed] [Google Scholar]
- Bargou R C, Emmerich F, Krappmann D, Bommert K, Mapara M Y, Arnold W, Royer H D, Grinstein E, Greiner A, Scheidereit C, Dörken B. Constitutive nuclear factor-κB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest. 1997;100:2961–2969. doi: 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sovak M A, Arsura M, Zanieski G, Kavanagh K T, Sonenshein G E. The inhibitory effects of transforming growth factor β1 on breast cancer cell proliferation are mediated through regulation of aberrant nuclear factor-κB/Rel expression. Cell Growth Differ. 1999;10:537–544. [PubMed] [Google Scholar]
- Wang W, Abbruzzese J L, Evans D B, Larry L, Cleary K R, Chiao P J. The nuclear factor-κ B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- Suh J, Payvandi F, Edelstein L C, Amenta P S, Zong W X, Gelinas C, Rabson A B. Mechanisms of constitutive NF-κB activation in human prostate cancer cells. Prostate. 2002;52:183–200. doi: 10.1002/pros.10082. [DOI] [PubMed] [Google Scholar]
- Kojima M, Morisaki T, Sasaki N, Nakano K, Mibu R, Tanaka M, Katano M. Increased nuclear factor-κB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer Res. 2004;24:675–681. [PubMed] [Google Scholar]
- Mayo M W, Baldwin A S., Jr The transcription factor NF-κB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000;1470:M55–M62. doi: 10.1016/s0304-419x(00)00002-0. [DOI] [PubMed] [Google Scholar]
- Herr I, Debatin K M. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–2614. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- Kaufmann S H, Earnshaw W C. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- Pierce J W, Schoenleber R, Jesmok G, Best J, Moore S A, Collins T, Gerritsen M E. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Keller S A, Hernandez-Hopkins D, Vider J, Ponomarev V, Hyjek E, Schattner E J, Cesarman E. NF-κB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood. 2006;107:3295–3302. doi: 10.1182/blood-2005-07-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrasch T, Kim J S, Muhlbauer M, Magness S T, Jobin C. Gnotobiotic IL-10−/−; NF-κ B(EGFP) mice reveal the critical role of TLR/NF-κ B signaling in commensal bacteria-induced colitis. J Immunol. 2007;178:6522–6532. doi: 10.4049/jimmunol.178.10.6522. [DOI] [PubMed] [Google Scholar]
- Fernandez-Majada V, Aguilera C, Villanueva A, Vilardell F, Robert-Moreno A, Aytes A, Real F X, Capella G, Mayo M W, Espinosa L, Bigas A. Nuclear IKK activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc Natl Acad Sci USA. 2007;104:276–281. doi: 10.1073/pnas.0606476104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Vargas H, Rodriguez-Pinilla S M, Julian-Tendero M, Sanchez-Rovira P, Cuevas C, Anton A, Rios M J, Palacios J, Moreno-Bueno G. Gene expression profiling of breast cancer cells in response to gemcitabine: NF-κB pathway activation as a potential mechanism of resistance. Breast Cancer Res Treat. 2007;102:157–172. doi: 10.1007/s10549-006-9322-9. [DOI] [PubMed] [Google Scholar]
- Garcia M G, Alaniz L, Lopes E C, Blanco G, Hajos S E, Alvarez E. Inhibition of NF-κB activity by BAY 11-7082 increases apoptosis in multidrug resistant leukemic T-cell lines. Leuk Res. 2005;29:1425–1434. doi: 10.1016/j.leukres.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Pickering B M, de Mel S, Lee M, Howell M, Habens F, Dallman C L, Neville L A, Potter K N, Mann J, Mann D A, Johnson P W, Stevenson F K, Packham G. Pharmacological inhibitors of NF-κB accelerate apoptosis in chronic lymphocytic leukemia cells. Oncogene. 2007;26:1166–1177. doi: 10.1038/sj.onc.1209897. [DOI] [PubMed] [Google Scholar]
- Shuford W W, Klussman K, Tritchler D D, Loo D T, Chalupny J, Siadak A W, Brown T J, Emswiler J, Raecho H, Larsen C P, Pearson T C, Ledbetter J A, Aruffo A, Mittler R S. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186:47–55. doi: 10.1084/jem.186.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shamkhani A, Birkeland M L, Puklavec M, Brown M H, James W, Barclay A N. OX40 is differentially expressed on activated rat and mouse T cells and is the sole receptor for the OX40 ligand. Eur J Immunol. 1996;26:1695–1699. doi: 10.1002/eji.1830260805. [DOI] [PubMed] [Google Scholar]
- Julius M H, Simpson E, Herzenberg L. A rapid method for the isolation of functional thymus-derived murine lymphocytes. Eur J Immunol. 1973;3:645–649. doi: 10.1002/eji.1830031011. [DOI] [PubMed] [Google Scholar]
- Unkeless J C. Characterization of a monoclonal antibody directed against mouse macrophage and lymphocyte Fc receptors. J Exp Med. 1979;150:580–596. doi: 10.1084/jem.150.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaung J, Jin M, Barron E, Spee C, Wawrousek E F, Kannan R, Hinton D R. α-Crystallin distribution in retinal pigment epithelium and effect of gene knockouts on sensitivity to oxidative stress. Mol Vis. 2007;13:566–577. [PMC free article] [PubMed] [Google Scholar]
- Fierro A F, Wurth G A, Zweifach A. Cross-talk with Ca(2+) influx does not underlie the role of extracellular signal-regulated kinases in cytotoxic T lymphocyte lytic granule exocytosis. J Biol Chem. 2004;279:25646–25652. doi: 10.1074/jbc.M400296200. [DOI] [PubMed] [Google Scholar]
- Arnaud M, Crouin C, Deon C, Loyaux D, Bertoglio J. Phosphorylation of Grb2-associated binder 2 on serine 623 by ERK MAPK regulates its association with the phosphatase SHP-2 and decreases STAT5 activation. J Immunol. 2004;173:3962–3971. doi: 10.4049/jimmunol.173.6.3962. [DOI] [PubMed] [Google Scholar]
- Cannons J L, Choi Y, Watts T H. Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol. 2000;165:6193–6204. doi: 10.4049/jimmunol.165.11.6193. [DOI] [PubMed] [Google Scholar]
- Zhu X, Subbaraman R, Sano H, Jacobs B, Sano A, Boetticher E, Munoz N M, Leff A R. A surrogate method for assessment of β(2)-integrin-dependent adhesion of human eosinophils to ICAM-1. J Immunol Methods. 2000;240:157–164. doi: 10.1016/s0022-1759(00)00192-7. [DOI] [PubMed] [Google Scholar]
- Barata J T, Silva A, Brandao J G, Nadler L M, Cardoso A A, Boussiotis V A. Activation of PI3K is indispensable for interleukin 7-mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med. 2004;200:659–669. doi: 10.1084/jem.20040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara T, Uchi H, Urabe K, Chen Q, Furue M, Moroi Y. Role of c-Jun N-terminal kinase on lipopolysaccharide induced maturation of human monocyte-derived dendritic cells. Int Immunol. 2004;16:1701–1709. doi: 10.1093/intimm/dxh171. [DOI] [PubMed] [Google Scholar]
- Vella A T, Dow S, Potter T A, Kappler J, Marrack P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:3810–3815. doi: 10.1073/pnas.95.7.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Li W Q, Aiello F B, Mazzucchelli R, Asefa B, Khaled A R, Durum S K. Cell biology of IL-7, a key lymphotrophin. Cytokine Growth Factor Rev. 2005;16:513–533. doi: 10.1016/j.cytogfr.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Hayden M S, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Pahl H L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Boatright K M, Salvesen G S. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Martin E, Capini C, Duggan E, Lutzky V P, Stumbles P, Pettit A R, O'Sullivan B, Thomas R. Antigen-specific suppression of established arthritis in mice by dendritic cells deficient in NF-κB. Arthritis Rheum. 2007;56:2255–2266. doi: 10.1002/art.22655. [DOI] [PubMed] [Google Scholar]
- Bowne W B, Srinivasan R, Wolchok J D, Hawkins W G, Blachere N E, Dyall R, Lewis J J, Houghton A N. Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med. 1999;190:1717–1722. doi: 10.1084/jem.190.11.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley M E, Wunderlich J R, Robbins P F, Yang J C, Hwu P, Schwartzentruber D J, Topalian S L, Sherry R, Restifo N P, Hubicki A M, Robinson M R, Raffeld M, Duray P, Seipp C A, Rogers-Freezer L, Morton K E, Mavroukakis S A, White D E, Rosenberg S A. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts T H. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- Martindale J L, Holbrook N J. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- Hildeman D A, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci USA. 2003;100:15035–15040. doi: 10.1073/pnas.1936213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Almeida S, Oliveira C R, Rego A C. Cytosolic and mitochondrial ROS in staurosporine-induced retinal cell apoptosis. Free Radic Biol Med. 2003;35:1500–1514. doi: 10.1016/j.freeradbiomed.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Kelly J A, Spolski R, Kovanen P E, Suzuki T, Bollenbacher J, Pise-Masison C A, Radonovich M F, Lee S, Jenkins N A, Copeland N G, Morse H C, III, Leonard W J. Stat5 synergizes with T cell receptor/antigen stimulation in the development of lymphoblastic lymphoma. J Exp Med. 2003;198:79–89. doi: 10.1084/jem.20021548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre C, Carvalho G, Tasdemir E, Braun T, Ades L, Grosjean J, Boehrer S, Metivier D, Souquere S, Pierron G, Fenaux P, Kroemer G. NF-κB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2007;26:4071–4083. doi: 10.1038/sj.onc.1210187. [DOI] [PubMed] [Google Scholar]
- Lenardo M J. Interleukin-2 programs mouse αβ T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- Arch R H, Gedrich R W, Thompson C B. Translocation of TRAF proteins regulates apoptotic threshold of cells. Biochem Biophys Res Commun. 2000;272:936–945. doi: 10.1006/bbrc.2000.2873. [DOI] [PubMed] [Google Scholar]
- Inoue J, Gohda J, Akiyama T, Semba K. NF-κB activation in development and progression of cancer. Cancer Sci. 2007;98:268–274. doi: 10.1111/j.1349-7006.2007.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy and cell death. Curr Top Dev Biol. 2007;78:217–245. doi: 10.1016/S0070-2153(06)78006-1. [DOI] [PubMed] [Google Scholar]