

Abstract
Escherichia coli (thyA ΔfolA) mutants are viable and can grow in minimal medium when supplemented with thymidine alone. Here we present evidence from in vivo and in vitro studies that the ydgB gene determines an alternative dihydrofolate reductase that is related to the trypanosomatid pteridine reductases. We propose to rename this gene folM.
Tetrahydrofolate (H4-folate) is the major C1 carrier in the synthesis of purines, thymidine, glycine, methionine, and pantothenate in bacteria and eukaryotes. In bacteria, H4-folate is also required for the synthesis of formylmethionyl tRNAfMet. Dihydrofolate (H2-folate) consists of dihydropterin linked to p-aminobenzoate and to one or more glutamate residues that are linked to the p-aminobenzoate moiety. The reduction of H2-folate to H4-folate is performed in most bacteria and eukaryotes by the enzyme dihydrofolate reductase (DHFR), which in Escherichia coli is coded for by the folA gene. In addition to its role in the de novo synthesis of H4-folate, DHFR recycles the H2-folate produced in most organisms by the enzyme thymidylate synthase (encoded by the thyA gene), which transfers a methylene group from methylene-H4-folate to dUMP with concomitant oxidation of H4-folate.
Formylmethionyl tRNAfMet is essential for initiation of protein synthesis in E. coli and cannot be provided exogenously. Surprisingly, it was found that folA deletion mutants (created in a thyA genetic background) are viable (1, 7, 8) and can grow, though slowly, in minimal medium supplemented with thymidine. Moreover, quantitative analysis of reduced folates in E. coli ΔfolA mutants (6) demonstrated the presence of various reduced folates, including CHO-H4-folate, CH3-H4-folate, and H4-folate. These findings imply the existence in E. coli of another enzyme that is able to carry out the de novo synthesis of H4-folate. Indeed, Vasudevan et al. (11) reported the purification from E. coli of a dihydropteridine reductase that was able to reduce H2-folate to form H4-folate. However, this observation was not further studied, and the gene encoding this protein was not identified.
Pteridine reductase (PTR1) is a short-chain dehydrogenase/reductase (SDR) that functions to salvage pterins in parasitic trypanosomatids (2, 10). Amplification of the PTR1 gene confers resistance to the protozoan parasite Leishmania against the DHFR inhibitor methotrexate (2). Biochemical analysis showed that PTR1 is able to catalyze the NADPH-dependent reduction of folates to H4-folate in two steps (10, 12). The three-dimensional structure of PTR1 was recently determined, and the active-site residues that interact with the substrates dihydrobiopterin and NADPH were identified. Among these residues are Asp 181, Tyr 194, and Lys 198, which make up the catalytic triad, and Arg 17, Ser 111, and Phe 113, which interact with the substrates (see Fig. 2) (5).
FIG. 2.

PCR analyses for the deletions of folA (A) and ydgB (B) genes. (A) PCR was performed with the primers eD-short-up and eD-short-down (Table 2). Genomic DNA of MM612 (lane 1) and MM512 (lane 2) served as templates for the PCR. (B) PCR was performed with the primers ydgB short up and ydgB short down (Table 1). Genomic DNA of MM777 (lane 1) and MG1655 (lane 2) served as templates. DNA size markers are shown (MW).
We performed a BLAST search of the E. coli MG1655 protein sequence database by using the Leishmania PTR1 protein as the query and identified several homologues whose sequences are significantly similar to those of the SDR family. One of these proteins, the ydgB gene product, contains each of the amino acid residues in PTR1 that are important for substrate binding and catalysis (Fig. 1). Using PCR, we created an NdeI restriction site at the 5′ end and an XbaI restriction site at the 3′ end of the ydgB gene. The amplified product was cloned into the vector pUC120, which had been modified to contain a His tag coding sequence positioned immediately downstream of the ATG initiation codon, followed by an NdeI site. The resulting plasmid, pFolM (Table 1), contains the ydgB gene placed under the control of the lacZ promoter and operator and was used to express the recombinant protein with a His tag at its N terminus.
FIG. 1.

Alignment of the Leishmania major PTR1 protein with the E. coli ydgB gene product. The arrows indicate the conserved amino acid residues that are involved in the catalytic activity of PTR1 (5).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Genotype or description | Source or Reference |
|---|---|---|
| MG1655 | E. coli K-12, wild type | |
| MM512 | MG1655 ΔthyA | 4 |
| MM612 | MG1655 ΔthyA ΔfolA::kan | This work |
| MM667 | MG1655 ΔthyA ΔfolA::kan/pFolM | This work |
| MM777 | MG1655 ΔydgB | This work |
| pFolM | pUC 120 containing the E. coli folM gene expressed from the lac promoter | This work |
| pKD46 | repA101(Ts) araBp-gam-bet-exo oriR101 bla | 3 |
| pCVD442 | oriR6K mobRP4 sacB Ampr | 9 |
| pMM712 | ydgB flanking regions cloned into pCVD442 | This work |
To determine whether the cloned ydgB gene can complement a ΔfolA mutation in vivo, we created a ΔthyA ΔfolA::kan double mutation in E. coli MG1655 such that each mutation resulted in a precise deletion of the DNA region corresponding to the structural gene. We started with strain MM512 ΔthyA, obtained previously (4), by using the method described by Mobley et al. (9). The ΔfolA mutation was introduced into MM512 by the PCR targeting method of Datsenko and Wanner (3), with the following modifications. The 1,100-bp DNA fragment upstream of the E. coli folA gene was amplified from genomic DNA by using the primers eD5up and eD3up, and the 1,000-bp DNA fragment downstream of the E. coli folA gene was amplified by using the primers eD5down and eD3down (Table 2). The two DNA fragments were cloned into pUC19, and the Tn903 kanamycin resistance cassette (kan) was inserted between the upstream and downstream regions of folA. This plasmid served as a template for PCR with the eD5up and eD3down primers. The PCR product was electroporated into E. coli MM512, which contains plasmid pKD46 (3). The transformants were incubated at room temperature overnight, and kanamycin-resistant colonies were selected on Luria-Bertani (LB) agar plates supplemented with the appropriate antibiotics. Transformants containing the folA deletion were screened by using PCR as shown in Fig. 2A. One of the clones that tested positive for the deletion was designated MM612. The pKD46 plasmid was cured from this strain by overnight incubation at 37°C.
TABLE 2.
Oligonucleotide primers
| Primer | Sequence | Location relative to the first nucleotide of:
|
|
|---|---|---|---|
| folA | ydgB | ||
| eD5up | 5′ATTCTAGACGCCATGCTGTGGCTGATTGC | −1131 to −1111 | |
| eD3up | 5′GCGTCGACCGATAAAAAAAATTGTCGCC | −9 to −30 | |
| eD5down | 5′ATGTCGACTTTTGTATAGAATTTACGGC | +480 to +499 | |
| eD3down | 5′ATGCATGCGAAACCCCGCTGGGCACCATGC | +1519 to +1498 | |
| eD-short-up | 5′GTTTACGCTTTACGTATAGTGG | −50 to −31 | |
| eD-short-down | 5′GTCGCATCCGGCGCTAGCC | +515 to +497 | |
| ydgB5up | 5′ATCTAGAGGTGCATCGGCTTTATTGTGG | −1023 to −1003 | |
| ydgB3up | 5′GGATCCCGTTATCTCCTTTGCTATCCAACG | −1 to −36 | |
| ydgB5 down | 5′GGATCCTGGCGGTCGTCATCTGCG | +701 to +718 | |
| ydgB3 down SacI | 5′GAGCTCGGGTAGCTAATTCCCACAATAATTCG | +1708 to +1683 | |
| ydgB short up | 5′CGACCTGGATGCTGGTGGG | +59 to +41 | |
| ydgB short down | 5′CGGCATTGAAGCCTTACGCG | +905 to +996 | |
Plasmid pFolM was introduced into MM612, and transformants were selected on LB agar plates containing 100 μg of ampicillin ml−1 to yield strain MM667. The ability of E. coli strains MG1655 (wild-type), MM512 (ΔthyA), MM612 (ΔthyA ΔfolA::kan), and MM667 (ΔthyA ΔfolA::kan/pFolM) to grow on M9 plates supplemented with 40 μg of thymidine ml−1 was examined. After 18 h of incubation at 37°C, colonies appeared in strains MG1655, MM512, and MM667. Colonies of strain MM612 appeared after 4 days under these conditions. These results demonstrate that the cloned ydgB can complement the ΔfolA mutation in vivo for normal growth.
Attempts were made to delete ydgB in strains MG1655, MM512, and MM612 by using the method described by Mobley et al. (9). Briefly, the 1-kb-long upstream flanking sequence was amplified by PCR by using primers ydgB5up and ydgB3up, and the 1-kb-long downstream flanking sequence was amplified by PCR by using primers ydgB5down and ydgB3down SacI. The amplified fragments were cloned together into the positive-selection suicide vector pCVD442. The resulting plasmid (pMM712) was electroporated into each of the three strains. Cells were plated on LB agar plates containing ampicillin (100 μg ml−1) and checked for the pMM712 integration by using PCR. The mero-diploid strains were grown overnight in LB medium, and 100 μl of the turbid culture was plated on LB plates containing 5% (wt/vol) sucrose and thymidine (40 μg ml−1) to facilitate the resolution of the alleles.
In strain MG1655, two colonies out of ten were found by PCR to retain the ΔydgB allele (Fig. 2B). In strain MM512, 15 colonies out of 23 retained the ΔydgB allele. In the case of strain MM612, all 47 colonies that lost pMM712 retained the ydgB allele. We conclude, therefore, that ydgB is “synthetic lethal” with folA. Strain MG1655, containing the ΔydgB mutation, was found to have no observable phenotype and can grow normally in M9 minimal medium. The trimethoprim MIC for the MG1655 strain and the MG1655 (ΔydgB) mutant was found to be 1.1 μg ml−1, while strain MG1655 (ΔydgB), carrying pFolM, is resistant even to 10 μg of trimethoprim ml−1.
Recombinant YdgB protein was purified as follows. MM667 cells were grown to an optical density at 600 nm of 0.6 in 500 ml of LB medium containing 100 μg of ampicillin ml−1 supplemented with thymidine at a final concentration of 40 μg ml−1. Isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 1 mM, and the culture was left to shake for an additional 4 h at 37°C. The cells were harvested, suspended in 20 ml of buffer containing 50 mM NaH2PO4, 300 mM NaCl, 15% glycerol (vol/vol), and 10 mM imidazole (pH 8), and disrupted by sonication. The supernatant was separated from the cellular debris by centrifugation for 10 min at 10,000 rpm (Sorvall SS-34 rotor), and the YdgB protein was purified according to the protocol described in the QIAexpressionist handbook. Briefly, 5 ml of the supernatant was incubated with 1 ml of QIAGEN Ni-NTA agarose for 1 h at 4°C. The suspension was then loaded onto a column and washed with buffer containing 50 mM NaH2PO4, 300 mM NaCl, 15% glycerol (vol/vol), and 20 mM imidazole (pH 8). The protein was eluted with buffer containing 50 mM NaH2PO4, 300 mM NaCl, 15% glycerol (vol/vol), and 250 mM imidazole (pH 8). The purity of the protein was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, as shown in Fig. 3.
FIG. 3.
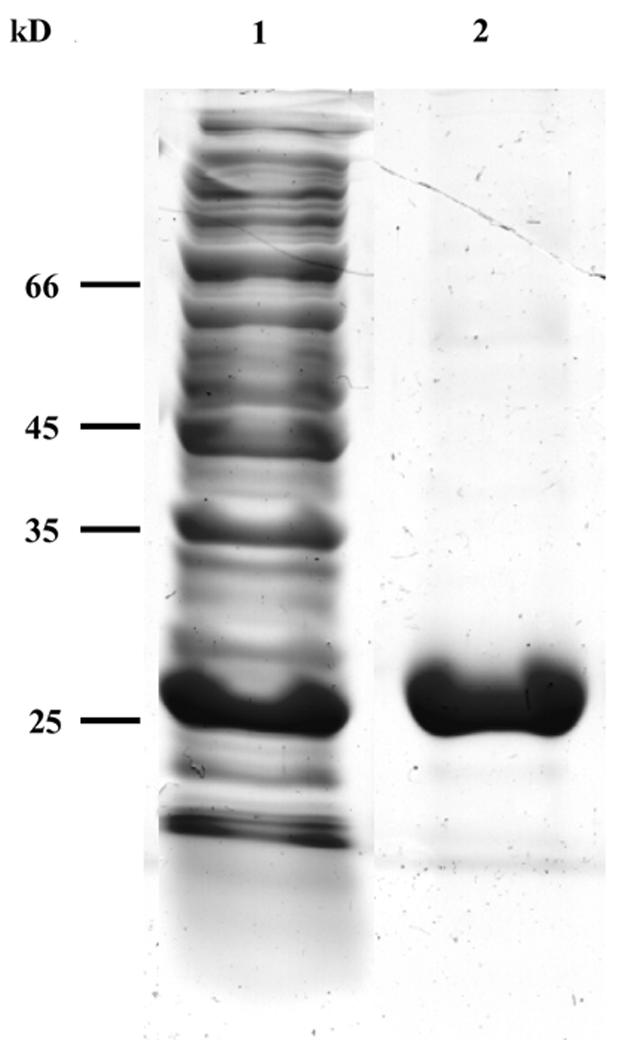
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of E. coli FolM. Expression and purification of FolM was carried out as described in the text. Protein extract of induced cells (lane 1) and the purified FolM after elution from the QIAGEN Ni-NTA agarose column (lane 2) are shown.
The enzymatic activity of the purified protein was measured with an Ultrospec 2100 Pro spectrophotometer (Amersham Pharmacia Biotech) equipped with the Swift II program. Enzymatic activity was determined in the pH range 4.7 to 7.0 and was found to be highest at pH 4.7 (data not shown). In order to avoid enzyme denaturation at low pH, routine measurements were performed in 0.1 M K2HPO4-KH2PO4 buffer (pH 6.0) containing 100 μM H2-folate and 100 μM NADPH. The change in the optical density of the solution at 340 nm was monitored. The extinction coefficient for the coupled oxidation-reduction of NADPH-H2-folate was taken as 12,300 M cm−1. The enzyme did not reduce folic acid to any appreciable extent in buffers with pH values ranging from 4.7 to 7.0 and could not use NADH as the reducing agent. The Km values for H2-folate and NADPH were determined at pH 6.0 to be 9.5 and 1.9 μM, respectively. The Vmax was determined to be 0.083 μmol min−1 mg−1 (which is about fourfold slower than for E. coli DHFR and L. major PTR1) (10). No inhibition of the enzymatic activity was observed with trimethoprim at concentrations up to 1.38 mM. However, the enzyme was inhibited by methotrexate, a competitive inhibitor, with a Ki of 5.9 μM. The purified enzyme was unable to reduce biopterin, and the rate of reduction of dihydrobiopterin was about 10% of that observed with H2-folate. Since the enzyme is most active in reducing H2-folate, we propose to rename the gene that encodes this reductase folM and the protein FolM. A puzzling feature of FolM is that while it possesses each of the three essential active-site amino acid residues needed for folate and biopterin reduction in PTR1 (Asp181, Phe194, and Lys198), it is unable to reduce these compounds.
Two main factors suggest that FolM is unrelated to the previously reported dihydropteridine reductase (11). First, FolM is a member of the SDR enzyme family, whereas dihydropteridine reductase is claimed to be a flavoprotein. Second, the reported N-terminal amino acid sequence of dihydropteridine reductase does not fit that of FolM.
With regard to the role of FolM, it is still unclear in what physiological conditions it functions. Although the E. coli ΔthyA ΔfolA mutant is viable and grows in minimal medium supplemented with thymidine alone, its growth rate is substantially reduced unless folM is overexpressed. It may be relevant that SDRs possessing the same conserved amino acids that were instrumental in identifying the folM gene are found in a large number of bacteria, including Shigella flexneri, Shewanella oneidensis, Pseudomonas aeruginosa, Xanthomonas campestris, Xylella fastidiosa, Magnetococcus sp., Brucella melitensis, Sinorhizobium meliloti, and Caulobacter crescentus. Evidently the folM gene is widespread, and a study of the properties of these bacteria with respect, for example, to their sensitivity to trimethoprim may shed light on this subject.
Acknowledgments
We thank Gerald Cohen for critical reading of the manuscript.
This work was supported by grants from the United States-Israel Binational Science Foundation and from the Israeli Science Foundation.
REFERENCES
- 1.Ahrweiler, P. M., and C. Frieden. 1988. Construction of a fol mutant strain of Escherichia coli for use in dihydrofolate reductase mutagenesis experiments. J. Bacteriol. 170:3301-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bello, A. R., B. Nare, D. Freedman, L. Hardy, and S. M. Beverley. 1994. PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc. Natl. Acad. Sci. USA 91:11442-11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giladi, M., G. Bitan-Banin, M. Mevarech, and R. Ortenberg. 2002. Genetic evidence for a novel thymidylate synthase in the halophilic archaeon Halobacterium salinarum and in Campylobacter jejuni. FEMS Microbiol. Lett. 216:105-109. [DOI] [PubMed] [Google Scholar]
- 5.Gourley, D. G., A. W. Schuttelkopf, G. A. Leonard, J. Luba, L. W. Hardy, S. M. Beverley, and W. N. Hunter. 2001. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 8:521-525. [DOI] [PubMed] [Google Scholar]
- 6.Hamm-Alvarez, S. F., A. Sancar, and K. V. Rajagopalan. 1990. The presence and distribution of reduced folates in Escherichia coli dihydrofolate reductase mutants. J. Biol. Chem. 265:9850-9856. [PubMed] [Google Scholar]
- 7.Herrington, M. B., and N. T. Chirwa. 1999. Growth properties of a folA null mutant of Escherichia coli K12. Can. J. Microbiol. 45:191-200. [PubMed] [Google Scholar]
- 8.Howell, E. E., P. G. Foster, and L. M. Foster. 1988. Construction of a dihydrofolate reductase-deficient mutant of Escherichia coli by gene replacement. J. Bacteriol. 170:3040-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mobley, H. L., K. G. Jarvis, J. P. Elwood, D. I. Whittle, C. V. Lockatell, R. G. Russell, D. E. Johnson, M. S. Donnenberg, and J. W. Warren. 1993. Isogenic P-fimbrial deletion mutants of pyelonephritogenic Escherichia coli: the role of α Gal(1-4) β Gal binding in virulence of a wild-type strain. Mol. Microbiol. 10:143-155. [DOI] [PubMed] [Google Scholar]
- 10.Nare, B., L. W. Hardy, and S. M. Beverley. 1997. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 272:13883-13891. [DOI] [PubMed] [Google Scholar]
- 11.Vasudevan, S. G., B. Paal, and W. L. Armarego. 1992. Dihydropteridine reductase from Escherichia coli exhibits dihydrofolate reductase activity. Biol. Chem. Hoppe-Seyler 373:1067-1073. [DOI] [PubMed] [Google Scholar]
- 12.Wang, J., E. Leblanc, C. F. Chang, B. Papadopoulou, T. Bray, J. M. Whiteley, S. X. Lin, and M. Ouellette. 1997. Pterin and folate reduction by the Leishmania tarentolae H locus short-chain dehydrogenase/reductase PTR1. Arch. Biochem. Biophys. 342:197-202. [DOI] [PubMed] [Google Scholar]