

Abstract
Sequence-selective recognition of DNA inside cells by oligonucleotides would provide valuable insights into cellular processes and new leads for therapeutics. Recent work, however, has shown that noncoding RNA transcripts overlap chromosomal DNA. These RNAs provide alternate targets for oligonucleotides designed to bind promoter DNA, potentially overturning previous assumptions about mechanism. Here, we show that antigene locked nucleic acids (agLNAs) reduce RNA levels of targeted genes, block RNA polymerase and transcription factor association at gene promoters, and bind to chromosomal DNA. These data suggest that the mechanism of LNAs involves recognition of chromosomal DNA and that LNAs are bona fide antigene molecules.
Chromosomal DNA is an important target for molecular recognition by synthetic agents (1). Research over thirty years has examined triple-helix forming agents (2), oligomers that bind through Watson-crick base pairing (3), and small molecule pyrimidine polyamides (4). These compounds share a common assumption—that recognition would occur through binding to DNA.
Recently, double stranded antigene RNA (agRNAs) (we use the term “antigene” to distinguish these short RNAs from similar duplexes that are complementary to mRNA) that target transcription start sites for gene promoters have been shown to modulate gene expression (5-8). Expression increases in a cell lines where endogenous expression is relatively low, and decreases in a cell line where endogenous expression is relatively high.
We had initially hypothesized that agRNAs were directly interacting with chromosomal DNA inside cells. However, we discovered that the interaction was mediated by the protein argonaute 2 (AGO2) (6). AGO2 is known to promote formation of RNA-RNA hybrids (9), rather than DNA-RNA hybrids, bringing the likelihood of DNA-RNA binding into question.
We then investigated the molecular target for agRNAs (8). We discovered that DNA was not the only potential target, there was also a noncoding RNA transcript overlapping the promoter that originated within the target gene and was transcribed in an antisense orientation. We showed that agRNAs were binding to this transcript, recruited AGO protein to the transcript, and that reduced expression of the transcript reversed the action of activating agRNAs. Together, these data suggested that the noncoding RNA, not chromosomal DNA, was the direct target for agRNAs.
The presence of noncoding RNA transcripts that overlap genes is a widespread phenomenon. Over 20% of genes have antisense transcripts and over 80% of the genome is transcribed into RNA (11). The purpose of these transcripts is a major unresolved question for biology. What is certain is that these transcripts complicate previously easy assumptions about the mechanism of action of antigene agents originally designed to target chromosomal DNA.
LNA nucleotides contain a methylene bridge connecting the 2′-oxygen of the ribose with the 4′-carbon (11,12) (Figure 1a). This bridge reduces the conformational flexibility of the ribose and lowers the entropic penalty paid upon hybridization. When placed within DNA or RNA, LNA bases have proven to be effective tools for tailoring the hybridization affinity of oligonucleotides, with a single LNA substitution raising Tm values by as much as 5-10 °C (13). LNAs are being tested in clinical trials (14) and appear to be a promising strategy for clinical development.
Figure 1.

A) Chemical structures of LNA and ENA® bases. B-D) depicts qPCR data showing the effect of adding LNA and ENA® oligomers on mRNA levels of endogenous PR and AR genes. Cells were treated with A) LNA at 50 nM targeting PR-B, B) ENA® at 50 nM targeting PR-B, and C) Dose Response curve of LNA AR1 showing the relative levels of AR mRNA. Error bars represent the standard deviation among ≥3 replicates.
We recently showed that single-stranded antigene LNAs (agLNAs) that target gene promoters can block gene expression (15). Inhibition is potent and the protocols for achieving inhibition are simple. Further progress with agLNAs, however, requires an understanding of their mechanism and, fundamentally, whether they are recognizing chromosomal DNA or cellular noncoding RNA.
We introduced agLNAs that are complementary to either progesterone receptor (PR) (16) or androgen receptor (AR) (17) promoters (Supplemental Table 1) into T47D human breast cancer cells. qPCR (Supplemental Table 2) revealed a reduction in levels of PR (Figure 1b, LNA PR2) or AR (Figure 1d, LNA AR1) when treated with complementary agLNAs, relative to addition of a noncomplementary control LNA. Addition of an LNA complementary to the nontemplate strand (LNA PR1) of the PR promoter only marginally reduced PR mRNA levels. Results were confirmed with additional primer sets (Supplemental Figure 1).
We also transfected cells with a related oligonucleotide chemistry, 2′-O,4′-C-ethylene-bridged nucleic acid (ENA®) (Figure 1a). ENA® contains an additional methyl group in the 2′-4′ carbon bridge and has been shown to increase Tm values (18). Addition of ENA® oligomers (Supplemental Table 1) to T47D cells also resulted in reduced RNA levels (Figure 1c). Reduction of RNA levels by agLNA and agENA® oligonucleotides is consistent with a mechanism that involves binding to chromosomal DNA and reduction of transcription.
Binding of compounds to transcription start sites would be expected to reduce association of RNA polymerase. To test this hypothesis we performed chromatin immunoprecipitation (ChIP) on cells treated with either agLNA or a noncomplementary control. RNase was added to all samples to restrict potential targets to DNA. For agLNAs targeting the PR or AR promoters we observed reduction of RNA polymerase association upon transfection of oligonucleotide (Figure 2a,b). Reduction of RNA polymerase levels were also observed upon introduction of the ENA®-modifed oligonucleotide into cells (Figure 2c). Reduction of RNA polymerase occupancy by oligonucleotides containing two different constrained nucleotides (LNA, ENA®) in two different genes (PR, AR) is consistent with recognition of chromosomal DNA.
Figure 2. Addition of LNA and ENA® oligomers reduces occupancy of RNA polymerase and SP1 transcription factor at gene promoters.

ChIP analysis using qPCR showing the relative occupancy of RNA polymerase II near the PR-B transcription start site following treatment with 50 nM of A) LNA or B) ENA®. C) Relative occupancy of transcription factor SP1 near the PR-B transcription start site following treatment with 50 nM of LNA. D) RNA polymerase II occupancy near the AR transcription start site following treatment with 50 nM of LNA AR24. Error bars represent the standard deviation among 3 replicates.
It is also possible that binding to chromosomal DNA will block binding of transcription factors. There is a well characterized binding site for the transcription factor SP1 within the PR promoter (19), so we decided to investigate the effect of agLNAs on SP1 occupancy. Using ChIP, we observed that addition of LNA PR2 reduced levels of SP1 on the PR promoter (Figure 2d), further supporting the hypothesis that recognition occurs through binding to DNA.
Our data on qPCR measurement of RNA levels, ChIP for RNA polymerase, and ChIP for SP1 strongly suggests that agRNAs silence transcription by binding to DNA. To further support the hypothesis that a physical association is occurring between LNA and DNA we synthesized LNA-biotin conjugates, introduced them into T47D cells, and examined whether biotin-avidin purification would isolate PR promoter chromosomal DNA. Sequences bound to the biotin-LNA conjugate were purified using avidin beads and then amplified by PCR. The amplification used did not employ a reverse transcriptase step and could not amplify RNA.
Cells were transfected with biotin-labeled LNA and cultured for four days. Using qPCR, we showed that the biotin-labeled LNA complementary to the template strand of the PR promoter inhibited PR expression similarly to analogous unlabeled LNAs (Figure 3a). The biotin-labeled RNA complementary to the nontemplate strand did not significantly reduce PR mRNA levels, similar to what we had observed for the analogous unmodified LNA (Figure 1a).
Figure 3. Biotin-labeled LNA inhibits PR gene expression and binds chromosomal DNA at the PR promoter.
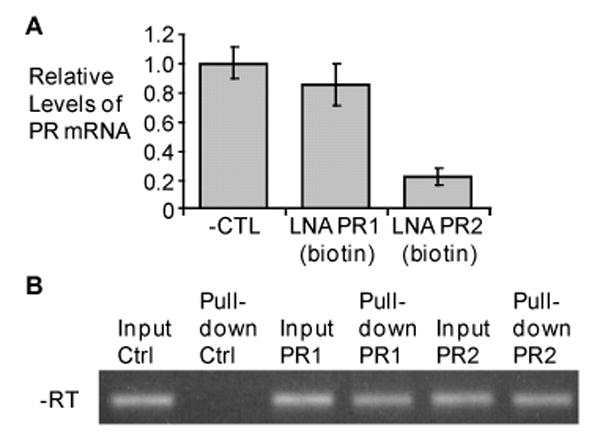
A) qPCR data showing the relative levels of mRNA following treatment with 3′-biotin modified LNAs or an negative LNA control. Error bars show the standard deviation among 3 replicates. B) Representative Agarose gel showing PCR-amplified DNA (reverse transcriptase was not performed) that associated with a biotin-modified LNA following transfection. ‘Input’ controls were taken prior to adding the nuclear lysate to avidin-coated beads and ensures that the lysate contains the DNA of interest. Bands labeled ‘Pull-down’ show PCR-amplified DNA following biotin pull-down and elution from avidin-coated beads. Primers corresponding to the PR promoter were used to amplify DNA. Bands were excised from gel, cloned, and sequenced to verify identity.
After demonstrating that the biotin-labeling did not effect inhibition of PR expression by the agLNA complementary to the template strand, we used the biotin-avidin purification to test whether the LNA was binding to PR promoter DNA (Figure 3b). The “input” sample was harvested prior to avidin purification and demonstrates that it is possible to amplify the PR promoter in our samples. When no LNA was added, qPCR analysis did not reveal the presence of any PR promoter DNA after purification.
When biotin-labeled LNA PR2 was present, PCR detected DNA corresponding to the PR-B promoter. This data shows that LNA PR2, which targets the template strand of DNA, associates with its chromosomal DNA target. LNA PR1 also associates with chromosomal DNA. LNA PR1 targets the non-template strand of DNA and does not reduce protein levels or mRNA levels (Figures 1 and 3) when introduced into T47D cells. Data showing that LNA PR1 binds to chromosomal DNA results suggests that the inability of LNA PR1 to inhibit gene expression is not due to a lack of DNA accessibility. We note that inhibition of transcription in cell-free systems by template strand targeted agents, but not similar compounds that target the nontemplate strand, has been observed for oligonucleotides (20) and peptide nucleic acids (21,22). One concern with experiments applying biotin-avidin purification protocols to living cells is that association may take place after cell lysis. Our lysis buffer (supplemental methods), however, contained divalent cation concentrations that should minimize the likelihood of DNA breathing and discourage strand invasion (23).
Why do agRNAs bind to noncoding transcripts while agLNAs bind to chromosomal DNA? The most likely explanation is that recognition by agRNAs is facilitated by argonaute proteins that have evolved to promote RNA:RNA rather than RNA:DNA recognition. LNAs, by contrast, are well suited to bind duplex DNA without assistance from cellular proteins because of their high affinity hybridization and resistance to digestion by nucleases.
Taken together, our data support the conclusion that the mechanism for agLNA-mediated inhibition of gene expression involves binding of LNAs to chromosomal DNA. RNA levels are reduced, association of RNA polymerase and SP1 transcription factor are decreased, and the LNA binds to chromosomal DNA. These results are significant because they suggest that the presence of a noncoding RNA is not necessary for gene inhibition by agLNAs. The ability to directly bind DNA also suggests that agLNAs have potential uses that go beyond inhibition of gene expression. They may be sensitive intracellular probes for transcription factor binding, and our experiments with SP1 provide one example of the potential for this approach. Chromosome-bound LNAs might also be useful agents for introducing or correcting gene mutations.
Supplementary Material
Details of LNA/ENA® oligomers, cell culture, RNA assays, and ChIP. This material is available free of charge via the internet at http:/pubs.acs.org.
Acknowledgments
We would like to thank Mitsubishi-Kagaku Foods Corporation (MFC), Tokyo, Japan, for generously providing the ENA® amidites required for these experiments. ENA is a registered trademark of Mitsubishi-Kagaku Foods.
Footnotes
This work was supported by the National Institutes of Health (NIGMS 60642 and NIGMS 73042) and the Robert A. Welch Foundation (I-1244).
References
- 1.Kaihatsu K, Janowski BA, Corey DR. Chem Biol. 2004;11:749–758. doi: 10.1016/j.chembiol.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 2.Kalish JM, Glazer PM. Ann New York Acad Sci. 2005;1058:151–161. doi: 10.1196/annals.1359.023. [DOI] [PubMed] [Google Scholar]
- 3.Janowski BA, Kaihatsu K, Huffman KE, Schwartz JC, Ram R, Hardy D, Mendelson CR, Corey DR. Nat Chem Biol. 2005;1:210–215. doi: 10.1038/nchembio724. [DOI] [PubMed] [Google Scholar]
- 4.Nickols NG, Jacobs CS, Farkas ME, Dervan PB. ACS Chem Biol. 2007;2:561–571. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janowski BA, Huffman KE, Schwartz JC, Ram R, Hardy D, Shames DS, Minna JD, Corey DR. Nat Chem Biol. 2005;1:216–222. doi: 10.1038/nchembio725. [DOI] [PubMed] [Google Scholar]
- 6.Janowski BA, Huffman KE, Schwartz JC, Ram R, Nordsell R, Shames DS, Minna JD, Corey DR. Nat Struct Mol Biol. 2006;13:787–792. doi: 10.1038/nsmb1140. [DOI] [PubMed] [Google Scholar]
- 7.Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Nat Chem Biol. 2007;3:166–173. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- 8.Schwartz JC, Younger ST, Nguyen N, Hardy DB, Corey DR, Janowski BA. Nat Struct Mol Biol. 2008;15:842–848. doi: 10.1038/nsmb.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 10.Wahlstedt C. Drug Discov Today. 2006;11:503–508. doi: 10.1016/j.drudis.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 11.Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, Olsen CE, Wengel J. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 12.Obika S, Nanbu D, Hari Y, Andoh J, Morio K, Doi T, Imanishi T. Tet Lett. 1998;39:5401–5404. [Google Scholar]
- 13.Vester B, Wengel J. Biochemistry. 2004;43:13233–13241. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 14.Koch T, Orum H. In: Antisense Drug Technology. 2nd. Crooke S, editor. 2007. pp. 519–564. [Google Scholar]
- 15.Beane RL, Ram R, Gabillet S, Arar K, Monia B, Corey DR. Biochemistry. 2007;46:7572–7580. doi: 10.1021/bi700227g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H, Chambon P. EMBO J. 1990;9:1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tilley WD, Marcelli M, McPhaul MJ. J Biol Chem. 1990;265:13776–13781. [PubMed] [Google Scholar]
- 18.Morita K, Takagi M, Hasegawa C, Kaneko M, Tsutsumi S, Sone J, Ishikawa T, Imanishi T, Koizumi M. Bioorg Med Chem. 2003;11:2211–2226. doi: 10.1016/s0968-0896(03)00115-9. [DOI] [PubMed] [Google Scholar]
- 19.Schultz JR, Petz LN, Nardulli AM. Mol Cell Endocrinol. 2003;201:165–175. doi: 10.1016/s0303-7207(02)00415-x. [DOI] [PubMed] [Google Scholar]
- 20.Milne L, Xu Y, Perrin DM, Sigman DS. Proc Natl Acad Sci U S A. 2000;97:3136–3141. doi: 10.1073/pnas.050544597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanvey JC, Peffer NJ, Bisi JE, Thomson SA, Cadilla R, Josey JA, Ricca DJ, Hassman CF, Bonham MA, Au KG, et al. Science. 1992;258:1481–1485. doi: 10.1126/science.1279811. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen PE, Egholm M, Buchardt O. Gene. 1994;149:139–145. doi: 10.1016/0378-1119(94)90422-7. [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Ishihara T, Corey DR. Nucl Acid Res. 2000;28:3332–3338. doi: 10.1093/nar/28.17.3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of LNA/ENA® oligomers, cell culture, RNA assays, and ChIP. This material is available free of charge via the internet at http:/pubs.acs.org.