

Abstract
Aerobic growth of Streptococcus pneumoniae results in production of amounts of hydrogen peroxide (H2O2) that may exceed 1 mM in the surrounding media. H2O2 production by S. pneumoniae has been shown to kill or inhibit the growth of other respiratory tract flora, as well as to have cytotoxic effects on host cells and tissue. The mechanisms allowing S. pneumoniae, a catalase-deficient species, to survive endogenously generated concentrations of H2O2 that are sufficient to kill other bacterial species is unknown. In the present study, pyruvate oxidase (SpxB), the enzyme responsible for endogenous H2O2 production, was required for survival during exposure to high levels (20 mM) of exogenously added H2O2. Pretreatment with H2O2 did not increase H2O2 resistance in the mutant, suggesting that SpxB activity itself is required, rather than an H2O2-inducible pathway. SpxB mutants synthesized 85% less acetyl-phosphate, a potential source of ATP. During H2O2 exposure, ATP levels decreased more rapidly in spxB mutants than in wild-type cells, suggesting that the increased killing of spxB mutants was due to more rapid ATP depletion. Together, these data support the hypothesis that S. pneumoniae SpxB contributes to an H2O2-resistant energy source that maintains viability during oxidative stress. Thus, SpxB is required for resistance to the toxic by-product of its own activity. Although H2O2-dependent hydroxyl radical production and the intracellular concentration of free iron were similar to that of Escherichia coli, killing by H2O2 was unaffected by iron chelators, suggesting that S. pneumoniae has a novel mechanism to avoid the toxic effects of the Fenton reaction.
Streptococcus pneumoniae (the pneumococcus) commonly colonizes the nasopharynx of humans and is a leading cause of meningitis, septicemia, otitis media, and community-acquired pneumonia (22, 39). S. pneumoniae exists primarily as a commensal, with invasive disease resulting from a failure of host defenses (19). S. pneumoniae, like all streptococci, possesses a mainly fermentative metabolism and lacks the cytochromes and heme-containing proteins involved in aerobic respiration. Despite its predominantly aerobic lifestyle in the oxygen rich environment on the airway surface, it lacks many proteins that have been shown to protect against oxidative stress in other bacterial species, such as the global regulators OxyR and PerR or the hydrogen peroxide scavengers catalase and NADH peroxidase (27, 59). The mechanism by which S. pneumoniae is able to grow under high oxygen tension without many of the defenses commonly found in aerobic organisms is unknown. S. pneumoniae contains two oxidases (NADH oxidase and pyruvate oxidase) which have been shown to be required for growth in vivo (2, 56). NADH oxidase performs the four-electron reduction of O2 to H2O, thereby acting to limit the damaging effects that O2 and its by-products have on the organism (2). However, pyruvate oxidase catalyzes a two-electron reduction of O2, thereby forming the potentially damaging compound, H2O2 (56):
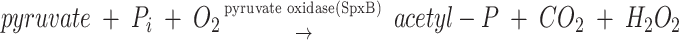 |
Hydrogen peroxide diffuses rapidly through cell membranes, and its concentration in culture supernatants of S. pneumoniae can therefore exceed 1.0 mM, a level approximately 103-fold higher than the concentration that is sufficient to inhibit the growth of Escherichia coli cells that have been rendered deficient in their ability to scavenge H2O2 (43, 52, 53). Although endogenous hydrogen peroxide production has been shown to increase mutation rates in S. pneumoniae, its growth is generally not inhibited by endogenously generated H2O2 (43). In contrast, concentrations of H2O2 between 0.1 and 1.0 mM have been shown to have toxic effects on many other species of bacteria (14, 30, 44, 48). Indeed, H2O2 produced by S. pneumoniae in vitro is sufficient to kill or inhibit other common inhabitants of the respiratory tract, such as Haemophilus influenzae, Neisseria meningitidis, and Moraxella catarrhalis, during coculture (44). Production of H2O2 by S. pneumoniae has also been shown to have cytotoxic effects on human epithelial cells in culture and host tissue in animal models of pneumococcal disease (5, 15, 24). However, it is unclear what factors allow S. pneumoniae, which does not express catalase, to survive endogenously generated concentrations of H2O2 that can kill or inhibit catalase-expressing bacteria or eukaryotic cells.
H2O2 has been shown to cause lethality in E. coli through a bimodal pattern with mode I killing being maximal between concentrations of 1.0 and 3.0 mM. Mode I killing is thought to occur mainly through DNA damage from hydroxyl radicals (OH·) produced via the Fenton reaction (29, 30) as follows: H2O2 + Fe2+ → Fe3+ + OH· + OH−. The hydroxyl radical is a highly reactive substance, and it is thought that this reaction is most damaging when it occurs in the immediate vicinity of the DNA (29). Accordingly, the amount of ferrous iron (Fe2+) available to associate with DNA is believed to be a rate-limiting factor in Fenton-reaction killing (29). Mode II killing occurs at higher concentrations of H2O2 (up to at least 50 mM) but has been show to occur at a slower rate (30). The mechanism of mode II killing has not been determined but is believed to be distinct from DNA damage (30). One possibility is that mode II killing involves inactivation of housekeeping enzymes, perhaps through oxidation of active site thiols (28).
We initiated these studies to determine what mechanism allows S. pneumoniae to escape lethal DNA damage at millimolar concentrations of H2O2. Both naturally occurring and constructed spxB mutants have been shown to produce approximately 100-fold less H2O2 than does the wild type (44, 56). Surprisingly, these spxB mutants exhibited 102- to 103-fold-lower survival after a 30-min exposure to high levels (20 mM) of exogenously added H2O2. The present study was designed to determine the mechanism by which these mutants are more sensitive to H2O2, as well as the factors that allow S. pneumoniae to survive levels of H2O2 that are toxic to many other bacteria.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
S. pneumoniae strains used in the present study are described in Table 1. S. pneumoniae was grown in tryptic soy medium (TS) or Todd-Hewitt medium plus 0.2% yeast extract (TH+Y) at 37°C. Unless otherwise noted, bacteria were grown to mid-log phase (A620 = 0.4) in static liquid culture. Next, 5,000 U of bovine liver catalase (Worthington Biochemical, Freehold, N.J.) was applied to the surface of TS plates containing 1.5% agar prior to plating, followed by incubation in candle extinction jars. Anaerobic growth conditions were obtained by using the BBL GasPak System (Becton Dickinson, Cockeysville, Md.). Colony morphology was determined on transparent medium under magnification and oblique, transmitted illumination as previously described (62). Unless otherwise stated, chemicals and reagents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, Mo.).
TABLE 1.
Hydrogen peroxide production by various S. pneumoniae strains
| Strain | Genotype and/or relevant characteristics | Mean amt of H2O2 generated (mM)a ± SD | Source or reference |
|---|---|---|---|
| P125 | Type 2 phase variant | 1.49 ± 0.18 | 63 |
| P126 | Type 2 phase variant | 1.07 ± 0.08 | 63 |
| P835 | Type 4 phase variant | 1.05 ± 0.10 | TIGR4 genome strain |
| P836 | Type 4 phase variant | 2.02 ± 0.10 | TIGR4 genome strain |
| D39 | Type 2 | 1.83 ± 0.20 | 3 |
| P878 | D39 spxB::TnphoA(erm) | <0.025 | 56 |
| P1221 | P878 + pMU1328::300+spxB | 2.03 ± 0.16 | This study |
| R6x | Type 2 unencapsulated | 1.78 ± 0.12 | 60 |
| P1167 | R6x spxB::TnphoA(erm) | <0.025 | This study |
| P1185 | P1167 + pMU1328::300+spxB | 2.14 ± 0.18 | This study |
| Rx1 | Type 2 unencapsulated | 0.89 ± 0.11 | 2 |
| P1234 | Rx1 spxB::TGA | <0.025 | 2 |
That is, the H2O2 concentration present in culture supernatants after the aeration of ca. 5 × 107 washed, log-phase cells for 30 min in 800 μl of TS in a 1.6-ml Eppendorf tube at 37°C.
Strain construction.
Genomic DNA was purified as previously described (42). A region of DNA containing the spxB open reading frame, the 300 bp immediately upstream of the translational start site, and unique restriction enzyme sites at either end was amplified from D39 chromosomal DNA by using the PCR as previously described (PCR vector). Gel-purified PCR fragments were digested with EcoRI and XbaI and ligated into the pMU1328 E. coli-streptococcal shuttle vector (1) that had been digested with EcoRI and XbaI. The resulting plasmid (pMU1328::300+spxB) was transformed into strain P878 or P1167 as previously described (34). The sequences of the primers used for amplification were as follows: forward EcoRI, 5′-CGG AAT TCC TGA CCA GTT CCC TTC TG-3′; and reverse XbaI, 5′-CGC TCT AGA TTA TTT AAT TGC GCG TGA TTG-3′ (restriction sites are underlined). DNA manipulations were performed by standard techniques (50). The accession number for spxB is L39074.
Hydrogen peroxide sensitivity assays.
Bacteria were grown until mid-log phase, and duplicate 100-μl aliquots of each culture were added to 100 μl of medium or 100 μl of medium containing 40 mM H2O2 that had been diluted from a 30% (9.8 M) stock solution, followed by incubation at 37°C for 30 min. Serial dilutions from each tube were then prepared in ice-cold phosphate-buffered saline, and duplicate aliquots were spotted onto TS agar plates containing 5,000 U of bovine liver catalase. In some experiments, the iron chelator desferrioxamine (DF; 20 mM) or 2′,2′ dipyridyl (1 mM) was added 15 min prior to challenge with H2O2. The percent survival was calculated by dividing the CFU of cultures after exposure to H2O2 by the CFU of the control tube without H2O2.
SpxB-His6 purification and antibody purification.
A PCR product containing the spxB open reading frame and unique restriction enzyme sites was amplified from strain D39 chromosomal DNA. Gel-purified PCR fragments were digested with NheI and SalI and ligated into the pET28a expression vector (Novagen, Inc., Madison, Wis.) which had been digested with NheI and SalI. The resulting plasmid (pET28a::spxB) was transformed into E. coli strain BL21, creating strain E549. His-tagged S. pneumoniae pyruvate oxidase (SpxB) was expressed in the presence of IPTG (isopropyl-β-d-thiogalactopyranoside) and purified by using an activated His-Bind resin column (Novagen). The sequences of the primers used for amplification were as follows: forward, 5′-CGG CTA GCA TGA CTC AAG GGA AAA TTA C-3′; and reverse, 5′-CGC GTC GAC TTA TTT AAT TGC GCG TGA TTG-3′ (restriction sites are underlined). Polyclonal rabbit serum to the recombinant SpxB protein was commercially prepared (Rockland, Inc., Gilbertsville, Pa.).
Western blotting.
Bacteria were harvested in mid-log phase, washed in cold PBS, sonicated, and treated at 100°C for 5 min in gel loading buffer (50 mM Tris-Cl [pH 6.8], 100 mM β-mercaptoethanol, 10% glycerol, 2% sodium dodecyl sulfate [SDS], and 1% bromophenol blue) prior to separation by SDS-10% polyacrylamide gel electrophoresis. Equal loading was confirmed by measurement of total protein in whole-cell sonicates by using the Micro BCA protein assay (Pierce Chemical Co., Rockford, Ill.). After transfer to Immobilon-P membranes (Millipore Co., Bedford, Mass.), immunoblotting was carried out with an antiserum raised against SpxB-His6 and detected with an antiserum to rabbit immunoglobulin G conjugated to either alkaline phosphatase or horseradish peroxidase as described previously (64).
Coculture experiments.
Aliquots (2 ml) of mid-log-phase cultures of wild-type and spxB mutant strains that had been diluted 1/10 in TS were combined and cultured together for at least three generations before they were tested for H2O2 resistance as described above. Duplicate aliquots were spotted onto TS plates or TS plates containing 1 μg of erythromycin/ml to select against growth of wild-type bacteria. Viable counts of the spxB mutant strain were determined from the TS plates containing erythromycin, and counts of the wild-type strain were determined by subtracting the number of colonies on selective plates from the number on nonselective plates.
Anaerobic growth.
Stationary-phase cultures were diluted 1/10 in 4 ml of TS medium that had been rendered free of oxygen by incubating them in the GasPak anaerobic system (Becton Dickinson) for 24 h. After growth to mid-log phase, cultures were tested for H2O2 resistance as described above. For aerobic induction experiments, 200-μl aliquots of anaerobic cultures were removed from the GasPak anaerobic system, diluted to 400 μl with either TS or TS containing 10 μg of chloramphenicol/ml, and incubated in 1.6-ml tubes for 15 min at 37°C. After 15 min of aerobic incubation, 100-μl aliquots were then tested for H2O2 resistance as described above.
Hydrogen peroxide production assays.
We added 20 μl of a solution consisting of 3 mg of ABTS [2,2′azinobis(3-ethylbenzthiazolinesulfonic acid)]/ml and 0.2 mg of horseradish peroxidase/ml in 0.1 M sodium phosphate buffer (pH 7.0) to 180-μl aliquots of each supernatant to be tested (20). The reaction was allowed to proceed for 20 min at room temperature, and then the A560 was measured. Values were calculated by comparison to a standard curve that was generated by using known concentrations of H2O2.
ATP assays.
ATP was assayed according to a method adapted from Fukui et al. (18). A total of 50 μl of a log-phase culture was added to 950 μl of distilled H2O at 100°C, and the mixture was boiled for 5 min. Samples were cooled to room temperature, and 50-μl aliquots added to 50 μl of reconstituted CellTiter-Glo reagent (Promega Corp., Madison, Wis.). Plates were mixed on an orbital shaker for 10 min at room temperature, and luminescence was determined by using a Wallac Microbeta Trilux luminescence counter (Perkin-Elmer Life Sciences, Boston, Mass.). Values were calculated by comparison to a standard curve that was generated by using known concentrations of ATP. Aliquots of cultures were also assayed for protein by using a Micro BCA protein assay (Pierce).
AcP assays.
Acetyl-phosphate (AcP) was assayed according to a method adapted from Prüss and Wolfe (47). A total of 10 ml of cells was harvested by centrifugation at 2,000 × g for 15 min, washed, and resuspended in 250 μl of cold 10 mM sodium phosphate (pH 7.5)-10 mM MgCl2-1 mM EDTA. A total of 50 μl of cold 3 M HClO4 was added to each tube prior to incubation for 30 min on ice. The tubes were then centrifuged 2 min at 6,000 × g. The supernatants were next neutralized with saturated KHCO3 (∼250 μl/ml) and centrifuged as described above. A total of 50 mg of powdered activated charcoal/ml was then added to the supernatants to remove small adenylated compounds such as ADP and ATP. The tubes were vortex mixed and then incubated on ice for 15 min. The charcoal was removed by filtration through a 0.22-μm-pore-size filter. Aliquots of each extract were assayed for protein by using a Micro BCA protein assay (Pierce). Aliquots were also assayed to ensure that no ATP remained in the treated cell extracts. To the remaining aliquots, 1 mM MgCl2, 30 μM ADP, and 4 μg of acetate kinase/ml were added. The reaction tubes were incubated at 30°C for 90 min. ATP was then assayed by using the CellTiter-Glo kit as described above. Acetate kinase was omitted from one tube of each extract to correct for the ATP present in the ADP preparation (estimated to be 0.2%). Control extracts were also heated to 100°C for 5 min before the addition of acetate kinase to destroy AcP and exclude the possibility of adenylate kinase contamination in the enzyme preparation. For each assay, an aliquot of 0.5 mM AcP was subjected to the entire extraction procedure as a quantification standard.
Total iron assay.
Cultures of E. coli (AB1157) or S. pneumoniae (D39) were grown in 1 liter of TS with or without shaking, respectively. AB1157 is a previously described wild-type strain of E. coli (52). When the optical density at 620 nm (OD620) reached 0.3, the cells were then centrifuged and washed twice with 20 mM Tris-Cl (pH 7.4) buffer containing 1 mM EDTA. The cell pellets were then washed once in Tris buffer without EDTA and resuspended in 3 ml of Tris buffer. The suspensions were then passed through a French press before being analyzed for iron content by inductively coupled plasma-optical emission spectroscopy (OES Optima 2000 DV [Perkin-Elmer]) at the Microanalysis Laboratory, University of Illinois at Urbana Champaign. Aliquots of cell lysates were also analyzed for protein concentration by using the Micro BCA protein assay (Pierce). Total iron concentrations were normalized to total protein content.
Intracellular free iron measurement by EPR.
S. pneumoniae cultures (R6x) were grown in 1 liter of TS without shaking. When the OD620 reached 0.3 to 0.4, the cells were harvested, and the pellets were resuspended in 9 ml of the same medium. One milliliter of 0.2 M DF was added, and the culture was incubated at 37°C for 15 min. The cells were then centrifuged, washed twice with 5 ml of cold 20 mM Tris-Cl (pH 7.4) buffer, and then resuspended in 400 μl of cold Tris buffer containing 10% glycerol. Next, 200 μl of the cell suspension was loaded into a quartz electron paramagnetic resonance (EPR) tube, frozen in dry ice, and stored at −80°C until analysis. E. coli (AB1157) samples were prepared the same way except that the cultures were grown with shaking. Ferric sulfate standards were mixed with DF and prepared in the same Tris-glycerol buffer. The concentration of iron in the standard samples was determined using an absorption coefficient at 420 nm of 2.865 cm−1. The EPR signals were measured with a Varian Century E-112 X-band spectrophotometer equipped with a Varian TE102 cavity and temperature controller. The spectrometer settings were as follows: field center, 1,570 G; receiver gain, 5,000 G; field sweep, 400 G; modulation amplitude, 12.5 G; temperature, 15 K; and power, 10 mW. The measured EPR signals were normalized to intracellular protein that was liberated upon cell lysis by using a French press.
Spin trapping.
Formation of hydroxyl radicals were detected by a spin trapping system containing α-(4-pyridyl-1-oxide)-N-tert-butyl nitrone (4-POBN; Aldrich, Milwaukee, Wis.) and ethanol (31, 35). S. pneumoniae (R6x) or a previously described catalase-deficient E. coli strain (JI367 [katG katE]) were grown in 1 liter of TS without shaking or with shaking, respectively (52). A catalase-deficient strain of E. coli was used so that H2O2 would not be instantly scavenged by the thick slurry of cells. Cells were harvested at an OD620 of 0.3 to 0.4, and the pellets were washed twice with Chelex (Sigma)-treated Hanks balanced salt solution (HBSS). The cells were resuspended in 0.5 ml of HBSS. The reaction mixtures included 0.1 ml of the cell suspension, 100 μM diethylenetriaminepentaacetic acid (DETAPAC), 10 mM 4-POBN, 170 mM ethanol, 2 mM H2O2, and HBSS for a final volume of 1 ml. After 10 min of incubation, the suspension was transferred to an EPR flat cell, and the EPR spectra were monitored at room temperature. The spectrometer settings were as follows: field center, 3,393 G; field sweep, 100 G; modulation frequency, 100 kHz; modulation amplitude, 1 G; receiver gain, 63,000; and power, 20 mW.
Statistical analysis.
Statistical significance was determined by using the two-tailed Student t test.
RESULTS
Resistance to hydrogen peroxide is proportional to the production of hydrogen peroxide.
In a previous study, levels of SpxB were found to differ among phenotypic phase variants of S. pneumoniae, as measured by two-dimensional protein gel electrophoresis (41). This finding suggested that the production of H2O2, one of the products of pyruvate oxidase activity, might also vary between phase variants. The H2O2 concentration present in supernatants of cultures grown aerobically for 30 min was determined. As expected, the variants expressing more SpxB were also found to produce higher concentrations of H2O2 than did their counterparts (Table 1) (41). We determined whether sensitivity to H2O2 was related to the production of H2O2 by focusing on two pairs of phase variants from either an unencapsulated derivative of the serotype 2 D39 strain or a serotype 4 clinical isolate. Cultures were exposed to 20 mM H2O2 for 30 min, and survival was assessed by colony counts. In both cases, the variant expressing increased amounts of SpxB also produced more H2O2 (range, 40 to 90% increase) (Table 1). For both pairs of phase variants, the variant producing higher concentrations of H2O2 was also found to have approximately 20-fold-greater survival in the presence of elevated H2O2 (Fig. 1A). This suggested that there was an association between the production of and sensitivity to H2O2.
FIG. 1.

Effect of hydrogen peroxide production and SpxB expression on hydrogen peroxide resistance. (A) After growth to mid-log phase, cultures of type 2 phase variants P125 (░⃞) and P126 (□) or type 4 phase variants P836 (░⃞) and P835 (□) were incubated in TS containing 20 mM H2O2 for 30 min. (B) After growth to mid-log phase, cultures of wild type (░⃞), spxB mutant (□), or spxB mutant complemented with pMU1328::300+spxB (▨) were incubated in TH+Y (D39) or TS (R6x) containing 20 mM H2O2 for 30 min. Values are the average of three independent determinations in duplicate and represent the change in CFU expressed as a percentage of the control culture without hydrogen peroxide ± the standard deviation (SD). Symbols: †, below the limit of detection; ✽, P < 0.05. (C) Relative expression of SpxB in different S. pneumoniae strains. Cell lysates of D39 (lane 1), P878 (lane 2), P1221 (lane 3), R6x (lane 4), P1167 (lane 5), and P1185 (lane 6) were electrophoresed on a 10% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and immunoblotted with an antibody to SpxB. Size markers are given in kilodaltons.
Resistance to hydrogen peroxide is proportional to pyruvate oxidase expression.
We then tested the hypothesis that a strain unable to produce H2O2 would also have a defect in H2O2 resistance. Strain P878, a previously constructed mutant of strain D39 containing a truncated spxB gene, produces undetectable levels (<0.025 mM) of H2O2 (Table 1) (56). Consistent with the differences seen in H2O2 resistance between pairs of phase variants, the spxB mutant exhibited ca. 102 to 103-fold greater killing by high levels (20 mM) of exogenously added H2O2 compared to its parent strain (Fig. 1B). A similar effect was seen in strain P1167, a strain created by transformation of the unencapsulated strain R6x with the spxB-null mutation from P878 (Fig. 1B).
We next determined whether loss of the spxB gene itself was responsible for increased hydrogen peroxide sensitivity. Strain P1221 was created by complementation of strain P878 with the low-copy plasmid pMU1328::300+spxB, which contains the spxB gene under control of its native promoter. Complementation of the mutation restored SpxB expression and H2O2 resistance (Fig. 1B and C). In fact, both SpxB expression and H2O2 resistance were higher in P1221 than in its parent strain, further demonstrating that H2O2 resistance correlated with H2O2 production.
The phoA fusion that truncates and inactivates spxB in the P878 and P1167 strains is unlikely to have a polar effect, since Northern blots show that transcripts of this gene are monocistronic (10). Furthermore, strain P1234, which has the same defect in H2O2 resistance, was constructed with an in-frame stop codon in spxB, so any polar effects can be excluded. In addition, the resistance defect of spxB mutants does not result from a defect in H2O2 scavenging, since neither the wild type nor spxB mutants demonstrated any ability to scavenge exogenously added H2O2 (data not shown). Lastly, when wild-type or spxB mutant S. pneumoniae strains exposed to 20 mM H2O2 were washed and lysed, similar intracellular concentrations of H2O2 were present, confirming that the phenotype of the spxB mutant does not result from an inability to exclude H2O2 from their intracellular space (data not shown).
Oxygen but not protein synthesis is required for induction of hydrogen peroxide resistance.
Pyruvate oxidase has been shown to require the presence of oxygen for its activity (9, 54). We therefore tested the hypothesis that the protective effect of pyruvate oxidase against hydrogen peroxide requires oxygen. Rx1, an unencapsulated strain of S. pneumoniae, grown anaerobically and then exposed to oxygen for 15 min before challenge with 20 mM H2O2, exhibited significantly increased survival compared to those grown without exposure to oxygen (Fig. 2). In contrast, there was relatively little effect of oxygen on the H2O2 resistance of P1234, a previously described spxB-null mutant constructed by insertion of a premature stop codon into the spxB gene of strain Rx1 (Fig. 2) (2). This result demonstrated that resistance to H2O2 required the presence of oxygen, as well as pyruvate oxidase expression.
FIG. 2.

Effect of oxygen or inhibition of protein synthesis on resistance to hydrogen peroxide. S. pneumoniae wild-type Rx1 (░⃞) or spxB mutant P1234 (□) was grown in GasPak anaerobic jars (<0.01% O2, 10% CO2) and divided into three aliquots, which were then aerated for 15 min, aerated for 15 min in the presence of 10 μg of chloramphenicol/ml, or grown anaerobically as indicated. These cultures were then exposed for 30 min to TS containing 20 mM H2O2. Values are the average of three independent determinations in duplicate and represent the change in CFU expressed as a percentage of the control culture containing no hydrogen peroxide ± the SD. †, Below the limit of detection.
In order to determine whether the induction of H2O2 resistance by oxygen requires de novo protein synthesis, we tested survival in the presence of 20 mM H2O2 with or without the addition of growth-inhibitory amounts of chloramphenicol during a 15-min oxygen exposure. Chloramphenicol had no effect on the H2O2 resistance of wild-type S. pneumoniae Rx1 or the spxB mutant, P1234, suggesting that de novo protein synthesis was not required for oxygen-dependent induction of resistance (Fig. 2). Together, the data confirmed that pyruvate oxidase activity was necessary for wild-type levels of H2O2 resistance. Moreover, growth for several generations in subinhibitory levels of H2O2 (0.5 to 1.0 mM) or coculture with wild-type H2O2-producing bacteria for several generations did not induce an increase in survival of spxB mutants (data not shown). This suggested that a product of pyruvate oxidase activity other than H2O2 is important for survival in the presence of elevated H2O2.
Pyruvate oxidase mutants have a defect in constitutive AcP production and aerobic ATP production.
Since pyruvate oxidase in lactic acid bacteria is known to produce AcP in addition to H2O2 and CO2, we compared the ability of the spxB mutant and wild type to synthesize AcP (9, 54). There was ca. 85% less AcP in extracts from the spxB mutant, P878, compared to the D39 parent or the complemented strain, P1221 (Fig. 3). AcP can donate its phosphate group to the formation of ATP via acetate kinase, thereby increasing ATP production under aerobic conditions (12). Accordingly, we hypothesized that a defect in the aerobic production of AcP would result in an inability to increase ATP production in the presence of oxygen. ATP production during either anaerobic or aerobic growth was assayed in D39 wild-type, P878 spxB mutant, and P1221 spxB complemented strains. Under anaerobic growth conditions, all of the strains produced similar levels of ATP (Fig. 4). However, when cultures of each strain were assayed for ATP after aeration for 15 min, D39 and P1221 showed a 60 to 70% increase in ATP concentration, whereas the ATP concentration in P878 was essentially unchanged (Fig. 4). This confirmed that pyruvate oxidase is required to increase ATP production upon aerobic conditions.
FIG. 3.

Effect of SpxB expression on AcP concentration in S. pneumoniae. Following growth to mid-log phase in TH+Y, lysates of wild-type S. pneumoniae strain D39 (░⃞), spxB mutant P878 (□), or spxB complemented strain P1221 (▨) were assayed for AcP content. Values are the average of three independent determinations ± the SD. ✽✽, P < 0.005.
FIG. 4.

Effect of oxygen exposure on the ATP concentration in S. pneumoniae. After growth to mid-log phase in TH+Y, cells were diluted twofold and grown statically (anaerobic) or aerated (aerobic) for 15 min. ATP content of wild-type D39 (░⃞), spxB mutant P878 (□), or spxB complemented strain P1221 (▨) were assayed. Values are the average of three independent determinations in duplicate ± the SD. ✽✽, P < 0.005 (for P878 compared to other strains when grown aerobically).
ATP levels are more sensitive to oxidative stress in spxB mutants than in wild type.
To determine whether SpxB plays a role in maintaining the ATP pool under conditions of oxidative stress, we assayed ATP levels in S. pneumoniae upon exposure to inhibitory levels of H2O2. ATP levels were assayed in D39 wild-type, P878 spxB mutant, and P1221 spxB complemented strains after a 30-min exposure to either 2 or 4 mM hydrogen peroxide and then compared to control cultures without added H2O2. As predicted, ATP levels in strain P878 exhibited a significantly larger decrease upon exposure to either 2 or 4 mM H2O2 than D39 or P1221 (Fig. 5). This confirmed that SpxB contributes to the maintenance of ATP pools during oxidative stress.
FIG. 5.

Effect of various sublethal H2O2 concentrations on the ATP content of S. pneumoniae. Strain D39 (wild type), P878 (spxB mutant), and P1221 (spxB complemented) were incubated at 37°C in TH+Y or TH+Y containing the indicated concentrations of H2O2. After 15 min, the ATP content of D39 (░⃞), P878 (□), or P1221 (▨) were assayed (see Materials and Methods). Values represent the change in ATP level corrected for colony counts and are expressed as a percentage of the control culture containing no H2O2. Values are the average of four independent determinations in duplicate ± the SD. ✽, P < 0.05; ✽✽, P < 0.005.
ATP levels and viability decrease more rapidly in spxB mutants upon exposure to lethal concentrations of hydrogen peroxide than in the wild type.
We next tested the hypothesis that SpxB would also increase the stability of the ATP pool during exposure to lethal concentrations of H2O2. D39 wild-type, P878 spxB mutant, and P1221 spxB complemented strains were assayed for ATP content at various time points during a 10-min exposure to 20 mM hydrogen peroxide. Although ATP levels decreased in all strains over time, the rate of decrease was more rapid and substantial in strain P878 compared to the D39 and P1221 strains (Fig. 6A). Within 1.25 min of exposure to H2O2, the ATP pool of P878 had decreased to ca. 8% of its starting level, whereas strains D39 and P1221 maintained their ATP pools at ca. 20 and 35% of starting levels, respectively (P value < 0.005). After 10 min of exposure to H2O2, the ATP pool of P878 had decreased to below 5% of its starting level, whereas strains D39 and P1221 maintained their ATP pools at ca. 11% and 16% of starting levels, respectively (P value < 0.05). Viability, as measured by colony counts, decreased more than 80% in P878 after 5 min of exposure, whereas D39 and P1221 exhibited less than a 20% decrease in viability at this time point (Fig. 6B). After 10 min of exposure, P878 viability decreased by more than 95%, whereas D39 and P1221 exhibited less than a 50% decrease (Fig. 6B). ATP was not detected in culture supernatants, suggesting that loss of ATP upon exposure to 20 mM H2O2 was not due to leakage of cytoplasmic contents (data not shown).
FIG. 6.

Effect of a lethal concentration of H2O2 or NEM on the survival and ATP content of S. pneumoniae. After growth to mid-log phase in TH+Y, cells were exposed to 20 mM H2O2 (A and B) or 10 mM NEM (C) in TH+Y at 37°C. At the times indicated, the ATP content (A and C) or colony counts (B) of the wild-type D39 (•), spxB mutant P878 (▪), or spxB complemented strain P1221 (▴) were assayed (see Materials and Methods). Values represent the average of three independent determinations in duplicate. The dashed line in panel A corresponds to the concentration of ATP at which ca. 90% loss of viability of P878 occurs, as shown in panel B.
ATP levels decrease more rapidly upon exposure to thiol-reactive reagents in spxB mutants than in wild type.
We then sought to determine whether inhibition of ATP production by H2O2 might occur as a result of thiol oxidation and whether thiol-reactive reagents might have a more deleterious effect on the ATP pool of an spxB mutant compared to strains with wild-type spxB expression. D39 wild-type, P878 spxB mutant, and the P1221 spxB complemented strains were assayed for ATP content during exposure to 10 mM N-ethylmaleimide (NEM), a compound known to oxidize protein thiol groups (21, 37). Although ATP levels decreased in all strains over time, the rate of decrease was again more rapid in P878 compared to strains D39 and P1221. Within 2.5 min of exposure to NEM, the ATP pool of P878 had decreased to ca. 10% of its starting level, whereas strains D39 and P1221 maintained their ATP pools at ca. 28 and 40% of starting levels, respectively (Fig. 6C). After 10 min of exposure to H2O2, the ATP pool of P878 had decreased to less than 5% of its starting level, whereas strains D39 and P1221 maintained their ATP pools at ca. 8 and 12% of starting levels, respectively.
Iron chelators fail to prevent killing by hydrogen peroxide in the wild type or spxB mutant.
The contribution of SpxB-mediated ATP production toward survival in millimolar concentrations of H2O2 does not explain the lack of killing by toxic intermediates generated from micromolar concentrations of H2O2. Since iron, particularly soluble iron (Fe2+), has been shown to increase the rate of H2O2 killing in E. coli via the Fenton reaction, we assayed whether chelating iron would decrease killing of wild-type or spxB mutant S. pneumoniae by 20 mM H2O2. Treatment with the iron chelators dipyridyl or DF had no effect on the survival of either strain in H2O2 (data not shown). This suggested that the Fenton reaction does not play a major role in the killing of S. pneumoniae by H2O2.
The free iron content of S. pneumoniae is similar to that of E. coli.
We next determined whether total or free iron levels in S. pneumoniae are unusually low, thereby explaining the lack of a protective effect from iron chelators compared to E. coli. Total iron content of cell extracts from S. pneumoniae R6x and E. coli AB1157 were determined by using inductively coupled plasma-optical emission spectroscopy and expressed as a function of total protein content. Total iron concentrations were approximately equal in S. pneumoniae and E. coli (Table 2). Moreover, when EPR was used to assay the free iron (Fe2+) concentrations in S. pneumoniae and E. coli, the concentrations were found to be 228 and 140 μM, respectively (Table 2).
TABLE 2.
Iron content of S. pneumoniae and E. coli
| Strain | Mean total iron contenta (ng/mg of protein) ± SD | Concn (μM)b of: |
||
|---|---|---|---|---|
| Fe3+ | Fe3+ with DF | Fe2+ (free iron) | ||
| S. pneumoniae R6x | 911 ± 11 | 40 | 268 | 228 |
| E. coli AB1157 | 760 ± 8 | 30 | 170 | 140 |
Total iron content was determined in washed cells by inductively coupled plasma-optical emission spectroscopy. Values are the averages of five determinations.
Free iron was assayed in intact, washed cells using electron paramagnetic resonance and normalized based on intracellular protein content. All concentration values are the average of seven scans and are representative of three independent determinations.
Hydroxyl radicals are formed in S. pneumoniae as in E. coli.
To determine whether the Fenton reaction occurs in S. pneumoniae, we measured the formation of hydroxyl radicals. The 4-POBN-ethanol spin-trapping system generates an intracellular 4-POBN-CH(CH3)OH spin adduct that is indicative of hydroxyl radical generation. Significant EPR signals were observed in E. coli suspensions treated with exogenous H2O2 (Fig. 7A). The signal was greatly suppressed if the cells were incubated with DF, thus confirming the role of intracellular iron in generating the hydroxyl radical. A similar result was observed with S. pneumoniae cells (Fig. 7B). Indeed, S. pneumoniae generated hydroxyl radicals even when exogenous H2O2 was not added. The addition of catalase or DF suppressed the signal, indicating that it arose from a Fenton reaction that was driven by endogenous H2O2. Thus, S. pneumoniae tolerates H2O2 despite the fact that it causes the continuous generation of intracellular hydroxyl radicals.
FIG. 7.

4-POBN-ethanol spin strapping of hydroxyl radical in E. coli and S. pneumoniae. EPR scans of E. coli JI367 (A) and S. pneumoniae R6x (B) incubated with the spin trap, 4-POBN. Scans are normalized to total protein released upon cell lysis. The signals are the average of 10 scans and are representative of four independent determinations. Where indicated, 20 mM DF was preincubated with cells for 5 min, and catalase was added at 1,850 U/ml. For the control, 100 μM FeSO4 was added to a DETAPAC-free reaction mixture.
DISCUSSION
Expression of SpxB increases resistance to H2O2 in S. pneumoniae.
Since S. pneumoniae exists primarily in an aerobic environment, one would expect it to encounter the toxic effects of oxygen metabolism, mainly H2O2 and O2•−. Compounding this dilemma is the fact that this organism produces millimolar concentrations of H2O2 during aerobic growth. The aim of the present study was the elucidation of the mechanism that allows survival in the presence of these high concentrations of H2O2. This was explored by investigating the possible connection between H2O2 production and resistance. Our results extend the findings of Overweg et al. by showing that phase variants exhibit not only differential SpxB expression and H2O2 production but also differential resistance to killing by H2O2 (41). Although one pair of variants examined in that study was unique in that a more opaque colony variant of the serotype 9V strain was later determined to have a frameshift mutation in spxB, differential production of SpxB and H2O2 and resistance to H2O2 between phase variants appears to be a common phenomenon (43). These differences may reflect the preference of different phase variants for different environments within the host, which presumably differ in the availability of oxygen (62). The role of SpxB expression in resistance to killing by H2O2 was confirmed by the demonstration that spxB mutants, which produces no detectable H2O2, had a significant decrease in H2O2 resistance and by demonstrating that complementation of the spxB gene fully restored both H2O2 production both H2O2 resistance. These results showed that SpxB expression is required for wild-type levels of resistance to H2O2 rather than merely correlating with it. A further implication is that pyruvate oxidase is required for survival in the presence of the toxic by-product of its own activity.
It should be noted that R6x and Rx1 were observed to have a lower resistance to H2O2 than D39, the strain from which they were derived. This may be due at least in part to null mutations they contain in the hexA locus, which confer a DNA mismatch repair defect and increased sensitivity to DNA damage (60). The more severe defect in Rx1 resistance to H2O2 may be due to lower expression of SpxB, as evidenced by its lower production of H2O2 (Table 1).
S. pneumoniae lacks an inducible response to H2O2.
Coculture of spxB mutant bacteria with wild-type bacteria or the addition of subinhibitory concentrations of H2O2 did not induce H2O2 resistance in the mutant. In addition, H2O2 resistance appeared to increase in proportion to H2O2 production, rather than being dramatically increased at a certain threshold concentration, as is usually the case with inducible stress responses. Together, these findings suggested that an H2O2-inducible genetic pathway is not involved in H2O2 resistance. This is in contrast to other bacteria, such as E. coli, Streptococcus pyogenes, or Bacillus subtilis, which exhibit 10- to 100-fold-greater survival in the presence of 5 to 10 mM H2O2 after brief pretreatment with 10 to 50 μM H2O2 (13, 14, 33). S. pneumoniae is also unique in that it does not appear to contain a homolog to either OxyR or PerR, the transcriptional regulators that have been shown to control the H2O2-inducible response in other bacteria (6, 11, 26, 49). This lack of an inducible response to H2O2 may reflect the possibility that S. pneumoniae is often exposed to micromolar or greater concentrations of H2O2 through its own metabolism.
We further explored the possibility of an inducible response to H2O2 by measuring resistance to killing by H2O2 after growth in the presence or absence of O2. If S. pneumoniae has an inducible response to H2O2, one would expect its resistance to increase in the presence of O2, because O2 is required for the production of H2O2 by SpxB. Indeed, wild-type cells grown with O2 had significantly more resistance to killing by H2O2 than those grown without O2. In contrast, there was no significant difference in the resistance to H2O2 of spxB mutant cultures, whether grown with or without O2. However, when cultures of both strains were shifted from anaerobic to aerobic conditions with or without chloramphenicol, a protein synthesis inhibitor, H2O2 resistance was not affected. This confirmed that de novo protein synthesis is not involved in the increase of H2O2 resistance observed during aerobic growth, suggesting that an inducible response is not involved.
spxB mutants do not have a defect in general stress response.
There was no significant difference in the abilities of wild-type and spxB mutant bacteria to survive heat, high osmolarity, low pH, UV radiation, or the superoxide-generating compound paraquat, suggesting that there is no overall stress response defect (data not shown). The observation that H2O2 resistance is decreased when wild-type S. pneumoniae is grown in the absence of oxygen suggests that SpxB does not act by decreasing the effective concentration of oxygen. Therefore, the H2O2 sensitivity of spxB mutants does not result from either the absence of H2O2 or an excess of O2. Likewise, the possibility that spxB mutants have increased levels of pyruvate seemed unlikely to cause a defect in H2O2 resistance since pyruvate is a known scavenger of H2O2 (7). Furthermore, the addition of pyruvate to either mutant or wild-type cultures slightly decreased H2O2-mediated killing, arguing that increased killing of spxB mutants is not due to toxic by-products resulting from the reaction of excess pyruvate and H2O2 (data not shown). Together, this suggested that a product of SpxB activity other than H2O2 acts to increase resistance.
SpxB mutants are defective in their ability to produce AcP.
We therefore focused on the possibility that spxB mutants have a defect in their ability to aerobically produce AcP, a potential source of ATP (12, 55). Pyruvate oxidase mutants did indeed have a defect in AcP production. This report is, to our knowledge, the first demonstration that SpxB accounts for the majority of AcP production in S. pneumoniae. This suggests that a spxB mutant cannot fully compensate for a lack of AcP via conversion of acetate or acetyl-coenzyme A. The concentration of AcP in wild-type S. pneumoniae appears to exceed that of ATP by at least 10-fold (data not shown). Therefore, S. pneumoniae may utilize AcP as an additional energy source by conversion to ATP via acetate kinase. Aeration has been shown to increase ATP production in Streptococcus sanguis, a closely related species that also expresses pyruvate oxidase (61).
SpxB mutants are defective in their ability to maintain ATP levels during oxidative stress.
The spxB mutant also had decreased ability to maintain ATP levels during sublethal or lethal H2O2 stress. This implies that conversion of AcP into ATP may be a compensatory mechanism when traditional sources of ATP become unavailable due to oxidative stress. A lack of ATP may occur due to inactivation of sugar transport or glycolysis, since both processes are known to be particularly sensitive to oxidative stress (4, 58). Killing of Streptococcus lactis due to hypochlorous acid (HOCl), a potent oxidizer, has been attributed to inactivation of sugar phosphotransferases and subsequent depletion of ATP (4). In addition, the toxic effects of H2O2 and HOCl on E. coli have been shown to kill through similar mechanisms that are distinct from membrane disruption or inhibition of protein synthesis (16, 36). The metabolism of S. pneumoniae, being a strictly fermentative organism, appears to depend on sugar metabolism. Therefore, inactivation of sugar phosphotransferases or glycolytic enzymes by H2O2 may cause the dramatic decrease in ATP levels. Both H2O2 and NEM are known to react efficiently with sulfhydryl groups, which are present in the active sites of sugar phosphotransferases and several glycolytic enzymes (21, 37, 58). Similarly, sugar transport in Streptococcus agalactiae has been shown to be inhibited by lactoperoxidase-thiocyanate-hydrogen peroxide, as well as by NEM (38). The turnover time for the entire ATP pool in Streptococcus mutans growing in glucose has been estimated at 4 s, indicating that a halt in metabolism would rapidly deplete ATP levels (18). In wild-type S. pneumoniae (D39) exposed to 20 mM H2O2 there was an approximately 80% decrease in ATP content within 1.25 min (Fig. 6A). This is similar to the drop in ATP observed during arsenate treatment or starvation of Streptococcus cremoris (40, 45).
ATP levels in spxB mutants decreased more rapidly upon exposure to the thiol reagent NEM than in the wild type (Fig. 6C). This finding supports the theory that H2O2 may inactivate sugar transporters, since NEM is thought to act mainly on thiol groups located at the cell surface (25). In contrast to 20 mM H2O2 exposure, significant killing does not occur during exposure to 10 mM NEM, despite the reduction of ATP levels to approximately the same level (data not shown). This illustrates that killing by H2O2 is not due to ATP reduction alone. Since it has significantly greater permeability through cell membranes, H2O2 may inactivate intracellular enzymes more efficiently than NEM. Inactivation of one or more housekeeping enzymes, in addition to a decrease in ATP levels, may be a fatal combination for the cell.
DNA damage is an additional factor that may cause the loss of viability by H2O2. In S. pneumoniae, ATP is known to be required for DNA repair via the action of such proteins as HexA, RecA, and MmsA (23, 46, 57). Nevertheless, killing of S. pneumoniae by 20 mM H2O2 occurs only after ATP depletion, implying that ATP depletion is at least partly the cause. In addition, the rapid kinetics of the ATP drop we saw in S. pneumoniae is more consistent with a halt in glycolysis than a response to DNA damage. Lastly, the growth of spxB mutants is inhibited at a lower concentration of H2O2 than is the growth of the wild type (data not shown), arguing that the defect in H2O2 resistance is explained by a metabolic deficiency rather than one of DNA protection or repair.
Our observations suggest that pyruvate oxidase and acetate kinase may be more resistant to H2O2, compared to other metabolic enzymes. Consistent with this hypothesis, pyruvate oxidase in another streptococcal species has been shown to retain activity in vitro in the presence of millimolar concentrations of H2O2 (8). An inability to produce AcP and hence maintain ATP levels in spxB mutants would leave them unable to repair damage and maintain viability. Therefore, we propose that spxB mutants are defective in their ability to produce AcP and that this compound is an important source of ATP during H2O2 stress.
Fenton chemistry does not play a major role in the oxidative killing of S. pneumoniae.
Our data show that killing by 20 mM H2O2 is due at least in part, to metabolic inhibition. However, other species of bacteria are killed by submillimolar levels of hydrogen peroxide through a mechanism thought to proceed via the Fenton reaction, which requires iron. Since S. pneumoniae is not inhibited at H2O2 concentrations of <1 mM, we assayed iron levels in S. pneumoniae to determine whether its relatively high resistance to H2O2 was due to a lack of this element. However, our data indicated that total iron levels in S. pneumoniae are similar to those in more H2O2-sensitive organisms, such as E. coli. In the present study, we found that iron chelators failed to increase the resistance of S. pneumoniae to H2O2, suggesting that the Fe2+ level is not a major factor in the toxicity of H2O2 to this organism. An inability of iron chelators to mitigate oxidative stress has also been reported for S. pyogenes (49). These results suggest that streptococci, or perhaps lactic acid bacteria in general, may be resistant to the deleterious effects of the Fenton reaction. When EPR was used to directly assay the Fe2+ concentration in S. pneumoniae, we determined that the concentrations of Fe2+ were as high as or higher than those in E. coli (Table 2). Using spin trapping, we also determined that hydroxyl radical production during exposure to H2O2 occurs at a rate in S. pneumoniae similar to that which occurs in E. coli (Fig. 7). Furthermore, the ability of DF to eliminate the EPR signal caused by OH· in S. pneumoniae suggests that the inability of this iron chelator to increase H2O2 resistance in S. pneumoniae is not merely due to a lack of permeability (Fig. 7B).
There are several possible explanations for the relative resistance of S. pneumoniae to killing by the Fenton reaction compared to E. coli. One possibility is that in S. pneumoniae the majority of the Fe2+ is sequestered away from DNA. This would protect the organism from DNA damage because OH• is so reactive that if formed away from the chromosome it will likely be reduced before it can come into contact with it. A candidate for this role in S. pneumoniae is Dpr, a homolog of E. coli Dps. In vitro, S. mutans Dpr has been shown to bind iron and prevent OH• formation in the presence of H2O2 (65). However, unlike E. coli Dps, S. mutans Dpr does not bind DNA and contains an extra α-helical domain (65). Although uncharacterized, the S. pneumoniae Dpr has 57.1% identity to S. mutans Dpr. Therefore, it is possible that in S. pneumoniae this protein plays an enhanced role in the protection of DNA compared to E. coli. Efforts to generate a dpr mutant in S. pneumoniae were unsuccessful, suggesting that it may have an essential role in this species. Another possible explanation is that S. pneumoniae simply has a greater ability to repair DNA or tolerate damage than other organisms such as E. coli.
Other differences in metabolisms may explain the ability of S. pneumoniae to grow in concentrations of H2O2 that are inhibitory to E. coli. For example, one of the effects of oxidative stress on the metabolism of E. coli is the leaching of iron from enzymatic [4Fe-4S]2+ clusters (32). A search of the TIGR4 and R6 whole genomes revealed that many E. coli proteins that contain labile iron-sulfur cluster proteins (such as those involved in the tricarboxylic acid cycle) are absent in S. pneumoniae. Furthermore, genomic analysis revealed that none of the S. pneumoniae proteins predicted to contain iron-sulfur clusters have all 4 consensus cysteines which coordinate a [4Fe-4S]2+ cluster (data not shown). For example, among the streptococcal species examined to date, the DNA repair proteins MutY and Endo III lack two of the four well-conserved cysteines which form the iron-sulfur cluster in these proteins in other bacterial species (51). Importantly, enzymes containing [2Fe-2S]2+ clusters are more resistant to oxidants (17).
In summary, S. pneumoniae may actually avoid many of the toxic effects of aerobic growth by retaining a metabolism that does not involve respiration and yet maximizes ATP production during aerobic conditions. Since the pathogenic streptococci, like all lactic acid bacteria, have very similar metabolisms, these findings may have broad implications for the resistance of related species to oxidative stress caused by either the host or the environment.
Acknowledgments
We thank M. C. Trombe for strains Rx1 and P1234. We also thank Deborah Bae for expert technical assistance.
This work was supported by grants from the U.S. Public Heath Service (AI38436 and AI44231) to J.N.W.
REFERENCES
- 1.Achen, M. G., B. E. Davidson, and A. J. Hillier. 1986. Construction of plasmid vectors for the detection of streptococcal promoters. Gene 45:45-49. [DOI] [PubMed] [Google Scholar]
- 2.Auzat, I., S. Chapuy-Regaud, G. Le Bras, D. Dos Santos, A. D. Ogunniyi, I. Le Thomas, J. R. Garel, J. C. Paton, and M. C. Trombe. 1999. The NADH oxidase of Streptococcus pneumoniae: its involvement in competence and virulence. Mol. Microbiol. 34:1018-1028. [DOI] [PubMed] [Google Scholar]
- 3.Avery, O. T., C. M. MacLeod, and M. McCarty. 1944. Studies on the nature of the chemical nature of the substance inducing transformation of pneumococcal types. J. Exp. Med. 79:137-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrette, W. C. J., D. M. Hannum, W. D. Wheeler, and J. K. Hurst. 1989. General mechanism for the bacterial toxicity of hypochlorous acid: abolition of ATP production. Biochemistry 28:9172-9178. [DOI] [PubMed] [Google Scholar]
- 5.Braun, J. S., J. E. Sublett, D. Freyer, T. J. Mitchell, J. L. Cleveland, E. I. Tuomanen, and J. R. Weber. 2002. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J. Clin. Investig. 109:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bsat, N., A. Herbig, L. Casillas-Martinez, P. Setlow, and J. D. Helmann. 1998. Bacillus subtilis contains multiple Fur homologues: identification of the iron uptake (Fur) and peroxide regulon (PerR) repressors. Mol. Microbiol. 29:189-198. [DOI] [PubMed] [Google Scholar]
- 7.Carlsson, J., and M.-B. K. Edlund. 1987. Pyruvate oxidase in Streptococcus sanguis under various growth conditions. Oral Microbiol. Immunol. 2:10-14. [DOI] [PubMed] [Google Scholar]
- 8.Carlsson, J., M. B. Edlund, and S. K. Lundmark. 1987. Characteristics of a hydrogen peroxide-forming pyruvate oxidase from Streptococcus sanguis. Oral Microbiol. Immunol. 2:15-20. [DOI] [PubMed] [Google Scholar]
- 9.Carlsson, J., and U. Kujala. 1985. Pyruvate oxidase activity dependent on thiamine phosphate, flavin adenine dinucleotide, and orthophosphate in Streptococcus sanguis. FEMS Microbiol. Lett. 25:53-56. [Google Scholar]
- 10.Chapuy-Regaud, S., F. Duthoit, L. Malfroy-Mastrorillo, P. Gourdon, N. D. Lindley, and M. C. Trombe. 2001. Competence regulation by oxygen availability and by Nox is not related to specific adjustment of central metabolism in Streptococcus pneumoniae. J. Bacteriol. 183:2957-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christman, M. F., G. Storz, and B. N. Ames. 1989. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proc. Natl. Acad. Sci. USA 86:3484-3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condon, S. 1987. Responses of lactic acid bacteria to oxygen. FEMS Microbiol. Rev. 46:269-280. [Google Scholar]
- 13.Demple, B., and J. Halbrook. 1983. Inducible repair of oxidative DNA damage in Escherichia coli. Nature 304:466-468. [DOI] [PubMed] [Google Scholar]
- 14.Dowds, B. C. A., and J. A. Hoch. 1991. Regulation of the oxidative stress response by the hpr gene in Bacillus subtilis. J. Gen. Microbiol. 137:1121-1125. [DOI] [PubMed] [Google Scholar]
- 15.Duane, P. G., J. B. Rubins, H. R. Weisel, and E. N. Janoff. 1993. Identification of hydrogen peroxide as a Streptococcus pneumoniae toxin for rat alveolar epithelial cells. Infect. Immun. 61:4392-4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dukan, S., and D. Touati. 1996. Hypochlorous acid stress in Escherichia coli: resistance, DNA damage, and comparision with hydrogen peroxide stress. J. Bacteriol. 178:6145-6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flint, D. H., J. F. Tuminello, and M. H. Emptage. 1993. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 268:22369-22376. [PubMed] [Google Scholar]
- 18.Fukui, K., K. Kato, T. Kodoma, H. Ohta, T. Shimamoto, and T. Shimono. 1988. Kinetic study of a change in intracellular ATP level associated with aerobic catabolism of ethanol by Streptococcus mutans. J. Bacteriol. 170:4589-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghaffar, F., I. Friedland, and G. McCracken, Jr. 1999. Dynamics of nasopharyngeal colonization by Streptococcus pneumoniae. Pediatr. Infect. Dis. J. 18:638-646. [DOI] [PubMed] [Google Scholar]
- 20.Gibson, C. M., T. C. Mallett, A. Claiborne, and M. G. Caparon. 2000. Contribution of NADH oxidase to the aerobic metabolism of Streptococcus pyogenes. J. Bacteriol. 182:448-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glazer, A. N., R. J. DeLange, and D. S. Sigman. 1975. Chemical modification of proteins. Elsevier Publishing Co., Inc., New York, N.Y.
- 22.Greenwood, B. 1999. The epidemiology of pneumococcal infection in children in the developing world. Philos. Trans. R. Soc. London Ser. B 354:777-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hedayati, M. A., S. E. Steffen, and F. R. Bryant. 2002. Effect of the Streptococcus pneumoniae MmsA protein on the RecA protein-promoted three-strand exchange reaction. J. Biol. Chem. 277:24863-24869. [DOI] [PubMed] [Google Scholar]
- 24.Hirst, R. A., K. S. Sikand, A. Rutman, T. J. Mitchell, P. W. Andrew, and C. O'Callaghan. 2000. Relative roles of pneumolysin and hydrogen peroxide from Streptococcus pneumoniae in inhibition of ependymal ciliary beat frequency. Infect. Immun. 68:1557-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollenbach, A. D., K. A. Dickson, and W. M. Washabaugh. 2002. Thiamin transport in Escherichia coli: the mechanism of inhibition by the sulfhydryl-specific modifier N-ethylmaleimide. Biochim. Biophys. Acta 1564:421-428. [DOI] [PubMed] [Google Scholar]
- 26.Horsburgh, M. J., M. O. Clements, H. Crossley, E. Ingham, and S. J. Foster. 2001. PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun. 69:3744-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoskins, J., W. E. Alborn, Jr., J. Arnold, L. C. Blaszczak, S. Burgett, B. S. DeHoff, S. T. Estrem, L. Fritz, D. J. Fu, W. Fuller, C. Geringer, R. Gilmour, J. S. Glass, H. Khoja, A. R. Kraft, R. E. Lagace, D. J. LeBlanc, L. N. Lee, E. J. Lefkowitz, J. Lu, P. Matsushima, S. M. McAhren, M. McHenney, K. McLeaster, C. W. Mundy, T. I. Nicas, F. H. Norris, M. O'Gara, R. B. Peery, G. T. Robertson, P. Rockey, P. M. Sun, M. E. Winkler, Y. Yang, M. Young-Bellido, G. Zhao, C. A. Zook, R. H. Baltz, S. R. Jaskunas, P. R. Rosteck, Jr., P. L. Skatrud, and J. I. Glass. 2001. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183:5709-5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imlay, J. A. 2002. How oxygen damages microbes: oxygen tolerance and obligate anaerobiosis. Adv. Microb. Physiol. 46:111-153. [DOI] [PubMed] [Google Scholar]
- 29.Imlay, J. A., S. M. Chin, and S. Linn. 1988. Toxic DNA damage by hydrogen peroxide through the fenton reaction in vivo and in vitro. Science 240:640-642. [DOI] [PubMed] [Google Scholar]
- 30.Imlay, J. A., and S. Linn. 1986. Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J. Bacteriol. 166:519-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janzen, E. G., Y. Y. Wang, and R. V. Shetty. 1978. Spin trapping with a-pyridyl-1-oxide-N-tert-butyl nitrones in aqueous solutions. A unique electron spin resonance spectrum for the hydroxyl radical adduct. J. Am. Chem. Soc. 100:2923-2925. [Google Scholar]
- 32.Keyer, K., and J. A. Imlay. 1996. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 93:13635-13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King, K. Y., J. A. Horenstein, and M. G. Caparon. 2000. Aerotolerance and peroxide resistance in peroxidase and PerR mutants of Streptococcus pyogenes. J. Bacteriol. 182:5290-5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacks, S., and R. D. Hotchkiss. 1960. A study of the genetic material determining an enzyme activity in pneumococcus. Biochim. Biophys. Acta 39:508-517. [DOI] [PubMed] [Google Scholar]
- 35.McCormick, M. L., G. R. Buettner, and B. E. Britigan. 1998. Endogenous superoxide dismutase levels regulate iron-dependent hydroxyl radical formation in Escherichia coli exposed to hydrogen peroxide. J. Bacteriol. 180:622-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McKenna, S. M., and K. J. A. Davies. 1988. The inhibition of bacterial growth by hypochlorous acid. Biochem. J. 254:685-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meadow, N. D., M. A. Kukuruzinska, and S. Roseman. 1985. The bacterial phosphoenolpyruvate:sugar phosphotransferase system, p. 523-559. In A. N. Martonosi (ed.), The enzymes of biological membranes, vol. 3. Plenum Press, Inc., New York, N.Y.
- 38.Mickelson, M. N. 1977. Glucose transport in Streptococcus agalactiae and its inhibition by lactoperoxidase-thiocyanate-hydrogen peroxide. J. Bacteriol. 132:541-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musher, D. M. 1992. Infections caused by Streptococcus pneumonaie: clinical spectrum, pathogenesis, immunity, and treatment. Clin. Infect. Dis. 14:801-809. [DOI] [PubMed] [Google Scholar]
- 40.Otto, R., J. Vije, B. ten Brink, B. Klont, and W. N. Konings. 1985. Energy metabolism in Streptococcus lactis during lactose starvation. Arch. Microbiol. 141:348-352. [Google Scholar]
- 41.Overweg, K., C. D. Pericone, G. G. C. Verhoef, J. N. Weiser, H. D. Meiring, A. P. J. M. De Jong, R. De Groot, and P. W. M. Hermans. 2000. Differential protein expression in phenotypic variants of Streptococcus pneumoniae. Infect. Immun. 68:4604-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearce, B. J., Y. B. Yin, and H. R. Masure. 1993. Genetic identification of exported proteins in Streptococcus pneumoniae. Mol. Microbiol. 9:1037-1050. [DOI] [PubMed] [Google Scholar]
- 43.Pericone, C. D., D. Bae, M. Shchepetov, T. McCool, and J. N. Weiser. 2002. Short-sequence tandem and nontandem DNA repeats and endogeous hydrogen peroxide production contribute to genetic instability of Streptococcus pneumoniae. J. Bacteriol. 184:4392-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pericone, C. D., K. Overweg, P. W. M. Hermans, and J. N. Weiser. 2000. Inhibitory and bactericidal effects of Streptococcus pneumoniae on other inhabitants of the upper respiratory tract. Infect. Immun. 68:3990-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poolman, B., E. J. Smid, and W. N. Konings. 1987. Kinetic properties of a phosphate-bond-driven glutamine-glutamine transport system in Streptococcus lactis and Streptoccus cremoris. J. Bacteriol. 169:2755-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Priebe, S. D., S. M. Hadi, B. Greenberg, and S. A. Lacks. 1988. Nucleotide sequence of the hexA gene for DNA mismatch repair in Streptococcus pneumoniae and homology of hexA to mutS of Escherichia coli and Salmonella typhimurium. J. Bacteriol. 170:190-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prüss, B., and A. Wolfe. 1994. Regulation of acetyl phosphate and degradation, and the control of flagellar expression in Escherichia coli. Mol. Microbiol. 12:973-984. [DOI] [PubMed] [Google Scholar]
- 48.Repine, J. E., R. B. Fox, and E. M. Berger. 1981. Hydrogen peroxide kills Staphylococcus aureus by reacting with staphylococcal iron to form hydroxyl radical. J. Biol. Chem. 256:7094-7096. [PubMed] [Google Scholar]
- 49.Ricci, S., R. Janulczyk, and L. Bjorck. 2002. The regulator PerR is involved in oxidative stress response and iron homeostasis and is necessary for full virulence of Streptococcus pyogenes. Infect. Immun. 70:4968-4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 51.Samrakandi, M. M., and F. Pasta. 2000. Hyperrecombination in Streptococcus pneumoniae depends on an atypical mutY homologue. J. Bacteriol. 182:3353-3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seaver, L. C., and J. A. Imlay. 2001. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183:7173-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seaver, L. C., and J. A. Imlay. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol. 183:7182-7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sedewitz, B., K. H. Schleifer, and F. Gotz. 1984. Purification and biochemical characterization of pyruvate oxidase from Lactobacillus plantarum. J. Bacteriol. 160:273-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smart, J. B., and T. D. Thomas. 1987. Effect of oxygen on lactose metabolism in lactic streptococci. Appl. Environ. Microbiol. 53:533-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spellerberg, B., D. R. Cundell, J. Sandros, B. J. Pearce, I. Idanpaan-Heikkila, C. Rosenow, and H. R. Masure. 1996. Pyruvate oxidase, as a determinant of virulence in Streptococcus pneumoniae. Mol. Microbiol. 19:803-813. [DOI] [PubMed] [Google Scholar]
- 57.Steffen, S. E., and F. R. Bryant. 2000. Purification and characterization of the RecA protein from Streptococcus pneumoniae. Arch. Biochem. Biophys. 382:303-309. [DOI] [PubMed] [Google Scholar]
- 58.Storz, G., and J. A. Imlay. 1999. Oxidative stress. Curr. Opin. Microbiol. 2:188-194. [DOI] [PubMed] [Google Scholar]
- 59.Tettelin, H., K. E. Nelson, I. T. Paulsen, J. A. Eisen, T. D. Read, S. Peterson, J. Heidelberg, R. T. DeBoy, D. H. Haft, R. J. Dodson, A. S. Durkin, M. Gwinn, J. F. Kolonay, W. C. Nelson, J. D. Peterson, L. A. Umayam, O. White, S. L. Salzberg, M. R. Lewis, D. Radune, E. Holtzapple, H. Khouri, A. M. Wolf, T. R. Utterback, C. L. Hansen, L. A. McDonald, T. V. Feldblyum, S. Angiuoli, T. Dickinson, E. K. Hickey, I. E. Holt, B. J. Loftus, F. Yang, H. Smith, J. Venter, B. A. Dougherty, D. A. Morrison, S. K. Hollingshead, and C. M. Fraser. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498-506. [DOI] [PubMed] [Google Scholar]
- 60.Tiraby, J. G., and M. S. Fox. 1973. Marker discrimination in transformation and mutation of pneumococcus. Proc. Natl. Acad. Sci. USA 70:3541-3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Beelen, P., J. S. van der Hoeven, M. H. de Jong, and H. Hoogendoorn. 1986. The effect of oxygen on the growth and acid production of Streptococcus mutans and Streptococcus sanguis. FEMS Microbiol. Ecol. 38:25-30. [Google Scholar]
- 62.Weiser, J. N., R. Austrian, P. K. Sreenivasan, and H. R. Masure. 1994. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect. Immun. 62:2582-2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weiser, J. N., Z. Markiewicz, E. I. Tuomanen, and J. H. Wani. 1996. Relationship between phase variation in colony morphology, intrastrain variation in cell wall physiology and nasopharyngeal colonization by Streptococcus pneumoniae. Infect. Immun. 64:2240-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weiser, J. N., N. Pan, K. L. McGowan, D. Musher, A. Martin, and J. C. Richards. 1998. Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J. Exp. Med. 187:631-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamamoto, Y., L. B. Poole, R. R. Hantgan, and Y. Kamio. 2002. An iron-binding protein, Dpr, from Streptococcus mutans prevents iron-dependent hydroxyl radical formation in vitro. J. Bacteriol. 184:2931-2939. [DOI] [PMC free article] [PubMed] [Google Scholar]