

Abstract
Sir2 enzymes catalyze the NAD+ dependent protein deacetylation and play critical roles in epigenetics, cell death and lifespan regulation. In spite of a current flurry of experimental studies, the catalytic mechanism for this unique and important class of enzymes remains elusive. Employing on-the-fly Born-Oppenheimer molecular dynamics simulations with the B3LYP/6-31G(d) QM/MM potential and the umbrella sampling method, we have characterized the initial step of the Sir2Tm catalyzed reaction, which is also the most controversial portion of its mechanism. Our results indicate that the nicotinamide cleavage reaction employs a highly dissociative and concerted displacement mechanism: the cleavage of the glycosidic bond is facilitated by the nucleophilic participation of the acetyl-lysine, and the dissociative transition state has a significant oxocarbenium ion character. During this step of the reaction, the Sir2Tm enzyme strongly stabilizes the covalent O-alkylamidate intermediate whereas its effect on the transition state is quite minimal. In addition, functional roles of key residues and motifs have been elucidated. This work further demonstrates the feasibility and applicability of the state-of-the-art ab initio QM/MM molecular dynamics approach in simulating enzyme reactions.
1 Introduction
Sirtuins, known as Sir2 (Silent information regulator 2), constitute a novel family of protein deacetylases dependent on nicotinamide-adenine dinucleotide (NAD+) and are highly conserved from bacteria to higher eukaryotes.1-4 These enzymes catalyze the deacetylation of a variety of proteins including histones, p53, α-tublin, FOXO proteins and NFκB. Yeast Sir2 (ySir2), the founding member of this family, deacetylates histones H3 and H4 which is required for silencing gene expression and maintaining genome integrity.5-7 Besides gene silencing, sirtuins have shown to be critically involved in regulating cell survival, apoptosis,8-10 DNA recombination,11, 12 and aging.13-15 For example, hyperactivation of Sir2 enzymes has been linked to increased lifespan, whereas deletion or inhibition of sirtuins leads to short lived species.16-18 The abnormal sir-tuin activity in mammals has been implicated in various age-related diseases, including diabetes, obesity and neurodegenerative disorders.19,20
In contrast to class I, II and IV histone deacetylases that utilize a zinc catalytic site to hydrolyze the amide bond and yield acetate,21-23 class III histone deacetylase sirtuin is a nicotinamide-adenine dinucleotide (NAD+) dependent enzyme, which cleaves the glycosidic bond and generates nicotinamide and novel metabolite O-acetyl-ADP-ribose (OAADPr).1-4 In the Sir2 enzyme complex, the acetyl-lysine side chain protrudes into a hydrophobic channel and terminates near the nicotinamide ribose of NAD+ to access the ribose of NAD+. Based on extensive biochemical24-32 and structural33-41 studies, the overall catalytic reaction has been suggested to proceed in two consecutive stages: the initial nicotinamide cleavage stage results in the formation of a positively charged O-alkylamidate intermediate; in the subsequent step, the acetyl group is transfered to the ADP-ribose to form a novel product OAADPr. The nicotinamide product is a non-competitive inhibitor of sirtuins, while the OAADPr may act as a second messenger, or a metabolite that links protein deacetylation to other cellular pathways.42
For the initial nicotinamide cleavage step, several different catalytic mechanisms have been proposed in the literatures,4 including: 1. a nucleophilic attack mechanism which involves the participation of His116 (in Sir2Tm indexing) and the employment of 2′-OH of NAD+ as the initial nucleophile;43 2. an enzyme nucleophile mechanism: an enzyme catalytic site residue acts as the nucleophile;33, 44, 45 3. an associative, SN2-like direct displacement mechanism, in which the cleavage of the nicotinamide is concerted with the attack of the acetyl-lysine;24, 30, 38, 40 4. a step-wise SN1-like mechanism in which an oxocarbenium intermediate is formed prior to the nucleophilic addition of the acetyl-lysine.24, 37 For the first two proposed mechanistic hypotheses, they have been convincingly ruled out based on the subsequent biochemical and structural studies: the mutagenesis study on the His11633 and the replacement of the 2′-OH of NAD+ by fluorine26 have indicated that neither His116 nor 2′-OH of NAD+ are involved in the initial step of the reaction; all the structures in complex with NAD+ determined for the Sir2 enzymes in recent years35-41 provided no evidence for the existence of an active site residue in the vicinity of C1′ of NAD+ to serve as a nucleophile; isotope labeling experiments24, 29 yielded results that do not support either scheme 1 or 2, but are consistent with the other two. On the other hand, although experimental studies have indicated the importance of the acetyl-lysine in the nicotinamide cleavage step,24, 29, 30 they cannot unambiguously distinguish between the latter two mechanisms, as shown in Figure 1. Furthermore, there is no direct experimental characterization of its transition state, and no theoretical studies on this unique and important class of enzymes.
Figure 1.

Illustration of SN1-like step-wise and SN2-like concerted mechanisms. In SN1-like mechanism, glycosidic bond is cleaved prior to the attack of carbonyl oxygen, whereas these two steps are synchronous in SN2-like mechanism.
In this work, we have filled this gap by characterizing the Sir2Tm catalyzed nicotinamide cleavage reaction with the state-of-the-art computational methods. Our calculation results indicate that this nicotinamide cleavage reaction employs a highly dissociative and concerted displacement mechanism. In addition, functional roles of key residues and motifs have been elucidated.
2 Methods
Our theoretical approaches center on Born-Oppenheimer molecular dynamics simulations with ab initio QM/MM potential46-56 and the umbrella sampling method.57-59 At each time step, atomic forces as well as the total energy of the QM/MM system are calculated with a pseudobond ab initio QM/MM approach54, 56, 60-62 on-the-fly, and Newton equations of motion are integrated. From a series of biased simulations, the potential of mean force (PMF) along the reaction coordinate is obtained with the weighted histogram analysis method (WHAM).63-65 This direct ab initio QM/MM MD approach, which has been implemented in our lab by interfacing Q-Chem66 with Tinker67 programs, provides an ab initio description of chemical bond forming and breaking processes and takes account of dynamics of the reaction active site and its environment on an equal footing. In spite of the conventional wisdom that such simulations would be computationally too expensive to be applicable for enzyme reactions, we have demonstrated this state-of-the-art approach to be feasible and successful in elucidating the catalytic power of histone lysine methyltransferase SET7/968 and providing insights into the methylation state specificity of histone lysine methylation.69 In this work, we have employed B3LYP/6-31G(d) QM/MM MD simulations to study the Sir2 enzyme, in which the QM subsystem has 65 atoms and 562 basis functions while the MM subsystem has more than 9000 atoms. We have employed 24 umbrella windows, and each window has been simulated for 30 ps. Therefore, in total we have performed 720 ps ab initio QM/MM MD simulations. This would represent a typical application that we would be able to carry out with the currently available computational resources.
The Sir2Tm enzyme reactant complex was prepared based on the crystal structure of 2H4F,40 the first reported crystal structure of Sir2 with both NAD+ and acetyl-lysine bound in the active site. The protonation states of charged residues were determined at constant pH = 7 based on the local environment via program pdb2pqr.70 The whole system was then reorientated with program Simulaid71 and solvated into a ∼ 60 × 60 × 84 Å3 TIP3P water rectangular box with a 10 Å buffer distance between the solvent box wall and the nearest solute atom. Seven sodium ions were added to neutralize the system. The resulting whole simulation system had 30938 atoms. After a series of minimizations and equilibrations, standard molecular dynamics simulations with periodic boundary condition were carried out for more than 25 ns. The trajectory was very stable after about 2 ns, and a snapshot at the 4 ns simulation time has been employed for subsequent QM/MM simulations. In the MD simulations, long-range electrostatic interactions were treated with particle mash Ewald (PME) method72, 73 and 12 Å cutoff was used for both van der Waals interactions and PME in real space. The pressure was coupled every 1 ps with isotropic position scaling and maintained at 1 atm. The Newton equations of motion were integrated with velocity Verlet algorithm. The shake algorithm74 was applied to constrain all bonds involving hydrogen atoms with tolerance of 10-5 Å. A time step of 2 fs was used, and Berendsen thermostat method75 has been used to control the system temperature at 300 K. All MD simulations were preformed with Amber9 molecular dynamics package,76 employing the Amber99SB77-79 force field for the protein and the TIP3P model80 for water molecules. Force field parameters for NAD+ were parameterized by Ryde81 and partial charges of non-standard residue acetyl-lysine were fitted with HF/6-31G(d) calculations and the RESP module in the Amber package.
With the snapshot taken from the MD trajectory, the QM/MM model was prepared by deleting the solvent molecules beyond 27 Å from the reaction center (C1′ of NAD+). The resulting system had 9143 atoms. The QM/MM partition is illustrated in Figure 2. Fragments directly participating in the reaction were defined as the QM subsystem, which had 65 atoms, including the nicotinamide, ribose ring and di-phosphate group portions of NAD+ as well as the side chain of acetyl-lysine, treated by B3LYP functional with 6-31G(d) basis set. The QM/MM boundary were treated with the pseudobond approach.60, 62 All other atoms were described by Amber99SB molecular mechanical force field77, 79 and the TIP3P model80 for water molecules. The prepared system was first minimized by QM/MM calculations, and then by choosing the difference of two bond lengths, C1′ - N1 and O - C1′, as the reaction coordinate: RC = dC1′-N1 - dO-C1′, an iterative minimization procedure with the reaction coordinate driving method61 had been employed to map out a minimum energy path. For each determined structure along the reaction path, the MM subsystem was further equilibrated by carrying out 500 ps MD simulations with the MM force field. Finally, the resulting snapshot was used as the starting structure for ab initio QM/MM MD simulations with umbrella sampling.57-59 A total of 24 simulation windows were employed, covering the reaction coordinate -2.0 ∼ 2.0. For each window, the total potential energy was biased with a harmonic potential of 40 kcal mol-1 Å-2, centered on the corresponding value of the reaction coordinate, and the configurations were collected for 20 ps for data analysis after an equilibration period of 10 ps. Thus, each simulation window had been sampled for 30 ps at B3LYP/6-31G(d) QM/MM level, and the total length of the ab initio QM/MM MD simulations for the whole reaction was 720 ps. In average, it took about 19 hours in wall time to run 1 ps B3LYP/6-31G(d) QM/MM MD simulation on a quad-core 2.33 GHz Intel Xeon cluster using 16 cores for this system. The probability distributions (e.g. histograms) along the reaction coordinate were determined for each window and pieced together with the weighted histogram analysis method (WHAM)63-65 to calculate the potential of mean force.
Figure 2.
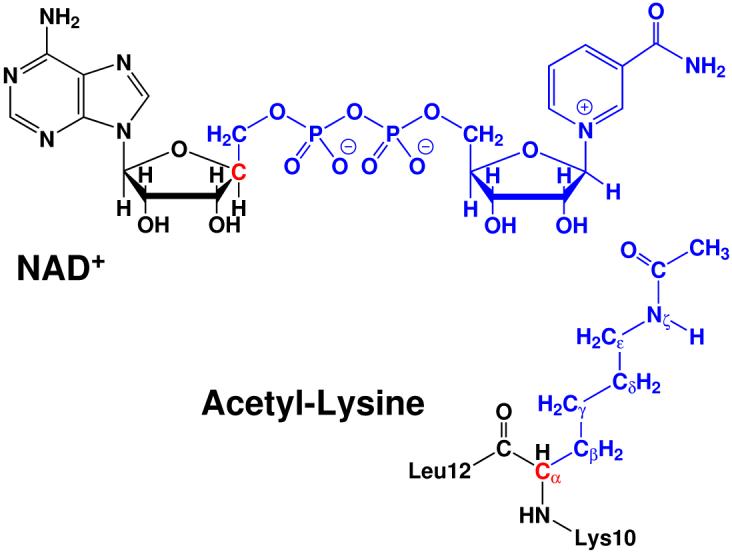
Illustration of the QM/MM partition. Color notations: black (MM subsystem); red (boundary carbon atoms in the QM subsystem described by improved pseudobond parameters62); blue (all other atoms in the QM subsystem).
All ab initio QM/MM calculations were performed with modified Q-Chem66 and Tinker67 programs. The time step was 1 fs, and Beeman algorithm82 was used to integrate the Newton equations of motion. In all QM/MM simulations, the spherical boundary condition had been applied so that atoms beyond 20 Å sphere centered on C1′ atom of NAD+ were fixed. The 18 and 12 Å cutoffs were employed for electrostatic and van der Waals interactions, respectively. There was no cutoff for electrostatic interactions between QM and MM subsystems. Berendsen thermostat method75 was used to control the system temperature at 300 K.
3 Results and Discussions
3.1 Catalytic Mechanism and Nature of Transition State
Since extensive experimental studies have already convincingly ruled out two mechanism hypotheses involving either 2′-hydroxyl of NAD+ or a catalytic site residue as the nucleophile, our computational studies exclusively focus on the remaining ambiguities regarding the nicotinamide cleavage reaction: a step-wise SN1-like mechanism or a concerted SN2-like mechanism, as shown in Figure 1. By exploring several different reaction coordinates which correspond to both step-wise and concerted schemes, we found that the step-wise scheme is unlikely. Using the difference between C1′ - N1 and O - C1′ as the reaction coordinate, which are the distances from the anomeric carbon to the leaving group and the incoming nucleophile, respectively, led to a reasonably smooth and continuous reaction path from the reactant to the α-1′-O-alkylamidate intermediate. Meanwhile, by mapping out a two-dimensional minimum energy surface along both C1′ - N1 and O - C1′ bonds, as shown in Figure 3, we further confirmed that there is no oxocarbenium intermediate formed in the reaction and it is appropriate to choose dC1′-N1 - dO-C1′ as the reaction coordinate. Thus, along the chosen reaction coordinate of dC1′-N1 - dO-C1′, we have carried out Born-Oppenheimer ab initio QM/MM molecular dynamics simulations with the umbrella sampling calculations to determine the free energy reaction profile.
Figure 3.
Two-dimensional minimum energy surface along C1′ - N1 and O - C1′ bonds. The reactant is on the lower left corner, the intermediate is on the upper right corner, and the transition state is located in the middle. The pink cycle line represents the minimum reaction path determined by employing dC1′-N1 - dO-C1′ as the reaction coordinate.
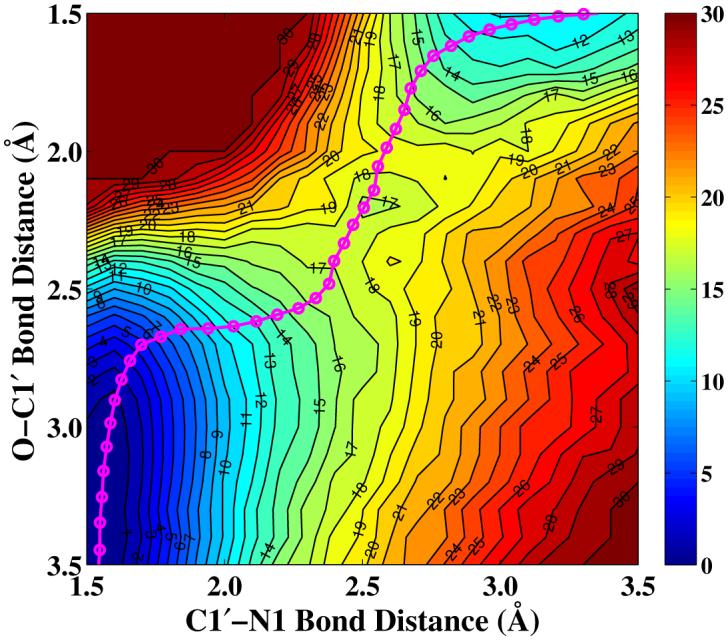
As shown in Figure 4, the computed potential of mean force for the nicotinamide cleavage reaction catalyzed by Sir2Tm converges very well. The calculated free energy of activation is 15.7 kcal/mol, which is consistent with 16.4 kcal/mol estimated from the reaction rate 6.7 s-1 of nicotinamide formation in Hst2.30 It should be noted that the experimentally measured kcat of 0.170 ± 0.006 s-1 for Sir2Tm39 is very similar to 0.2 ± 0.03 s-1 of Hst2.30 For the reaction rate of nicotinamide cleavage step, only the one for Hst2 has been measured experimentally,30 thus it was employed for the comparison here. Meanwhile, our calculated result indicated that the intermediate with the presence of the nicotinamide is not as stable as the reactant complex. This is quite reasonable considering that the nicotinamide cleavage step is known to be reversible24-26 and the nicotinamide release was suggested to be the rate-limiting step.29
Figure 4.
Potential of mean force for the Sir2Tm catalyzed nicotinamide cleavage reaction determined with B3LYP/6-31G(d) QM/MM MD simulations. Geometries at reactant, transition state, and intermediate in schematic representation and bond lengths are also shown.
To further confirm that there is no stable oxocarbenium state between the reactant and the intermediate, we have carried out the unrestrained ab initio QM/MM molecular dynamics simulations with 50 random snapshots around the transition state region. For each snapshot, two simulations have run, one with the original velocities and the other with the reversed ones. Among all simulations, there is no stable oxocarbenium state observed. For 50% of the snapshots (25 out of 50), one trajectory led to the reactant while the other went to the intermediate. These results further rule out the possibility of a step-wise mechanism, and confirm that our characterized transition state is meaningful.
At the reactant state, the distances of two bonds C1′ - N1 and O - C1′ are calculated to be 1.56 ± 0.04 and 3.26 ± 0.04 Å, respectively, consistent with the values of 1.49 and 3.18 Å in the crystal structure 2H4F.40 At the transition state, the corresponding values at the transition state are 2.60 ± 0.10 and 2.35 ± 0.10 Å, which are much longer than their covalent bond distances; the computed bond orders of C1′ - N1 and O - C1′ bonds with natural bond orbital (NBO) analysis are 0.10 ± 0.02 and 0.14 ± 0.04, respectively. Thus, the transition state for this nicotinamide cleavage reaction can be considered to be very loose and highly dissociative. We can see that from the reactant state to the transition state, not only does the glycosidic bond become almost broken, the nucleophilic attack distance has been significantly shortened, which is consistent with a concerted and SN 2-like mechanism.
The partial charges over acetyl-lysine, nicotinamide and ribose ring portions of NAD+ are computed for all the snapshots at the reactant, the transition state and the intermediate. As shown in Figure 5, both acetyl-lysine and nicotinamide undergo the charge migration through the ribose ring from the reactant to the transition state. At the transition state, the positive charge on the ribose ring significantly increases, whereas there has some amount of positive charge remaining on the nicotinamide. This further confirms the significant participation of the acetyl-lysine in the formation of the transition state, which is very consistent with the recent experimental result that the reaction rate of the nicotinamide cleavage reaction is dependent on the nucleophilicity of the acetyl-oxygen.30 Meanwhile, our results indicate that the dissociative transition state for the Sir2Tm enzyme also has a significant oxocarbenium ion character, which is to some extent similar to many other enzyme-catalyzed nicotinamide cleavage reactions.83-85
Figure 5.

Computed group partial charges of acetyl-lysine, nicotinamide, and ribose ring at reactant, transition state, and intermediate.
3.2 Catalytic Role of Enzyme Environment
To understand the role of enzyme environment in the nicotinamide cleavage reaction, we have calculated the van der Waals and electrostatic interactions between QM and MM subsystems with snapshots at the reactant, the transition state and the intermediate, respectively. Using the bin size of 0.05 Å, roughly 1100 snapshots have been picked up at each state. The charges for the QM subsystem were calculated in the presence of MM environment with B3LYP/6-31G(d) QM/MM calculations. Interestingly, as indicated in Figure 6, for this step of the reaction, the Sir2Tm enzyme strongly stabilizes the covalent α-1′-O-alkylamidate intermediate while its effect on the oxocarbenium ion transition state is quite minimal. It is not surprising since the nicotinamide cleavage is the first step of whole deacetylation, and the stabilization of the intermediate reduces the probability of the reverse reaction. In order to further elucidate how the intermediate is stabilized, we have calculated the individual residue contribution to the intermediate stabilization, as shown in Figure 7. The formula is: . The negative indicates that residue i stabilizes the intermediate, whereas the positive one is unfavorable. Such analyses have been demonstrated to be able to provide detailed insights into enzyme catalysis,23, 86-92 among which some computational hypotheses86, 88 have been confirmed by later experimental studies.93, 94 However, due to various inherent approximations employed in the energy decomposition calculations, such as the neglect of conformational change and dielectric screening, the calculated numbers can only be employed as qualitative indicators.
Figure 6.

Calculated electrostatic + van der Waals interaction energies between the QM subsystem and its environment at the reactant, the transition state and the intermediate. Note that the average energy distribution at the reactant is shifted to zero and the corresponding values are shifted for the transition state and the intermediate.
Figure 7.

Individual residue contribution to the intermediate stabilization. Negative value indicates that a residue stabilizes the intermediate, and vice versa. Note that the electrostatic interaction is dominant in the residue contribution.
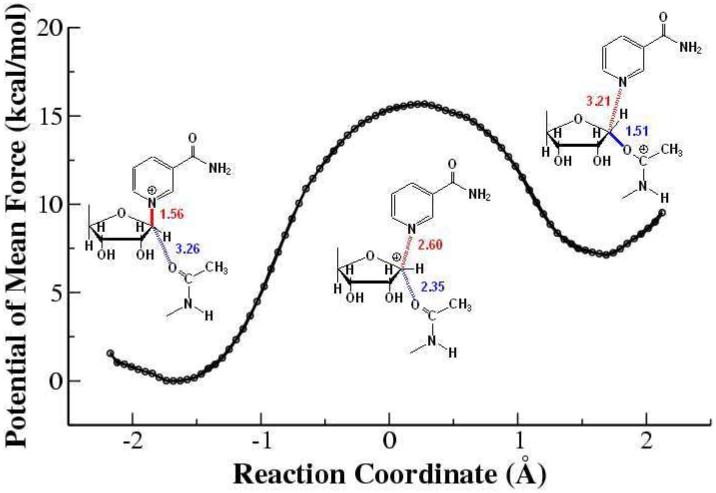
By analyzing the residue contribution, it is found that several motifs strictly conserved among sirtuin family play important catalytic roles in this initial nicotinamide cleavage reaction, as illustrated in Figure 8. The FGExL motif, which was previously suggested to be responsible for the acetyl-lysine binding,35 is found to also stabilize the O-alkylamidate intermediate mainly through the electrostatic interaction. The destabilization contribution of the adjacent residue Arg156 is mainly compensated by Glu164, which is a key residue in catalysis identified experimentally33 and in our current study. The salt bridge formed between these two residues would play an important structural role to stabilize the FGExL motif. Meanwhile, three highly invariant motifs in sirtuins, GAGxS, GIPxFR, and TQNIDxL, which make direct contacts with the NAD+,38 are also found to contribute considerably to the intermediate stabilization. As shown in Figure 9, the strictly conserved residue Arg34 in the GIPxFR motif forms two hydrogen bonds with two non-bridging oxygens of the di-phosphate portion of the NAD+. It was previously hypothesized to stabilize the O-alkylamidate intermediate,28 which is very consistent with our residue component analysis. Meanwhile, our results that Gln98, Asn99 and Ile100 in the TQNIDxL motif contribute to stabilize the intermediate and His116 has little effect in this first step of the reaction are in good agreement with the corresponding experimental mutagenesis results.28, 33, 34, 36, 37, 95
Figure 8.
Three dimensional enzyme-substrate complex structure. Some key motifs which significantly contribute to the substrate binding and enzyme catalysis are shown in color.
Figure 9.

The interactions of key residues with the NAD+ and the substrate acetyl-lysine. The left panel is shown in schematic representation, and the right is in actual three dimensional structure.
Among all residues, Figure 7 indicates that two negatively charged residues Asp101 and Asp32 most strongly disfavor the formation of the O-alkylamidate intermediate. This can be easily understood from the arrangement of these residues around the active site, as shown in Figure 9. As we discussed previously and illustrated in Figure 5, for this nicotinamide cleavage reaction, there is significant positive charge migration from the nicotinamide to the acetyl-lysine. Thus, negative charges located on the side of nicotinamide or positive charges on the side of acetyl-lysine would deter the charge transfer and destabilizes the O-alkylamidate intermediate, and vice versa. From Figure 9, we can see that Asp101 directly forms a hydrogen bond with the amide group of nicotinamide; while for Asp32, it lies above the ribose ring and is in close proximity of nicotinamide. Thus, both residues lead to much less favorable interaction with the intermediate than with the reactant. With the same physical reasoning, it is found that two charged residues distant from the active site, Asp49 on the side of acetyl-lysine and and Arg106 on the side of nicotinamide, may play an important catalytic role in this nicotinamide cleavage step, as indicated by Figure 7.
It should be noted that a residue destabilizing the intermediate does not necessarily mean that it is not important for the catalysis, since a residue can contribute to the enzyme function in many other ways, such as binding the substrate, facilitating the formation of the reactive conformation, lowering the barrier for subsequent reaction steps, stabilizing the local or global structure which is necessary for catalysis. For Asp32, it is not conserved among the sirtuin family proteins. In some of the Sir2 homologs, the corresponding one is a neutral residue Thr.4 Here we suggest that the mutation of Asp32 to Thr or Ser in Sir2Tm might result in the reaction rate enhancement for this nicotinamide cleavage step, but it may also weaken the binding of the substrate. For Pro31, its destabilization effect is mainly due to the electrostatic interaction between its backbone carboxyl oxygen and the nicotinamide portion of NAD+. Considering that Pro31 is widely conserved among the sirtuin protein family, it is likely to play an important structural role in the Sir2Tm enzyme.
The remaining question is the functional role of Asp101, which is strictly conserved within the Sir2 family4 and suggested to be a key residue in the C pocket for nicotinamide binding.39 Experimental studies indicated that the mutation of Asp101 to Asn would lead to not only a weaker binding of the substrate but also a significant loss in NAD+-dependent deacetylation activity.39 Since Asp101 directly forms a hydrogen bond with the amide group of the nicotinamide fragment which carries significant positive charge in the reactant complex, it is expected that the mutation would result in the weak binding of the substrate. However, it is unclear how the D101N mutation affects the deacetylation activity, since this mutation should facilitate the formation of the O-alkylamidate intermediate based on the consideration of electrostatic interaction alone. Here we have carried out two molecular dynamics simulations on the D101N mutant in complex with substrates acetyl-lysine and NAD+ with the same simulation protocol as the wild-type enzyme. For the wild-type enzyme, Asp101 makes two hydrogen bonds with the amide group of NAD+ and the backbone of Leu103 throughout the simulation. The torsion angle around the glycosidic bond in NAD+ is 36.8 ± 8.7°, which is in good agreement with 38.3° in the crystal structure.40 In the first simulation of the D101N mutant, the carboxyl oxygen hydrogen-bonding to the NAD+ was replaced with an amide group, as illustrated in Figure S1. We found that the NAD+ conformation changed immediately during the simulation. By superimposing the 5 ns snapshots of wild-type Sir2Tm and the D101N mutant for comparison, as shown in Figure 10, we can see that although the rest structures superimpose quite well, the nicotinamide ring adopts a very different conformation in the D101N mutant which leads to an unfavorable geometry arrangement for the acetyl-lysine to participate in the reaction as the nucleophile. In the second simulation of the D101N mutant, as illustrated in Figure S1, the carboxyl oxygen hydrogen-bonding to the Leu103 was replaced with an amide group. For this trajectory, Asn101 can maintain a weak hydrogen bond with NAD+ for about 8 ns, as shown in Figure 11, but later hydrogen bonds break and the torsion angle of the glycosidic bond in NAD+ significantly changes. These simulation results clearly indicate that the D101N mutation would lead to the disruption of key hydrogen bonds in the nicotinamide binding pocket, and the change of the binding conformation of NAD+. Thus, it would be reasonable to suggest that the changes in the binding of NAD+ in the active site due to the D101N mutation may lead to the decrease of its catalytic activity.
Figure 10.

Superimposition of the active sites of wild-type Sir2Tm (in blue) and its D101N mutant (in yellow, simulation I). Both snapshots are taken at 5 ns in MD simulations after equilibration.
Figure 11.

Time dependent trajectories of torsion angle of the glycosidic bond in NAD+ and some of the key hydrogen bonds in the nicotinamide binding pocket during 25 ns MD simulations in both wild-type Sir2Tm and its D101N mutant ( Simulation II), respectively.
4 Conclusions
By employing ab initio QM/MM molecular dynamics simulations, we have characterized the mechanism of nicotinamide cleavage reaction catalyzed by the histone deacetylase Sir2Tm. Our studies suggest that the nicotinamide cleavage reaction employs a highly dissociative and concerted displacement mechanism: the cleavage of glycosidic bond is facilitated by the nucleophilic participation of the acetyl-lysine. The transition state is very loose and dissociative, and has a significant oxocarbenium ion character. Furthermore, functional roles of key residues and motifs in the enzyme active site have been characterized. To provide such detailed mechanistic insights is not only of fundamental interest, but also of high medicinal importance since there are enormous current interests in the development of new mechanism-based sirtuin regulators. It should be noted that our simulation studies here focus on the Sir2Tm enzyme, thus some caution should be exercised to correlate these results to sirtuins as a whole. Meanwhile, this work further demonstrates the feasibility and applicability of the on-the-fly Born-Oppenheimer ab initio QM/MM molecular dynamics simulations in simulating enzyme reactions.
Supplementary Material
5 Acknowledgments
This work has been supported by NIH (R01-GM079223), NSF (CHE-CAREER-0448156) and NYU (Whitehead Fellowship). We thank NYU-ITS and NCSA for providing computational resources.
References
- [1].Blander G, Guarente L. Annu. Rev. Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- [2].Marmorstein R. Biochem. Soc. Trans. 2004;32:904–909. doi: 10.1042/BST0320904. [DOI] [PubMed] [Google Scholar]
- [3].Denu JM. Curr. Opin. Chem. biol. 2005;9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- [4].Sauve AA, Wolberger C, Schramm VL, Boeke JD. Annu. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- [5].Braunstein M, Rose AB, Holmes SG, Allis CD, Broach JR. Genes Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- [6].Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. Genes Dev. 1995;9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- [7].Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- [8].Luo JY, Nikolaev AY, Imai S, Chen DL, Su F, Shiloh A, Guarente L, Gu W. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- [9].Vaziri H, Dessain SK, Eagon EN, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- [10].Brunet A, et al. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- [11].Gottlieb S, Esposito RE. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- [12].McMurray MA, Gottschling DE. Science. 2003;301:1908–1911. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- [13].Kaeberlein M, McVey M, Guarente L. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lin SJ, Defossez PA, Guarente L. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- [15].Tissenbaum HA, Guarente L. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- [16].Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- [17].Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. PLoS. Biol. 2004;2:1381–1387. doi: 10.1371/journal.pbio.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kennedy BK, Smith ED, Kaeberlein M. Cell. 2005;123:548–550. doi: 10.1016/j.cell.2005.11.002. [DOI] [PubMed] [Google Scholar]
- [19].Guarente L, Picard F. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- [20].Mostoslavsky R, et al. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- [21].Biel M, Wascholowski V, Giannis A. Angew. Chem. Int. Ed. 2005;44:3186–3216. doi: 10.1002/anie.200461346. [DOI] [PubMed] [Google Scholar]
- [22].Holbert MA, Marmorstein R. Curr. Opin. Struct. Biol. 2005;15:673–680. doi: 10.1016/j.sbi.2005.10.006. [DOI] [PubMed] [Google Scholar]
- [23].Corminboeuf C, Hu P, Tuckerman ME, Zhang Y. J. Am. Chem. Soc. 2006;128:4530–4531. doi: 10.1021/ja0600882. [DOI] [PubMed] [Google Scholar]
- [24].Sauve AA, Celic I, Avalos J, Deng HT, Boeke JD, Schramm VL. Biochemistry. 2001;40:15456–15463. doi: 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- [25].Sauve AA, Schramm VL. Biochemistry. 2003;42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- [26].Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. J. Biol. Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- [27].Borra MT, Langer MR, Slama JT, Denu JM. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- [28].Khan AN, Lewis PN. J. Biol. Chem. 2006;281:11702–11711. doi: 10.1074/jbc.M511482200. [DOI] [PubMed] [Google Scholar]
- [29].Smith BC, Denu JM. Biochemistry. 2006;45:272–282. doi: 10.1021/bi052014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith BC, Denu JM. J. Am. Chem. Soc. 2007;129:5802–5803. doi: 10.1021/ja070162w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smith BC, Denu JM. Biochemistry. 2007;46:14478–14486. doi: 10.1021/bi7013294. [DOI] [PubMed] [Google Scholar]
- [32].Smith BC, Denu JM. J. Biol. Chem. 2007;282:37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- [33].Min J, Landry J, Sternglanz R, Xu RM. Cell. 2001;105:269–279. doi: 10.1016/s0092-8674(01)00317-8. [DOI] [PubMed] [Google Scholar]
- [34].Finnin MS, Donigian JR, Pavletich NP. Nat. Struct. Biol. 2001;8:621–625. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- [35].Avalos JL, Celic I, Muhammad S, Cosgrove MS, Boeke JD, Wolberger C. Mol. Cell. 2002;10:523–535. doi: 10.1016/s1097-2765(02)00628-7. [DOI] [PubMed] [Google Scholar]
- [36].Zhao K, Chai X, Marmorstein R. Structure. 2003;11:1403–1411. doi: 10.1016/j.str.2003.09.016. [DOI] [PubMed] [Google Scholar]
- [37].Zhao K, Harshaw R, Chai X, Marmorstein R. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8563–8568. doi: 10.1073/pnas.0401057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Avalos JL, Boeke JD, Wolberger C. Mol. Cell. 2004;13:639–648. doi: 10.1016/s1097-2765(04)00082-6. [DOI] [PubMed] [Google Scholar]
- [39].Avalos JL, Bever KM, Wolberger C. Mol. Cell. 2005;17:855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- [40].Hoff KG, Avalos JL, Sens K, Wolberger C. Structure. 2006;14:1231–1240. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- [41].Sanders BD, Zhao K, Slama JT, Marmorstein R. Mol. Cell. 2007;25:463–472. doi: 10.1016/j.molcel.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hoff KG, Wolberger C. Nat. Struct. Mol. Biol. 2005;12:560–561. doi: 10.1038/nsmb0705-560. [DOI] [PubMed] [Google Scholar]
- [43].Tanny JC, Moazed D. Proc. Natl. Acad. Sci. 2001;98:415–420. doi: 10.1073/pnas.031563798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Landry J, Slama JT, Sternglanz R. Biochem. BioPhys. Res. Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- [45].Tanner KG, Landry J, Sternglanz R, Denu JM. Proc. Natl. Acad. Sci. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Warshel A, Levitt M. J. Mol. Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- [47].Singh UC, Kollman PA. J. Comput. Chem. 1986;7:718–730. [Google Scholar]
- [48].Field MJ, Bash PA, Karplus M. J. Comput. Chem. 1990;11:700–733. [Google Scholar]
- [49].Gao J, Truhlar DG. Annu. Rev. Phys. Chem. 2002;53:467–505. doi: 10.1146/annurev.physchem.53.091301.150114. [DOI] [PubMed] [Google Scholar]
- [50].Cui Q, Karplus M. Adv. Protein Chem. 2003;66:315–372. doi: 10.1016/s0065-3233(03)66008-0. [DOI] [PubMed] [Google Scholar]
- [51].Shurki A, Warshel A. Adv. Protein Chem. 2003;66:249–313. doi: 10.1016/s0065-3233(03)66007-9. [DOI] [PubMed] [Google Scholar]
- [52].Friesner RA, Guallar V. Annu. Rev. Phys. Chem. 2005;56:389–427. doi: 10.1146/annurev.physchem.55.091602.094410. [DOI] [PubMed] [Google Scholar]
- [53].Mulholland AJ. Drug Discov. Today. 2005;10:1393–1402. doi: 10.1016/S1359-6446(05)03611-1. [DOI] [PubMed] [Google Scholar]
- [54].Zhang Y. Theor. Chem. Acc. 2006;116:43–50. [Google Scholar]
- [55].Senn HM, Thiel W. Top. Curr. Chem. 2007;268:173–290. [Google Scholar]
- [56].Hu H, Yang W. Annu. Rev. Phys. Chem. 2008;59:573–601. doi: 10.1146/annurev.physchem.59.032607.093618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Patey GN, Valleau JP. J. Chem. Phys. 1975;63:2334–2339. [Google Scholar]
- [58].Boczko EM, Brooks CL. J. Phys. Chem. 1993;97:4509–4513. [Google Scholar]
- [59].Roux B. Comput. Phys. Commun. 1995;91:275–282. [Google Scholar]
- [60].Zhang Y, Lee T, Yang W. J. Chem. Phys. 1999;110:46–54. [Google Scholar]
- [61].Zhang Y, Liu H, Yang W. J. Chem. Phys. 2000;112:3483–3492. [Google Scholar]
- [62].Zhang Y. J. Chem. Phys. 2005;122:024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- [63].Ferrenberg AM, Swendsen RH. Phys. Rev. Lett. 1988;61:2635–2638. doi: 10.1103/PhysRevLett.61.2635. [DOI] [PubMed] [Google Scholar]
- [64].Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- [65].Souaille M, Roux B. Comput. Phys. Commun. 2001;135:40–57. [Google Scholar]
- [66].Shao Y, et al. Q-Chem, Version 3.0. Q-Chem, Inc.; Pittsburgh, PA: 2006. [Google Scholar]
- [67].Ponder JW. TINKER, Software Tools for Molecular Design, Version 4.2. Jun, 2004. [Google Scholar]
- [68].Wang S, Hu P, Zhang Y. J. Phys. Chem. B. 2007;111:3758–3764. doi: 10.1021/jp067147i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hu P, Wang S, Zhang Y. J. Am. Chem. Soc. 2008;130:3806–3813. doi: 10.1021/ja075896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mezei M. Simulaid: Simulation Setup Utilites. Mount Sinai School of Medicine; New York, NY: 2008. http://atlas.physbio.mssm.edu/~mezei/simulaid. [Google Scholar]
- [72].Darden T, York D, Pedersen L. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- [73].Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- [74].Ryckaert JP, Ciccotti G, Berendsen HJC. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- [75].Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- [76].Case DA, Darden TA, T. E. Cheatham I, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER 9. University of California; San Francisco: 2006. [Google Scholar]
- [77].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- [78].Wang JM, Cieplak P, Kollman PA. J. Comp. Chem. 2000;21:1049–1074. [Google Scholar]
- [79].Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- [81].Ryde U. Protein Sci. 1995;4:1124–1132. doi: 10.1002/pro.5560040611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Beeman D. J. Comput. Phys. 1976;20:130–139. [Google Scholar]
- [83].Rising KA, Schramm VL. J. Am. Chem. Soc. 1997;119:27–37. [Google Scholar]
- [84].Berti PJ, Schramm VL. J. Am. Chem. Soc. 1997;119:12069–12078. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Scheuring J, Schramm VL. Biochemistry. 1997;36:8215–8223. doi: 10.1021/bi970379a. [DOI] [PubMed] [Google Scholar]
- [86].Liu H, Zhang Y, Yang W. J. Am. Chem. Soc. 2000;122:6560–6570. [Google Scholar]
- [87].Zhang Y, Kua J, McCammon JA. J. Phys. Chem. B. 2003;107:4459–4463. [Google Scholar]
- [88].Cisneros GA, Liu H, Zhang Y, Yang W. J. Am. Chem. Soc. 2003;125:10384–10393. doi: 10.1021/ja029672a. [DOI] [PubMed] [Google Scholar]
- [89].Cheng Y, Zhang Y, McCammon JA. Protein Sci. 2006;15:672–683. doi: 10.1110/ps.051852306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hu P, Zhang Y. J. Am. Chem. Soc. 2006;128:1272–1278. doi: 10.1021/ja056153+. [DOI] [PubMed] [Google Scholar]
- [91].Wang L, Yu X, Hu P, Broyde S, Zhang Y. J. Am. Chem. Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Xiao C, Zhang Y. J. Phys. Chem. B. 2007;111:6229–6235. doi: 10.1021/jp068657f. [DOI] [PubMed] [Google Scholar]
- [93].Poyner RR, Larsen TM, Wong SW, Reed GH. Arch. Biochem. Biophys. 2002;401:155–163. doi: 10.1016/S0003-9861(02)00024-3. [DOI] [PubMed] [Google Scholar]
- [94].Metanis N, Brik A, Dawson PE, Keinan E. J. Am. Chem. Soc. 2004;126:12726–12727. doi: 10.1021/ja0463841. [DOI] [PubMed] [Google Scholar]
- [95].North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. Mol. Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.