

Abstract
Five genes have been identified that contribute to Mendelian forms of Parkinson disease (PD); however, mutations have been found in fewer than 5% of patients, suggesting that additional genes contribute to disease risk. Unlike previous studies that focused primarily on sporadic PD, we have performed the first genomewide association study (GWAS) in familial PD. Genotyping was performed with the Illumina HumanCNV370Duo array in 857 familial PD cases and 867 controls. A logistic model was employed to test for association under additive and recessive modes of inheritance after adjusting for gender and age. No result met genomewide significance based on a conservative Bonferroni correction. The strongest association result was with SNPs in the GAK/DGKQ region on chromosome 4 (additive model: p = 3.4 × 10−6; OR = 1.69). Consistent evidence of association was also observed to the chromosomal regions containing SNCA (additive model: p = 5.5 × 10−5; OR = 1.35) and MAPT (recessive model: p = 2.0 × 10−5; OR = 0.56). Both of these genes have been implicated previously in PD susceptibility; however, neither was identified in previous GWAS studies of PD. Meta-analysis was performed using data from a previous case–control GWAS, and yielded improved p values for several regions, including GAK/DGKQ (additive model: p = 2.5 × 10−7) and the MAPT region (recessive model: p = 9.8 × 10−6; additive model: p = 4.8 × 10−5). These data suggest the identification of new susceptibility alleles for PD in the GAK/DGKQ region, and also provide further support for the role of SNCA and MAPT in PD susceptibility.
Background
Parkinson disease (PD [MIM 168600]) is the second most common neurodegenerative disease. Mutations in five genes have been identified to influence PD risk in fewer than 5% of those with PD (Pankratz and Foroud 2007). Three, PARK2 (parkin), PARK7 (DJ1), and PINK1, are typically transmitted with autosomal recessive inheritance and two, SNCA and LRRK2, are inherited in an autosomal dominant fashion. Mutations in all but LRRK2 are typically found in early onset PD.
Two genomewide association studies (GWAS) to identify susceptibility genes contributing to the risk for PD have been performed previously. The first employed a discordant sibling design with 443 families to identify a set of associated SNPs that were then confirmed with 332 cases and a similar number of controls (Maraganore et al. 2005). The second study utilized a case–control design and included 267 PD cases and 270 controls (Fung et al. 2006). Unfortunately, there was little overlap in results between the two studies, and a few independent studies published following Maraganore et al. have not confirmed the initially associated regions or SNPs [reviewed in (Myers 2006)].
Notably, both previous GWAS studies utilized primarily or exclusively sporadic PD participants. While the majority of people with PD do not report a family history of disease, 15–25% report a first degree relative with PD (Sellbach et al. 2006). It is likely that the genetic contribution to disease risk is greatest in this subset of patients with a positive family history of disease. Therefore, to maximize the power to detect genes affecting PD susceptibility, we performed a GWAS utilizing only PD patients with a family history of PD, primarily in a first degree relative. We hypothesize that the homogeneity with regards to family history of disease may provide us greater power to detect unique loci influencing familial PD.
Methods
Sample selection
PD cases negative for the LRRK2 G2019S mutation (n = 935) were selected from two ongoing studies of familial PD. Additional genes, such as PARK2 (parkin), PARK7 (DJ1), and NR4A2, were screened for many, but not all subjects (Foroud et al. 2003; Karamohamed et al. 2005; Nichols et al. 2004, 2007; Pankratz et al. 2006; Sun et al. 2006); no subjects were included who had known disease producing mutation(s). Both studies (PROGENI and GenePD) initially ascertained multiplex PD families consisting of at least a sibling pair, both of whom were reported to be affected with PD. In a small proportion (9%), the PD case may have had another affected relative rather than an affected sibling. On average, each PD participant had an additional 1.8 relatives who were reported to have PD. Only a single individual per family was genotyped ensuring sample independence. Both studies ascertained primarily Caucasian, non-Hispanic participants. PD cases underwent a uniform neurological evaluation that employed PD diagnostic criteria based broadly on the United Kingdom PD Society Brain Bank Criteria (Gibb and Lees 1988), although modified by both studies. A detailed description of the inclusion and exclusion criteria has been previously published for both the PROGENI (Pankratz et al. 2002) and GenePD (Maher et al. 2002) studies.
Control samples (n = 895) were obtained from the NINDS Human Genetics Resource Center at the Coriell Institute Coriell Cell Repositories (Camden, NJ); older individuals were preferentially selected in an effort to have the mean age at recruitment of the controls be similar to the mean age at onset of the PD cases. All selected control samples were reported to be Caucasian, non-Hispanic. Based on self-report, the control subjects did not have a personal history of PD, and none reported a positive family history of PD (family history data was available for 91% of controls). Appropriate written informed consent was obtained for all samples included in this study.
Microarray genotyping and quality assessment
Genotyping was performed by the Center for Inherited Disease Research (CIDR). DNA sources included blood (n = 905), lymphoblastoid cell lines (LDL, n = 895; all control samples) and whole genome amplified DNA (n = 30). Genotyping was performed using Illumina HumanCNV370 version1_C BeadChips (Illumina, San Diego, CA, USA) and the Illumina Infinium II assay protocol (Gunderson et al. 2006). In addition, intensity data was collected for 23,573 probes specifically designed to detect copy number variation (CNV). Allele cluster definitions for each SNP were determined using Illumina BeadStudio Genotyping Module version 3.1.14 and the combined intensity data from 96% of study samples (for details see Supplemental Methods I). The resulting cluster definition file was used on all study samples to determine genotype calls and quality scores. Genotype calls were made when a genotype yielded a quality score (Gencall value) of 0.25 or higher. The final raw dataset released by CIDR to the investigators and to dbGaP contained 344,301 SNPs with genotype calls and the 1,888 samples used in the current study (for details see Supplemental Methods I). Blind duplicate reproducibility was 99.98%. Data are available at dbGaP (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap; Accession number: phs000126.v1.p1).
Samples having genotypes for at least 98% of the SNPs were considered for inclusion in analyses. These samples were rigorously checked for cryptic relatedness, population stratification, and related issues (Fig. 1 and Supplemental Methods II), and consequently, a total of 106 samples were removed from further analyses. The final sample used in analyses included 857 PD cases and 867 controls (n = 1,724 individuals). A supplemental analysis using broader inclusion criteria was also performed with a sample of 902 PD cases and 881 controls (n = 1,783 individuals, this sample is described in Supplemental Methods III).
Fig. 1.
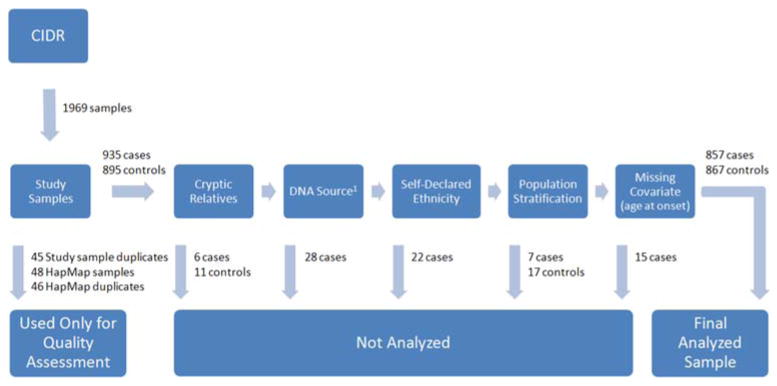
Sample processing. Diagram of sample processing from initial receipt of samples from CIDR to the final analyzed dataset. 1Samples from whole genome amplified (WGA) DNA had lower call rates, particularly near the telomeres, and for a subset of the SNPs the minor allele frequency estimates from WGA DNA differed significantly from that obtained from other sources of DNA (p < 1 × 10−7 for 65 markers)
SNPs with a call rate of 98% or greater were included for further quality control analyses (n = 336,537). SNPs were removed if: (1) the minor allele frequency was less than 0.01 in the combined case and control dataset (n = 7,667); (2) there were differential rates of missing genotypes in the cases and controls (n = 75) or males and females (n = 271), or (3) significant deviation from Hardy–Weinberg equilibrium was observed in the control sample (n = 906). Many markers failed multiple tests. The final dataset consisted of 328,189 SNPs that passed all quality control measures (94.6% of all attempted SNPs).
Statistical analysis
Logistic regression covarying for gender and age (age at evaluation for controls and onset for cases) was employed to test for the association of each SNP with PD susceptibility. An additive model was implemented, and because the additive model may not adequately identify recessive causal alleles (Lettre et al. 2007) and these cases were largely ascertained as affected sibling pairs, a recessive model was also implemented. Odds ratios and p values were computed to assess the strength of the association. All analyses were performed using PLINK (Purcell et al. 2007).
To further prioritize findings within our GWAS, we employed meta-analysis methods to combine p values from our study with those from the only comparable study that tested similar hypotheses. We obtained from dbGaP the publicly available genotype level data from the Fung et al. study. This study consisted of PD cases and controls available from the NINDS Human Genetics Resource Center at the Coriell Institute Coriell Cell Repositories (Camden, NJ). There was no overlap between the subjects used in Fung et al. and those included in this study. Genotyping for Fung et al. was performed with the Illumina Infinium I and the Infinium HumanHap300 SNP chip (Illumina, San Diego, CA, USA). A total of 408,803 unique SNPs were genotyped across these two arrays. We performed quality assessment similar to that performed for the data generated by CIDR for this study (Supplemental Methods IV) and removed eight samples (4 cases, 4 controls) due to quality control issues and three due to self reported African American or Hispanic ancestry. The final analytic sample included 262 cases and 260 controls.
In contrast with the initial report (Fung et al. 2006), we tested for association between the SNPs and disease susceptibility using a logistic regression model (both additive and recessive effects) incorporating an age (at evaluation for controls and onset for cases) and sex adjustment. For those SNPs genotyped in both studies (n = 310,160 markers), the p values obtained in each study for the two relevant models were combined as implemented in METAL (Abecasis and Willer 2007). Specifically, for each SNP a Z statistic was computed for each study based on the study specific p value and direction of the estimated effect. An overall Z statistic (and then corresponding p value) was computed as a weighted average of the study specific Z statistics, with the weights proportional to the square root of the number of individuals within each study. Given the substantially larger size of our sample, proportional weighting of each study (1,724 for this study; 537 for the Fung et al. study) was performed.
Role of the funding sources
The funding sources did not have any involvement in the collection, analysis, interpretation or writing of this report.
Results
The familial PD participants recruited from the two studies had quite similar demographic characteristics (Table 1). Gender and age (age at onset for cases and age at exam for controls) were each significantly associated with affection status (p < 1 × 10−15). The overall average call rate for the final analytic dataset was 0.9986 (standard deviation was 0.0023 when called by sample and 0.0040 when called by SNP).
Table 1.
Sample demographics
| PD Cases (n = 857) | PD Cases | Controls | Controls | ||
|---|---|---|---|---|---|
| PROGENI (n = 488) |
GenePD (n = 369) |
Fung et al. 2006 (n = 262) |
NINDS Coriell Repository (n = 867) |
Fung et al. 2006 (n = 263) |
|
| Average age at onset (cases) or at enrollment (controls) | 62.1 ± 10.4 | 61.6 ± 11.5 | 65.6 ± 7.0 | 54.8 ± 13.1 | 69.5 ± 8.7 |
| % Male | 60.5% | 57.8% | 61.1% | 39.9% | 48.7% |
| % with parent reported with PD | 35.5% | 23.0% | 8% | 0% | 0.4% |
Association results are summarized in Fig. 2 and Tables 2, 3. No result met genomewide significance based on a conservative Bonferroni correction for multiple testing (p < 1.5 × 10−7 based on 328,189 SNPs considered in this study). The strongest evidence of association (p ≤ 6 × 10−6) was obtained with three SNPs within a 112 kb region on chromosome 4p in the genes DGKQ and GAK (Fig. 3a). Multiple SNPs met a p < 1 × 10−4 threshold within a chromosomal region on chromosome 4q which encompassed SNCA, GPRIN3, and MMRN1 (Fig. 3b). This nearly 600 kb region has long range LD and previous studies have provided evidence supporting an association of SNCA with PD susceptibility (Maraganore et al. 2006; Winkler et al. 2007). Under the recessive model, the most significant SNP (p = 2 × 10−5) was near C17orf69 and the region of chromosome 17 that includes MAPT and a common inversion polymorphism (Fig. 3c). This SNP also provided evidence of association with the additive model.
Fig. 2.
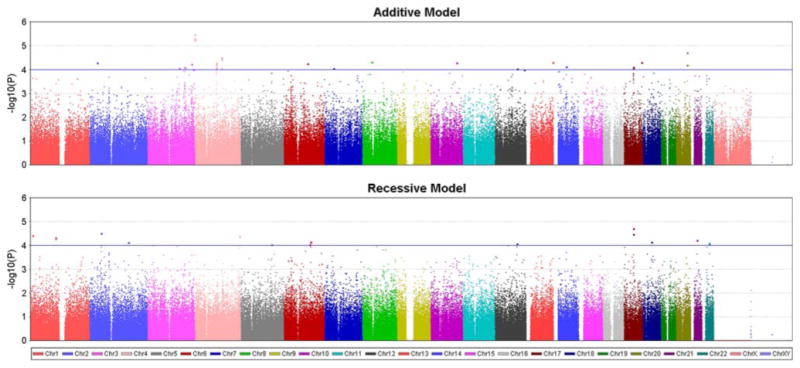
Additive and recessive models. Results from genomewide association analysis modeling two modes of inheritance (additive and recessive). The X-axis indicates the chromosomal position of each SNP while the Y-axis denotes the evidence of association [shown as −log(p value)]. The line indicates the inclusion threshold for the results presented in the Tables (p < 10−4)
Table 2.
Additive model: SNPs providing the strongest association results (p < 0.0001)
| # | Gene regiona | Chr | SNP | Minor allele | Positionb | MAF case | MAF control | Odds ratioc | p value | Meta-analysis p valued |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GAK/DGKQ | 4 | rs1564282 | T | 842313 | 0.131 | 0.087 | 1.70 | 6.0 × 10−6 | 6.7 × 10−7 |
| rs11248051 | T | 848332 | 0.132 | 0.087 | 1.70 | 5.2 × 10−6 | . | |||
| rs11248060 | T | 954359 | 0.145 | 0.097 | 1.69 | 3.4 × 10−6 | 2.5 × 10−7 | |||
| 2 | COX6CP2/PTPN1 | 20 | rs4811072 | G | 48519524 | 0.291 | 0.238 | 1.40 | 6.5 × 10−5 | 4.7 × 10−4 |
| rs1997791 | G | 48529835 | 0.297 | 0.238 | 1.43 | 1.9 × 10−5 | 5.9 × 10−5 | |||
| 3 | LOC729075 | 4 | rs2654735 | C | 112618062 | 0.391 | 0.461 | 0.75 | 9.0 × 10−5 | 1.3 × 10−3 |
| rs1806506 | A | 112686700 | 0.386 | 0.459 | 0.74 | 3.7 × 10−5 | 6.3 × 10−4 | |||
| rs11729080 | A | 112723321 | 0.135 | 0.187 | 0.66 | 3.3 × 10−5 | 1.8 × 10−2 | |||
| 4 | LOC727725/ZMAT4 | 8 | rs4736788 | T | 40947586 | 0.223 | 0.276 | 0.70 | 4.9 × 10−5 | 9.9 × 10−4 |
| rs10094981 | C | 40950451 | 0.223 | 0.276 | 0.70 | 4.8 × 10−5 | 9.7 × 10−4 | |||
| 5 | HRNBP3 | 17 | rs898528 | T | 74678398 | 0.296 | 0.368 | 0.74 | 4.9 × 10−5 | 1.7 × 10−4 |
| 6 | LAMP1 | 13 | rs12871648 | C | 113018663 | 0.376 | 0.316 | 1.36 | 5.0 × 10−5 | 9.4 × 10−4 |
| 7 | LTBP1 | 2 | rs4670322 | G | 33309246 | 0.323 | 0.256 | 1.38 | 5.1 × 10−5 | 2.0 × 10−3 |
| 8 | Gene desert | 10 | rs11592212 | C | 110407383 | 0.085 | 0.052 | 1.80 | 5.2 × 10−5 | 6.5 × 10−3 |
| 9 | SNCA/GPRIN3/MMRN1 | 4 | rs4106153 | C | 90463499 | 0.167 | 0.219 | 0.70 | 9.2 × 10−5 | 1.1 × 10−3 |
| rs1504489 | T | 90477611 | 0.362 | 0.424 | 0.75 | 8.4 × 10−5 | 5.1 × 10−3 | |||
| rs356229 | G | 90825620 | 0.435 | 0.369 | 1.35 | 5.5 × 10−5 | 3.5 × 10−4 | |||
| rs356188 | G | 90910560 | 0.177 | 0.225 | 0.70 | 8.4 × 10−5 | 3.5 × 10−4 | |||
| rs3775478 | G | 91061863 | 0.102 | 0.069 | 1.69 | 6.1 × 10−5 | 1.6 × 10−4 | |||
| 10 | PRDM13/MCHR2 | 6 | rs4431442 | G | 100320236 | 0.322 | 0.262 | 1.39 | 5.6 × 10−5 | 3.2 × 10−3 |
| 11 | VPS8 | 3 | rs10937194 | G | 186201412 | 0.192 | 0.241 | 0.70 | 5.9 × 10−5 | 6.5 × 10−4 |
| 12 | CGRRF1/SAMD4A | 14 | rs4901519 | C | 54088930 | 0.115 | 0.152 | 0.65 | 7.6 × 10−5 | 1.3 × 10−4 |
| 13 | C17orf69/PLEKHM1/MAPT | 17 | rs11012 | A | 40869224 | 0.143 | 0.196 | 0.68 | 8.8 × 10−5 | 4.8 × 10−5 |
| rs1724425 | T | 41137530 | 0.387 | 0.449 | 0.75 | 7.8 × 10−5 | 8.2 × 10−5 | |||
| 14 | LEKR1 | 3 | rs12638253 | C | 158108785 | 0.447 | 0.524 | 0.75 | 8.3 × 10−5 | 5.0 × 10−4 |
| 15 | POU6F2 | 7 | rs9655034 | T | 39258636 | 0.483 | 0.414 | 1.33 | 8.8 × 10−5 | 5.1 × 10−4 |
| 16 | TMEM108 | 3 | rs1197313 | T | 134583142 | 0.393 | 0.451 | 0.75 | 8.9 × 10−5 | 3.8 × 10−4 |
| 17 | LOC728328/PCTK2 | 12 | rs7312607 | C | 95350301 | 0.487 | 0.434 | 1.33 | 9.3 × 10−5 | . |
| 18 | FGF12 | 3 | rs9859577 | T | 193571219 | 0.125 | 0.172 | 0.67 | 9.9 × 10−5 | . |
| 19 | LOC652429/TMEM132B | 12 | rs2108521 | C | 124901417 | 0.219 | 0.271 | 0.72 | 1.0 × 10−4 | 4.4 × 10−4 |
Bold indicates entries where results from the meta-analysis had smaller p values than the initial results from this study, a period (.) indicates that the marker was not genotyped in the Fung et al. study, a “gene desert” is defined here as being more than 500 kb from any gene listed in RefSeq
Genes taken from the NCBI mRNA reference sequences collection (RefSeq)
From NCBI Build 36 reference
Odds ratios were computed for the minor allele
Meta-analysis with Fung et al.
Table 3.
Recessive model: SNPs providing the strongest association results (p < 0.0001)
| # | Gene regiona | Chr | SNP | Minor allele | Positionb | MAF case | MAF control | Odds ratioc | p value | Meta-analysis p valued |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C17orf69/MAPT | 17 | rs1724422 | G | 41133096 | 0.403 | 0.462 | 0.58 | 3.4 × 10−5 | 6.0 × 10−5 |
| rs1724425 | T | 41137530 | 0.387 | 0.449 | 0.56 | 2.0 × 10−5 | 9.8 × 10−6 | |||
| 2 | FSHR | 2 | rs7578654 | C | 49363677 | 0.470 | 0.423 | 1.73 | 3.1 × 10−5 | 9.3 × 10−4 |
| 3 | PIK3CD | 1 | rs4240910 | C | 9673044 | 0.515 | 0.449 | 1.65 | 3.9 × 10−5 | 5.4 × 10−6 |
| 4 | LOC728284/F11 | 4 | rs2889188 | G | 187552805 | 0.268 | 0.233 | 2.49 | 4.3 × 10−5 | 5.1 × 10−4 |
| 5 | Gene desert | 1 | rs11184419 | C | 105439944 | 0.333 | 0.362 | 0.53 | 5.3 × 10−5 | 3.1 × 10−4 |
| rs4128942 | C | 105447249 | 0.331 | 0.362 | 0.52 | 4.8 × 10−5 | 2.7 × 10−4 | |||
| 6 | GPXP2 | 21 | rs969988 | A | 27474523 | 0.469 | 0.421 | 1.71 | 6.2 × 10−5 | 5.1 × 10−4 |
| 7 | LOC643954/HS3ST5 | 6 | rs1519686 | T | 114553816 | 0.245 | 0.223 | 2.62 | 7.1 × 10−5 | 4.4 × 10−5 |
| 8 | SYT4/RIT2 | 18 | rs4890430 | A | 38951528 | 0.447 | 0.387 | 1.72 | 7.1 × 10−5 | 7.4 × 10−5 |
| 9 | FIGN | 2 | rs2083482 | A | 164146021 | 0.504 | 0.465 | 1.62 | 7.6 × 10−5 | 6.8 × 10−6 |
| 10 | SYN3 | 22 | rs1159220 | T | 31410753 | 0.391 | 0.423 | 0.58 | 8.2 × 10−5 | 4.7 × 10−2 |
| rs3788483 | C | 31414345 | 0.393 | 0.425 | 0.58 | 9.1 × 10−5 | 4.9 × 10−2 | |||
| 11 | LOC283398/TMCC3 | 12 | rs10859725 | C | 93468003 | 0.206 | 0.173 | 2.93 | 8.5 × 10−5 | . |
| 12 | CDC2L6 | 6 | rs6912010 | A | 111003337 | 0.283 | 0.245 | 2.18 | 9.2 × 10−5 | 1.9 × 10−3 |
| 13 | LOC728637/ACSL6 | 5 | rs1355095 | G | 131276668 | 0.165 | 0.211 | 0.23 | 9.4 × 10−5 | 5.4 × 10−4 |
| 14 | LOC651011/OXSM/NGLY1 | 3 | rs9310784 | C | 25905208 | 0.403 | 0.462 | 0.20 | 1.0 × 10−4 | 6.0 × 10−5 |
Bold indicates entries where results from the meta-analysis had smaller p values than the initial results from this study, a period (.) indicates that the marker was not genotyped in the Fung et al. study, a “gene desert” is defined here as being more than 500 kb away from any gene listed in RefSeq
Genes taken from the NCBI mRNA reference sequences collection (RefSeq)
From NCBI Build 36 reference
Odds ratios were computed for the minor allele
Meta-analysis with Fung et al.
Fig. 3.
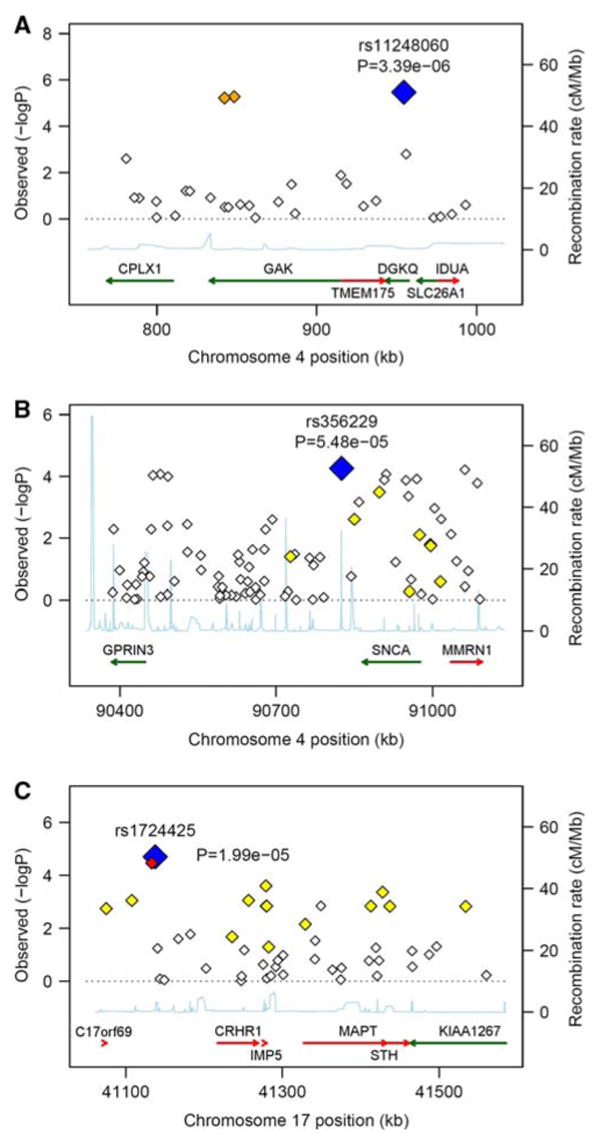
Evidence of association in particular chromosomal regions. Along the X-axis is the physical position in the region (in kB) with known genes shown in their orientation The left Y-axis denotes the association test result as −log(p value) corresponding to diamonds in the figure. The blue diamond identifies the primary SNP result labeled with an rs# and p value. The color of additional diamonds depicts the pairwise linkage disequilibrium with the primary SNP: red indicates r2 > 0.8, orange 0.5 < r2 < 0.8, yellow 0.2 < r2 < 0.5, white r2 < 0.2. r2 values were obtained from the control sample genotyped as part of this study using Haploview (Barrett et al. 2005). The right Y-axis indicates the recombination rate, obtained from the available HapMap data in the CEPH Caucasians, and shown within the figure by the solid blue line. a Additive results around GAK/DGKQ; b additive results around GPRIN3/SNCA/MMRN1; c Recessive results around C17orf69/MAPT/IMP5/STH
To further prioritize our association results, we performed a meta-analysis combining association results (direction of effect and p values) from this study with those from Fung et al. Under an additive model, the evidence for association increased for the DGKQ/GAK and the C17orf69/MAPT regions (Table 2). No other regions or genes identified using the additive model (Table 2) had smaller p values following meta-analysis. In contrast, several chromosomal regions had lower p values when meta-analyses were performed combining results generated under the recessive model of disease inheritance. These included the C17orf69/MAPT region, PIK3CD, LOC643954/HS3ST5 and FIGN (Table 3).
To ensure that SNPs providing modest but consistent evidence of association in both case–control studies were not overlooked, we summarize the top meta-analysis results for both the additive (Table 4) and recessive (Table 5) models. In addition to the GAK/DGKQ region and the MAPT region, a region on chromosome 2 encompassing SNPs in LY75/PLA2R1 also had smaller p values under the additive model when meta-analysis was performed.
Table 4.
Meta-analysis—additive model: SNPs providing the strongest association results (p < 0.0001)
| # | Gene regiona | Chr | SNP | Minor allele | Positionb | MAF case | MAF control | Odds ratioc | p value | Meta-analysis p valued |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | DGKQ/GAK | 4 | rs1564282 | T | 842313 | 0.131 | 0.087 | 1.70 | 6.0 × 10−6 | 6.7 × 10−7 |
| 4 | rs11248060 | T | 954359 | 0.145 | 0.097 | 1.69 | 3.4 × 10−6 | 2.5 × 10−7 | ||
| 2 | LOC728667/C14orf165 | 14 | rs12050360 | T | 23372513 | 0.195 | 0.247 | 0.73 | 4.2 × 10−4 | 9.7 × 10−6 |
| 3 | LOC387882 | 12 | rs11112522 | G | 104345578 | 0.136 | 0.181 | 0.72 | 8.1 × 10−4 | 1.3 × 10−5 |
| 4 | PIK3CD | 1 | rs4240910 | C | 9673044 | 0.515 | 0.449 | 1.31 | 2.3 × 10−4 | 1.6 × 10−5 |
| 5 | GRB10 | 7 | rs1978208 | G | 50752942 | 0.161 | 0.197 | 0.75 | 2.2 × 10−3 | 1.6 × 10−5 |
| 6 | LY75/PLA2R1 | 2 | rs12692575 | G | 160478277 | 0.250 | 0.197 | 1.34 | 7.9 × 10−4 | 2.3 × 10−5 |
| 2 | rs1995950 | G | 160508874 | 0.243 | 0.193 | 1.33 | 1.4 × 10−3 | 4.4 × 10−5 | ||
| 2 | rs3792161 | T | 160517469 | 0.244 | 0.194 | 1.33 | 1.4 × 10−3 | 2.9 × 10−5 | ||
| 2 | rs1511217 | C | 160523339 | 0.245 | 0.194 | 1.33 | 1.3 × 10−3 | 3.3 × 10−5 | ||
| 7 | SH3GL2 | 9 | rs2145656 | C | 17598784 | 0.213 | 0.172 | 1.36 | 9.5 × 10−4 | 3.0 × 10−5 |
| 8 | CAST | 5 | rs1559085 | C | 96104458 | 0.159 | 0.117 | 1.46 | 4.9 × 10−4 | 3.8 × 10−5 |
| 9 | DHFRP1 | 2 | rs7586694 | A | 82511859 | 0.299 | 0.338 | 0.77 | 1.0 × 10−3 | 7.9 × 10−5 |
| 2 | rs1549582 | C | 82542491 | 0.301 | 0.340 | 0.77 | 9.3 × 10−4 | 4.5 × 10−5 | ||
| 2 | rs2902376 | G | 82644916 | 0.270 | 0.306 | 0.78 | 1.7 × 10−3 | 7.5 × 10−5 | ||
| 2 | rs7608203 | G | 82706135 | 0.266 | 0.300 | 0.78 | 2.3 × 10−3 | 8.3 × 10−5 | ||
| 2 | rs6731289 | A | 82746305 | 0.228 | 0.270 | 0.75 | 5.8 × 10−4 | 3.9 × 10−5 | ||
| 10 | C17orf69/PLEKHM1/MAPT/IMP5/STH | 17 | rs11012 | A | 40869224 | 0.143 | 0.196 | 0.68 | 8.8 × 10−5 | 4.8 × 10−5 |
| 17 | rs393152 | G | 41074926 | 0.178 | 0.230 | 0.72 | 2.4 × 10−4 | 1.1 × 10−4 | ||
| 17 | rs1724425 | T | 41137530 | 0.387 | 0.449 | 0.75 | 7.8 × 10−5 | 8.2 × 10−5 | ||
| 17 | rs12185268 | G | 41279463 | 0.176 | 0.228 | 0.71 | 1.9 × 10−4 | 1.1 × 10−4 | ||
| 17 | rs12373139 | A | 41279910 | 0.175 | 0.228 | 0.71 | 1.5 × 10−4 | 4.2 × 10−5 | ||
| 17 | rs1981997 | A | 41412603 | 0.176 | 0.229 | 0.71 | 1.6 × 10−4 | 1.1 × 10−4 | ||
| 17 | rs8070723 | G | 41436901 | 0.177 | 0.229 | 0.71 | 1.6 × 10−4 | 1.1 × 10−4 | ||
| 11 | COX6CP2/PTPN1 | 20 | rs1997791 | G | 48529835 | 0.297 | 0.238 | 1.43 | 1.9 × 10−5 | 5.9 × 10−5 |
| 12 | LOC646309 | 10 | rs2151173 | A | 36911902 | 0.129 | 0.092 | 1.48 | 7.3 × 10−4 | 6.2 × 10−5 |
| 13 | GABRB3 | 15 | rs8041610 | C | 24559357 | 0.304 | 0.357 | 0.75 | 1.6 × 10−4 | 6.4 × 10−5 |
| 14 | RNF130 | 5 | rs3756616 | T | 179333453 | 0.339 | 0.396 | 0.76 | 4.3 × 10−4 | 6.5 × 10−5 |
| 15 | YWHAZP | 14 | rs8021486 | A | 43803554 | 0.225 | 0.179 | 1.39 | 2.3 × 10−4 | 6.9 × 10−5 |
| 16 | MCTP2 | 15 | rs4476132 | C | 92937185 | 0.347 | 0.296 | 1.31 | 5.7 × 10−4 | 7.7 × 10−5 |
| 17 | DPY19L3 | 19 | rs7245958 | G | 37673142 | 0.121 | 0.088 | 1.54 | 2.7 × 10−4 | 9.0 × 10−5 |
| 18 | C20orf79/SLC24A3 | 20 | rs6045766 | G | 18947826 | 0.413 | 0.357 | 1.28 | 8.6 × 10−4 | 9.2 × 10−5 |
| 19 | HLA-DQB1 | 6 | rs9275184 | C | 32762692 | 0.077 | 0.107 | 0.64 | 3.2 × 10−4 | 9.5 × 10−5 |
| 20 | SLC22A3 | 6 | rs884742 | T | 160727422 | 0.494 | 0.448 | 1.21 | 7.8 × 10−3 | 9.5 × 10−5 |
| 21 | LOC647123 | 7 | rs6957669 | A | 138038319 | 0.243 | 0.194 | 1.38 | 2.9 × 10−4 | 9.5 × 10−5 |
| 22 | LOC731917/CDH4 | 20 | rs6129005 | T | 59022667 | 0.305 | 0.345 | 0.78 | 1.3 × 10−3 | 9.6 × 10−5 |
| 23 | RIPK2 | 8 | rs39765 | A | 90872865 | 0.376 | 0.321 | 1.30 | 6.0 × 10−4 | 9.7 × 10−5 |
Genes taken from the NCBI mRNA reference sequences collection (RefSeq)
From NCBI Build 36 reference
Odds ratios were computed for the minor allele
Meta-analysis with Fung et al.
Table 5.
Meta-analysis—recessive model: SNPs providing the strongest association results (p < 0.0001)
| # | Gene regiona | Chr | SNP | Minor allele | Positionb | MAF case | MAF control | Odds ratioc | p value | Meta-analysis p valued |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PIK3CD | 1 | rs4240910 | C | 9673044 | 0.515 | 0.449 | 1.65 | 3.9 × 10−5 | 5.4 × 10−6 |
| 2 | FIGN | 2 | rs2083482 | A | 164146021 | 0.504 | 0.465 | 1.62 | 7.6 × 10−5 | 6.8 × 10−6 |
| 3 | C17orf69/MAPT | 17 | rs1724422 | G | 41133096 | 0.403 | 0.462 | 0.58 | 3.4 × 10−5 | 6.0 × 10−5 |
| 17 | rs1724425 | T | 41137530 | 0.387 | 0.449 | 0.56 | 2.0 × 10−5 | 9.8 × 10−6 | ||
| 4 | MYBBP1A | 17 | rs3816686 | G | 4401915 | 0.227 | 0.196 | 2.83 | 1.2 × 10−4 | 3.1 × 10−5 |
| 5 | LOC643954/HS3ST5 | 6 | rs11752866 | C | 114455213 | 0.338 | 0.302 | 1.96 | 1.2 × 10−4 | 7.2 × 10−5 |
| 6 | rs1519686 | T | 114553816 | 0.245 | 0.223 | 2.62 | 7.1 × 10−5 | 4.4 × 10−5 | ||
| 6 | LOC387941/SOX21 | 13 | rs9524596 | G | 94166840 | 0.417 | 0.450 | 0.69 | 4.1 × 10−3 | 4.4 × 10−5 |
| 7 | DHFRP1 | 2 | rs1549582 | C | 82542491 | 0.301 | 0.340 | 0.53 | 2.5 × 10−4 | 4.5 × 10−5 |
| 2 | rs7586694 | A | 82511859 | 0.299 | 0.338 | 0.52 | 2.1 × 10−4 | 8.7 × 10−5 | ||
| 2 | rs1365951 | T | 82519187 | 0.341 | 0.375 | 0.59 | 6.9 × 10−4 | 7.7 × 10−5 | ||
| 8 | ZNF345 | 19 | rs826278 | A | 42058972 | 0.313 | 0.296 | 1.78 | 1.4 × 10−3 | 5.4 × 10−5 |
| 9 | FLJ46257 | 22 | rs386300 | G | 46488923 | 0.349 | 0.365 | 0.59 | 7.7 × 10−4 | 5.4 × 10−5 |
| 10 | LOC728274 | 4 | rs4974767 | A | 4031034 | 0.446 | 0.407 | 1.46 | 3.8 × 10−3 | 5.5 × 10−5 |
| 11 | LOC730572/RIPK2 | 8 | rs39770 | T | 90905380 | 0.393 | 0.337 | 1.68 | 7.5 × 10−4 | 6.0 × 10−5 |
| 12 | MAP3K7 | 6 | rs1498249 | C | 92586434 | 0.183 | 0.166 | 2.91 | 1.8 × 10−4 | 8.7 × 10−5 |
| 6 | rs2454275 | C | 92605303 | 0.183 | 0.166 | 2.98 | 1.7 × 10−4 | 6.3 × 10−5 | ||
| 13 | SYT4/RIT2 | 18 | rs4890430 | A | 38951528 | 0.447 | 0.387 | 1.72 | 7.1 × 10−5 | 7.4 × 10−5 |
| 18 | rs1878680 | C | 38979393 | 0.396 | 0.345 | 1.71 | 4.4 × 10−4 | 2.0 × 10−4 | ||
| 14 | SBF2 | 11 | rs780382 | A | 9838051 | 0.363 | 0.411 | 0.63 | 1.5 × 10−3 | 7.6 × 10−5 |
| 15 | FNDC1 | 6 | rs12526577 | G | 159554596 | 0.470 | 0.427 | 1.61 | 2.2 × 10−4 | 7.7 × 10−5 |
| 16 | HS322B1A | 22 | rs140289 | T | 22666327 | 0.266 | 0.231 | 2.41 | 1.2 × 10−4 | 8.8 × 10−5 |
| 17 | CLEC1B | 12 | rs521040 | G | 10039117 | 0.266 | 0.279 | 0.49 | 3.5 × 10−4 | 8.9 × 10−5 |
| 18 | PDCL3 | 2 | rs4261746 | A | 100588062 | 0.459 | 0.428 | 1.47 | 3.0 × 10−3 | 9.8 × 10−5 |
Genes taken from the NCBI mRNA reference sequences collection (RefSeq)
From NCBI Build 36 reference
Odds ratios were computed for the minor allele
Meta-analysis with Fung et al.
Discussion
We performed the largest GWAS to date in PD. Unlike previous studies, we focused exclusively on cases having a positive family history of PD, which we hypothesize reflects an increased genetic contribution to disease risk. Using this approach, we detected consistent evidence of association to several chromosomal regions. Notably, we detected association to SNPs within or near two candidate genes previously associated with PD: SNCA and MAPT. Neither of these genes was identified in the two previous GWAS studies of PD.
SNCA was the first gene in which mutations were identified as causing PD (Polymeropoulos et al. 1997). It is thought that aberrant aggregation of the α-synuclein protein results in cell damage and ultimately neuronal death. Subsequent analyses have showed that point mutations (Kruger et al. 1998; Zarranz et al. 2004) as well as gene duplications (Chartier-Harlin et al. 2004; Ibanez et al. 2004) and triplications (Singleton et al. 2003) can result in PD; however, mutations in SNCA are a quite rare cause of autosomal dominant PD. More recently, several studies reported that variation in the promotor region of SNCA, specifically the dinucleotide repeat polymorphism known as Rep1, acts as a susceptibility factor for PD, increasing the risk for disease (Kruger et al. 1999; Maraganore et al. 2006). Association has also been reported at the 3′ end of the gene (Mueller et al. 2005), and a 3′ SNP (rs356219) was identified to be associated with SNCA mRNA levels in substantia nigra and cerebellum (Fuchs et al. 2008) The evidence of association we detected (p < 1 × 10−4) with several SNPs in SNCA is within intron 4 and the 3′ region of the gene. The rs356229 SNP that we report with a minor allele increasing risk of PD (OR = 1.35) exhibits modest LD with rs356219 (HapMap CEPH D′ = 0.65, r2 = 0.39). The evidence that alpha-synuclein levels in the brain are influenced by genetic variability in the 3′ region of the gene (Fuchs et al. 2008) and the LD between the reported SNPs in SNCA provide a link between our GWAS results and SNCA gene expression.
MAPT encodes microtubule-associated protein tau, which regulates microtubule dynamics and assembles microtubules into parallel arrays within axons. Aggregation of tau is a pathological hallmark of several neurodegenerative disorders collectively known as tauopathies, including Pick disease and Alzheimer disease, as well as several disorders with parkinsonian features such as progressive supranuclear palsy, corticobasal degeneration, and fronto-temporal dementia with parkinsonism. Linkage of PD to the MAPT region was previously reported (Scott et al. 2001) and several studies have indicated that a large haplotype block containing MAPT is associated with a small but significant increase in risk for PD (Healy et al. 2004; Tobin et al. 2008; Zabetian et al. 2007; Zhang et al. 2005). The deleterious haplotype (H1) and the protective haplotype (H2) actually represent groups of subhaplotypes that arose from an inversion of 900 kb on chromosome 17 several million years ago (Stefansson et al. 2005); however, associations with these subhaplotypes have not been replicated (Zabetian et al. 2007). The SNPs that define the parent haplotypes of H1 and H2 are in complete linkage disequilibrium with each other (r2 = 1), indicating that the functional variation could be anywhere within this large 900 kb region and not necessarily within the MAPT gene. Complex permutations of alternative splicing lead to many different isoforms of tau; so if the association with H1 is due to variation that were to upset this delicate balance of isoforms, it may help to explain the variety of different neurodegenerative phenotypes that exhibit tau pathology.
Within this MAPT region, which exhibits wide ranging LD, are several additional genes including C17orf69, CRHR1 and IMP5. A SNP between C17orf69 and CRHR1 provided the strongest evidence of association using the recessive model and had an even smaller p value when included as part of our meta-analysis. Evidence of association to this region was also strengthened when meta-analysis was performed using the additive model. Minor alleles of SNPs genotyped in this study that tag the H2 haplotype include rs12185268/G, rs12373139/A, rs1981997/A, and rs8070723/G, all of which were highlighted in the results of the additive meta-analysis (Table 4). Both SNPs in IMP5 identified in the meta-analysis (Table 4) are missense polymorphisms. Given the complex LD structure within this chromosomal region, it is not yet clear whether it harbors multiple susceptibility genes (or alleles) within this region or, conversely, whether the evidence of association with multiple SNPs in different genes reflects a single susceptibility allele. We favor the former hypothesis, although further genotyping and analysis are clearly warranted to resolve this issue. Nonetheless, both the primary GWA analysis and meta-analysis support the existing hypothesis that the complex genomic region around MAPT is related to PD risk.
In order to evaluate replication of our top findings and to identify SNPs with modest p values that may nonetheless be true associations, we performed a meta-analysis. The focus of the present study is a comparison of PD cases and controls, a design also employed by Fung et al. In contrast, a previous GWAS by Maraganore and colleagues (Maraganore et al. 2005) initially employed a discordant sibling design. As noted by others (Defazio et al. 2006), a discordant sibling design is less powerful than a case–control design since the unaffected sibling may have still inherited susceptibility alleles that as a result of incomplete penetrance are not expressed. Therefore, we thought it most appropriate to include in our meta-analysis only the study of Fung et al. which like our own study was an analysis of unrelated cases and controls. We considered combining the genotypic datasets from Fung et al. with our study and testing for association on the combined dataset; however, due to the potential variation introduced by genotyping in differing laboratories with unique control samples and protocols and the different ascertainment scheme of the cases (familial vs. sporadic), we elected to perform a conservative meta-analysis using the results of association tests performed in each study separately. The meta-analysis results have provided support for association to several novel genes and regions not previously reported in GWAS of PD.
To prioritize among these novel genes and regions, we carefully reviewed the evidence for association from nearby SNPs, any published literature about the function of the gene or its potential role in PD susceptibility, and the meta-analysis results. The evidence for a possible association with the LD block region containing GAK (cyclin G associated kinase, a cell cycle regulator) and DGKQ (diacylglycerol kinase, theta) increased following meta-analysis. GAK is a particularly promising candidate because it is one of 137 genes shown to be differentially expressed in PD, with a 1.56-fold change in expression in the substantia nigra pars compacta of PD patients as compared to controls (Grunblatt et al. 2004). No SNPs within the other 136 differentially expressed genes (or within 50 kb of these genes) highlighted in this expression study (Grunblatt et al. 2004) were significantly associated with PD susceptibility in our sample (p < 0.0001). Less is known about DGKQ; however, it is thought to be involved in the phosphatidylinositol signaling system (KEGG pathway ID: hsa04070) and is expressed in the brain. The gene PIK3CD, identified among top recessive model results, is involved in the same pathway as DGKQ. There is another gene (TMEM175) in between GAK and DGKQ; however, while there were SNPs genotyped in this gene, none showed suggestive evidence of association with PD (Fig. 3a). Nevertheless, it is possible that a disease risk modifying variant could be present in any of the genes in this region.
For the SNPs presented in Tables 2, 3, we performed a secondary analysis in a broader set of individuals encompassing 902 cases (PROGENI, n = 491; GenePD, n = 411) and 881 controls (see Supplemental Methods III and Supplemental Table 1) including 40 cases and 14 controls of Hispanic or Asian descent and 19 cases from whole genome amplified samples. Results were largely similar to those obtained in the primary sample (see Supplemental Tables 2A, B).
One limitation of our study is the difference in ascertainment that resulted in differences in the age and gender distribution between our case and control populations. Because the age at exam for the controls was on average 7 years younger than the average age of onset of the cases, it is possible that a small number of the controls might develop PD as they age. However, the lifetime risk of PD is only approximately 1%; therefore, if a few controls were to develop PD, this would have little effect on the power of the current study. As with any association study, the greatest concern is the possibility of population stratification within cases and controls. We have employed stringent criteria and did not detect evidence suggesting that any of the first 10 MDS components (a proxy for population stratfication) were significantly associated with disease status in the final sample. These results indicate that the sample is relatively homogenous and unlikely to be biased due to admixture.
The results obtained from this study do not meet genomewide significance based on a conservative Bonferroni correction for multiple testing (1.5 × 10−7). Although our sample size is more than twice the size of previous GWAS studies, we still have limited power to detect, at a genomewide significant level, the small to moderate effect sizes often seen in susceptibility alleles for complex diseases such as PD. It is likely that some of the true association results will not lie among the most significant association results. We, therefore, turned to other lines of evidence to discern which among our strongest association results are most likely to be true positive results. Notably, two of our strongest association results were in the regions that include SNCA and MAPT; both genes have been previously reported as associated with PD susceptibility and therefore independent replication has been demonstrated in the existing literature. Meta-analysis demonstrates consistency of the DGKQ/GAK region in two independent studies.
It is possible that genes related to familial PD may be different than sporadic PD and vice versa. Finding an appropriate sample to directly replicate our association results is hindered by the dearth of samples enriched for familial PD. Future directions include the recruitment and analysis of an independent sample of familial PD patients and collaborating with investigators that have already collected large samples of sporadic PD that can be used for replication. In addition we will perform analyses utilizing CNVs. The methodology for best calling CNVs is still evolving and we will apply new and existing algorithms to ensure we obtain consistent, robust results prior to dissemination of findings.
In summary, we have performed the largest GWAS to date in PD. We have limited our PD cases to only those with a family history of PD, thereby potentially increasing the contribution of genetic risk factors. Using this case–control design, we detected evidence of association to two chromosomal regions that encompassed previously reported genes: SNCA and MAPT. In addition, we found consistent evidence of association to DGKQ/GAK. Further analyses are warranted in these and additional chromosomal regions nominated in this study to evaluate the evidence of association in both familial and sporadic PD cohorts.
Supplementary Material
Electronic supplementary material: The online version of this article (doi:10.1007/s00439-008-0582-9) contains supplementary material, which is available to authorized users.
Acknowledgments
This project was supported by R01 NS37167, R01 NS036711, the Robert P. & Judith N. Goldberg Foundation, the Bumpus Foundation and the Harvard NeuroDiscovery Center. This study used samples from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds), as well as clinical data. DNA samples contributed by the Parkinson Institute—Istituti Clinici di Perfezionamento, Milan, Italy were from the “Human genetic bank of patients affected by PD and parkinsonisms”, supported by Italian Telethon grant n. GTB07001 and by the “Fondazione Grigioni per il Morbo di Parkinson”. Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. We particularly thank Justin Paschall from the NCBI dbGaP staff for his assistance in developing the dataset available at dbGaP. The data generated from this case–control study are available at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession number: phs000126.v1.p1. Data from the Fung et al. study were obtained from dbGaP. Funding support for “NINDS-Genome-Wide Genotyping in Parkinson's Disease: First Stage Analysis and Public Release of Data” was provided by intramural programs of the National Institute on Aging and the National Institute on Neurological Disorders and Stroke (NINDS) and the genotyping of samples was provided by NINDS. The dataset used for the meta-analyses described in this manuscript were obtained from the NINDS Database found at http://www.ncbi.nlm.nih.gov/sites/entrez?db=gap through dbGaP accession number phs000089.v1.p1. The following are members of the PROGENI Steering Committee. University of Tennessee Health Science Center: R. F. Pfeiffer; University of Rochester: F. Marshall, D. Oakes, A. Rudolph, A. Shinaman; Columbia University Medical Center: K. Marder; Indiana University School of Medicine: P. M. Conneally, T. Foroud, C. Halter; University of Kansas Medical Center: K. Lyons; Eli Lilly & Company: E. Siemers; Medical College of Ohio: L. Elmers; University of California, Irvine: N. Hermanowicz. The following are members of the GenePD Steering Committee. University of Virginia Health System: G. F. Wooten; UMDNJ-Robert Wood Johnson Medical School: L. Golbe; Center for Human Genetic Research, Massachusetts General Hospital, Harvard Medical School: J. F. Gusella; Boston University School of Medicine: R. H. Myers. We thank the subjects for their participation in this research study.
PSG-PROGENI Investigators and Coordinators
Albany Medical College: S. Factor, D. Higgins, S. Evans; Barrow Neurological Institute: H. Shill, M. Stacy, J. Danielson, L. Marlor, K. Williamson; Baylor College of Medicine: J. Jankovic, C. Hunter; Beth Israel Deaconess Medical Center: D. Simon, P. Ryan, L. Scollins; Beth Israel Medical Center: R. Saunders-Pullman, K. Boyar, C. Costan-Toth, E. Ohmann; Brigham & Women's Hospital: L. Sudarsky, C. Joubert; Brown University (Memorial Hospital of RI): J. Friedman, K. Chou, H. Fernandez, M. Lannon; Cleveland Clinic Florida-Weston: N. Galvez-Jimenez, A. Podichetty, K. Thompson; Clinical Neuroscience Center: P. Lewitt, M. DeAngelis; Colorado Neurological Institute: C. O'Brien, L. Seeberger, C. Dingmann, D. Judd; Columbia University Medical Center: K. Marder, J. Fraser, J. Harris; Creighton University: J. Bertoni, C. Peterson; Evanston Northwestern Healthcare: M. Rezak, G. Medalle; Hotel-Dieu Hospital-Chum: S. Chouinard, M. Panisset, J. Hall, H. Poiffaut; Hunter Homes McGuire Veterans Medical Center: V. Calabrese, P. Roberge; Indiana University School of Medicine: J. Wojcieszek, J. Belden; Institute For Neurodegenerative Disorders: D. Jennings, K. Marek, S. Mendick; Johns Hopkins University: S. Reich, B. Dunlop; London Health Sciences Centre: M. Jog, C. Horn; Mayo Clinic Jacksonville: R. Uitti, M. Turk; McFarland Neurosciences: T. Ajax, J. Mannetter; Medical College of Georgia: K. Sethi, J. Carpenter, B. Dill, L. Hatch, K. Ligon, S. Narayan; Medical College of Wisconsin: K. Blindauer, K. Abou-Samra, J. Petit; Medical University of Ohio: L. Elmer, E. Aiken, K. Davis, C. Schell, S. Wilson; Mount Sinai School of Medicine: M. Velickovic, W. Koller (deceased), S. Phipps; North Shore-LIJ Health System: A. Feigin, M. Gordon, J. Hamann, E. Licari, M. Marotta-Kollarus, B. Shannon, R. Winnick; Northwestern University: T. Simuni, A. Videnovic, A. Kaczmarek, K. Williams, M. Wolff; Ochsner Clinic Foundation: J. Rao, M. Cook; Ohio State University: M. Fernandez, S. Kostyk, J. Hubble, A. Campbell, C. Reider, A. Seward; Oregon Health & Science University: R. Camicioli, J. Carter, J. Nutt, P. Andrews, S. More-house, C. Stone; Ottawa Hospital Civic Site: T. Mendis, D. Grimes, C. Alcorn-Costa, P. Gray, K. Haas, J. Vendette; Pacific Neuroscience Medical Group: J. Sutton, B. Hutchinson, J. Young; Saskatoon Dist Health Board Royal Univ Hosp: A. Rajput, A. Rajput, L. Klassen, T. Shirley; Scott & White Hospital/Texas A&M University: B. Manyam, P. Simpson, J. Whetteckey, B. Wulbrecht; The Parkinson's & Movement Disorder Institute: D. Truong, M. Pathak, K. Frei, N. Luong, T. Tra, A. Tran, J. Vo; Toronto Western Hospital, University Health: A. Lang, G. Kleiner-Fisman, A. Nieves, L. Johnston, J. So; UMDNJ-School of Osteopathic Medicine: G. Podskalny, L. Giffin; University of Alabama at Birmingham: P. Atchison, C. Allen; University of Alberta: W. Martin, M. Wieler; University of Calgary: O. Suchowersky, M. Klimek; University of California Irvine: N. Hermanowicz, S. Niswonger; University of California San Diego: C. Shults (deceased), D. Fontaine; University of California San Francisco: M. Aminoff, C. Christine, M. Diminno, J. Hevezi; University of Chicago: A. Dalvi, U. Kang, J. Richman, S. Uy, J. Young; University of Cincinnati: A. Dalvi, A. Sahay, M. Gartner, D. Schwieterman; University of Colorado Health Sciences Center: D. Hall, M. Leehey, S. Culver, T. Derian; University of Connecticut: T. Demarcaida, S. Thurlow; University of Iowa: R. Rodnitzky, J. Dobson; University of Kansas Medical Center: K. Lyons, R. Pahwa, T. Gales, S. Thomas; University of Maryland School of Medicine: L. Shulman, S. Reich, W. Weiner, K. Dustin; University of Miami: K. Lyons, C. Singer, W. Koller (deceased), W. Weiner, L. Zelaya; University of Minnesota: P. Tuite, V. Hagen, S. Rolandelli, R. Schacherer, J. Kosowicz; University of New Mexico: P. Gordon, J. Werner; University of Puerto Rico School of Medicine: C. Serrano, S. Roque; University of Rochester: R. Kurlan, D. Berry, I. Gardiner; University of South Florida: R. Hauser, J. Sanchez-Ramos, T. Zesiewicz, H. Delgado, K. Price, P. Rodriguez, S. Wolfrath; University of Tennessee Health Science Center: R. Pfeiffer, L. Davis, B. Pfeiffer; University of Texas Southwestern Medical Center: R. Dewey, B. Hayward, A. Johnson, M. Meacham, B. Estes; Wake Forest University School of Medicine: F. Walker, V. Hunt, C. O'Neill; Washington University: B. Racette, L. Good, M. Rundle.
PROGENI Molecular Genetic Laboratory
Division of Human Genetics, Cincinnati Children's Hospital Medical Center: William C. Nichols, Michael W. Pauciulo, Diane K. Marek, Veronika E. Elsaesser.
GenePD Investigators and Coordinators
University Southern California School of Medicine: M. Lew; University of Calgary: O. Suchowersky; University of Lübeck, Germany: C. Klein; UMDNJ-Robert Wood Johnson Medical School: L. Golbe, M. H. Mark; Massachusetts General Hospital, Harvard Medical School: J. Growdon, N. Huggins; University of Virginia Health System: G. F. Wooten; University of Alabama at Birmingham : R. Watts; University of Toronto: M. Guttman; Washington University School of Medicine: B. Racette, J. Perlmutter; Barrow Neurological Institute: L. Marlor, Sun Health Research Institute: H. Shill; University of Miami: C. Singer; Parkinson Institute, Istituti Clinici di Perfezionamento, Milano, Italy: S. Goldwurm, G. Pezzoli; Boston University School of Medicine: M. H. Saint-Hilaire, T. Massood; Cleveland Clinic Foundation: K. Baker, I. Itin; University of Louisville School of Medicine: I. Litvan; University of Sydney ANZAC Research Institute, Concord Hospital, Sydney, Australia: G. Nicholson, A. Corbett; Struthers Parkinson's Center, Minneapolis: M. Nance; Port City Neurology, Scarborough, ME: E. Drasby; Parkinson's Disease and Movement Disorder Center of Boca Raton: S. Isaacson; Newcastle University, Newcastle upon Tyne, UK: D. Burn, P. Chinnery; General Regional Hospital Bolzano, Bolzano, Italy: P. Pramstaller; University of Arkansas for Medical Sciences: J. Al-hinti; Aarhus University Hospital, Aarhus, Denmark: A. Moller, K. Ostergaard; University of Arizona: S. Sherman; Auckland City Hospital, Auckland, New Zealand: R. Roxburgh, B. Snow; University of Kentucky College of Medicine: J. Slevin, F. Cambi.
GenePD Molecular Genetics Laboratories
Center for Human Genetic Research, Massachusetts General Hospital, Harvard Medical School: J. F. Gusella, M. E. McDonald, M. Sun, L. Mysore, M. A. Anderson, D. Lucente; Neurogenetics Laboratory, Boston University School of Medicine: S. Williamson, M. W. Nagle, R. H. Myers.
Contributor Information
Nathan Pankratz, Indiana University School of Medicine, Health Information and Translational Sciences Building—HS 4000, 410 West 10th Street, Indianapolis, IN 46202-3002, USA, e-mail: npankrat@iupui.edu.
Jemma B. Wilk, Department of Neurology, Boston University School of Medicine, 715 Albany Street, E-304, Boston, MA 02118-2526, USA
Jeanne C. Latourelle, Department of Neurology, Boston University School of Medicine, 715 Albany Street, E-304, Boston, MA 02118-2526, USA
Anita L. DeStefano, Boston University School of Public Health, Boston, MA 02118, USA
Cheryl Halter, Indiana University School of Medicine, Health Information and Translational Sciences Building—HS 4000, 410 West 10th Street, Indianapolis, IN 46202-3002, USA.
Elizabeth W. Pugh, Johns Hopkins University School of Medicine, Baltimore, MD 21224, USA
Kimberly F. Doheny, Johns Hopkins University School of Medicine, Baltimore, MD 21224, USA
James F. Gusella, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA
William C. Nichols, Cincinnati Children's Hospital Medical Center, Cincinnati, OH 45229, USA
Tatiana Foroud, Indiana University School of Medicine, Health Information and Translational Sciences Building—HS 4000, 410 West 10th Street, Indianapolis, IN 46202-3002, USA, e-mail: tforoud@iupui.edu.
Richard H. Myers, Department of Neurology, Boston University School of Medicine, 715 Albany Street, E-304, Boston, MA 02118-2526, USA, e-mail: rmyers@bu.edu
References
- Abecasis G, Willer C. Metal—Meta Analysis Helper 2007 [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Defazio G, Martino D, Aniello MS, Masi G, Gigante A, Bhatia K, Livrea P, Berardelli A. Planning genetic studies on primary adult-onset dystonia: sample size estimates based on examination of first-degree relatives. J Neurol Sci. 2006;251:29–34. doi: 10.1016/j.jns.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008;22:1327–1334. doi: 10.1096/fj.07-9348com. [DOI] [PubMed] [Google Scholar]
- Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, Okun MS, Mandel RJ, Fernandez HH, Foote KD, Rodriguez RL, Peckham E, De Vrieze FW, Gwinn-Hardy K, Hardy JA, Singleton A. Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. doi: 10.1136/jnnp.51.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunblatt E, Mandel S, Jacob-Hirsch J, Zeligson S, Amariglo N, Rechavi G, Li J, Ravid R, Roggendorf W, Riederer P, Youdim MB. Gene expression profiling of parkinsonian substantia nigra pars compacta; alterations in ubiquitin-proteasome, heat shock protein, iron and oxidative stress regulated proteins, cell adhesion/cellular matrix and vesicle trafficking genes. J Neural Transm. 2004;111:1543–1573. doi: 10.1007/s00702-004-0212-1. [DOI] [PubMed] [Google Scholar]
- Gunderson KL, Steemers FJ, Ren H, Ng P, Zhou L, Tsan C, Chang W, Bullis D, Musmacker J, King C, Lebruska LL, Barker D, Oliphant A, Kuhn KM, Shen R. Whole-genome genotyping. Methods Enzymol. 2006;410:359–376. doi: 10.1016/S0076-6879(06)10017-8. [DOI] [PubMed] [Google Scholar]
- Healy DG, Abou-Sleiman PM, Lees AJ, Casas JP, Quinn N, Bhatia K, Hingorani AD, Wood NW. Tau gene and Parkinson's disease: a case-control study and meta-analysis. J Neurol Neurosurg Psychiatry. 2004;75:962–965. doi: 10.1136/jnnp.2003.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Karamohamed S, Golbe LI, Mark MH, Lazzarini AM, Suchowersky O, Labelle N, Guttman M, Currie LJ, Wooten GF, Stacy M, Saint-Hilaire M, Feldman RG, Liu J, Shoemaker CM, Wilk JB, DeStefano AL, Latourelle JC, Xu G, Watts R, Growdon J, Lew M, Waters C, Vieregge P, Pramstaller PP, Klein C, Racette BA, Perlmutter JS, Parsian A, Singer C, Montgomery E, Baker K, Gusella JF, Herbert A, Myers RH. Absence of previously reported variants in the SCNA (G88C and G209A), NR4A2 (T291D and T245G) and the DJ-1 (T497C) genes in familial Parkinson's disease from the GenePD study. Mov Disord. 2005;20:1188–1191. doi: 10.1002/mds.20515. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kruger R, Vieira-Saecker AM, Kuhn W, Berg D, Muller T, Kuhnl N, Fuchs GA, Storch A, Hungs M, Woitalla D, Przuntek H, Epplen JT, Schols L, Riess O. Increased susceptibility to sporadic Parkinson's disease by a certain combined alpha-synuclein/apoli-poprotein E genotype. Ann Neurol. 1999;45:611–617. doi: 10.1002/1531-8249(199905)45:5<611::aid-ana9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Lettre G, Lange C, Hirschhorn JN. Genetic model testing and statistical power in population-based association studies of quantitative traits. Genet Epidemiol. 2007;31:358–362. doi: 10.1002/gepi.20217. [DOI] [PubMed] [Google Scholar]
- Maher NE, Golbe LI, Lazzarini AM, Mark MH, Currie LJ, Wooten GF, Saint-Hilaire M, Wilk JB, Volcjak J, Maher JE, Feldman RG, Guttman M, Lew M, Waters CH, Schuman S, Suchowersky O, Lafontaine AL, Labelle N, Vieregge P, Pramstaller PP, Klein C, Hubble J, Reider C, Growdon J, Watts R, Montgomery E, Baker K, Singer C, Stacy M, Myers RH. Epidemiologic study of 203 sibling pairs with Parkinson's disease: the GenePD study. Neurology. 2002;58:79–84. doi: 10.1212/wnl.58.1.79. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PV, Frazer KA, Cox DR, Ballinger DG. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wullner U, Meitinger T, Gasser T. Multiple regions of alpha-synuclein are associated with Parkinson's disease. Ann Neurol. 2005;57:535–541. doi: 10.1002/ana.20438. [DOI] [PubMed] [Google Scholar]
- Myers RH. Considerations for genomewide association studies in Parkinson disease. Am J Hum Genet. 2006;78:1081–1082. doi: 10.1086/504730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WC, Elsaesser VE, Pankratz N, Pauciulo MW, Marek DK, Halter CA, Rudolph A, Shults CW, Foroud T. LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology. 2007;69:1737–1744. doi: 10.1212/01.wnl.0000278115.50741.4e. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Uniacke SK, Pankratz N, Reed T, Simon DK, Halter C, Rudolph A, Shults CW, Conneally PM, Foroud T. Evaluation of the role of Nurr1 in a large sample of familial Parkinson's disease. Mov Disord. 2004;19:649–655. doi: 10.1002/mds.20097. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Foroud T. Genetics of Parkinson disease. Genet Med. 2007;9:801–811. doi: 10.1097/gim.0b013e31815bf97c. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C, Conneally PM, Foroud T. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet. 2002;71:124–135. doi: 10.1086/341282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N, Pauciulo MW, Elsaesser VE, Marek DK, Halter CA, Rudolph A, Shults CW, Foroud T, Nichols WC. Mutations in LRRK2 other than G2019S are rare in a north American-based sample of familial Parkinson's disease. Mov Disord. 2006;21:2257–2260. doi: 10.1002/mds.21162. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WK, Nance MA, Watts RL, Hubble JP, Koller WC, Lyons K, Pahwa R, Stern MB, Colcher A, Hiner BC, Jankovic J, Ondo WG, Allen FH, Jr, Goetz CG, Small GW, Masterman D, Mastaglia F, Laing NG, Stajich JM, Slotterbeck B, Booze MW, Ribble RC, Rampersaud E, West SG, Gibson RA, Middleton LT, Roses AD, Haines JL, Scott BL, Vance JM, Pericak-Vance MA. Complete genomic screen in Parkinson disease: evidence for multiple genes. JAMA. 2001;286:2239–2244. doi: 10.1001/jama.286.18.2239. [DOI] [PubMed] [Google Scholar]
- Sellbach AN, Boyle RS, Silburn PA, Mellick GD. Parkinson's disease and family history. Parkinsonism Relat Disord. 2006;12:399–409. doi: 10.1016/j.parkreldis.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, Desnica N, Hicks A, Gylfason A, Gudbjartsson DF, Jonsdottir GM, Sainz J, Agnarsson K, Birgisdottir B, Ghosh S, Olafsdottir A, Cazier JB, Kristjansson K, Frigge ML, Thorgeirsson TE, Gulcher JR, Kong A, Stefansson K. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–137. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, Labelle N, Growdon JH, Singer C, Watts RL, Goldwurm S, Pezzoli G, Baker KB, Pramstaller PP, Burn DJ, Chinnery PF, Sherman S, Vieregge P, Litvan I, Gillis T, MacDonald ME, Myers RH, Gusella JF. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63:826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- Tobin JE, Latourelle JC, Lew MF, Klein C, Suchowersky O, Shill HA, Golbe LI, Mark MH, Growdon JH, Wooten GF, Racette BA, Perlmutter JS, Watts R, Guttman M, Baker KB, Goldwurm S, Pezzoli G, Singer C, Saint-Hilaire MH, Hendricks AE, Williamson S, Nagle MW, Wilk JB, Massood T, Laramie JM, DeStefano AL, Litvan I, Nicholson G, Corbett A, Isaacson S, Burn DJ, Chinnery PF, Pramstaller PP, Sherman S, Al-hinti J, Drasby E, Nance M, Moller AT, Ostergaard K, Roxburgh R, Snow B, Slevin JT, Cambi F, Gusella JF, Myers RH. Haplotypes and gene expression implicate the MAPT region for Parkinson disease: the GenePD Study. Neurology. 2008;71:28–34. doi: 10.1212/01.wnl.0000304051.01650.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C. Alpha-synuclein and Parkinson disease susceptibility. Neurology. 2007;69:1745–1750. doi: 10.1212/01.wnl.0000275524.15125.f4. [DOI] [PubMed] [Google Scholar]
- Zabetian CP, Hutter CM, Factor SA, Nutt JG, Higgins DS, Griffith A, Roberts JW, Leis BC, Kay DM, Yearout D, Montimurro JS, Edwards KL, Samii A, Payami H. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson's disease. Ann Neurol. 2007;62:137–144. doi: 10.1002/ana.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhang J, Song Y, Chen H, Fan D. The tau gene haplotype h1 confers a susceptibility to Parkinson's disease. Eur Neurol. 2005;53:15–21. doi: 10.1159/000082956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Electronic supplementary material: The online version of this article (doi:10.1007/s00439-008-0582-9) contains supplementary material, which is available to authorized users.