

Abstract
Central pain syndrome (CPS) is defined as pain associated with a lesion of the CNS and is a common consequence of spinal cord injuries. We generated a rodent model of CPS by making unilateral electrolytic or demyelinating lesions centered on the spinothalamic tract in rats. Thermal hyperalgesia and mechanical allodynia occurred in both hind paws and forepaws by 7 d postlesion and were maintained >31 d. Field potentials in the ventral posterior lateral nucleus (VPL) in thalamic brain slices from lesioned animals displayed an increased probability of burst responses. Ethosuximide, a T-type calcium channel blocker, eliminated busting in lesioned thalamic slices and attenuated lesion-induced hyperalgesia and allodynia. We conclude that CPS in this model results from an increase in the excitability of thalamic nuclei that have lost normal ascending inputs as the result of a spinal cord injury and suggest that ethosuximide will relieve human CPS by restoring normal thalamic excitability.
Keywords: thalamus, excitability, denervation, deafferentation, spinothalamic, calcium channels
Introduction
Central Pain Syndrome (CPS) is defined as pain resulting from a lesion or pathology of the CNS. The majority of patients with CPS have suffered a spinal cord injury, and as many as 80% of spinal injury patients go on to develop CPS (Bonica, 1991; Yezierski, 2000; Cannavero and Bonicalzi, 2007), including some with complete transection. CPS is also a significant complication for almost half of patients with multiple sclerosis (Bonica, 1991), particularly those with spinal involvement. Stroke survivors also develop CPS, typically if the thalamus is affected. The cause of CPS remains unknown.
CPS patients experience severe and excruciating pain originating from areas in and around the primary sensory loss produced by the lesion (Tasker et al., 1992). Many patients also suffer from allodynia, a perception that a normally nonpainful stimulus is painful, and hyperalgesia, an increased sensitivity to weakly painful stimuli (Greenspan et al., 2004). The onset of CPS is usually delayed weeks or months from the original injury (Tasker et al., 1991). CPS patients are highly resistant to opioid analgesia and other pharmaceutical or surgical therapies. The severity of the pain and the lack of effective treatments are demonstrated by the very high incidence of suicide among CPS sufferers (Gonzales, 1995).
CPS has long been thought to originate in the thalamus (Dejerine and Roussy, 1906). Electrophysiological recordings demonstrate that the behavior of thalamic cells and circuits is pathologically altered in CPS. In human CPS patients, the receptive fields of thalamic relay cells in the ventral posterior lateral thalamus (VPL) are absent in regions representing the areas of sensory loss, but are greatly expanded in nearby regions or remapped to distant and unexpected body surfaces (Lenz et al., 1987, 1989). Sensory modalities are also reorganized such that a weak peripheral stimulus that would normally produce a thermal sensation now induces pain (Lenz et al., 1998). EEG recordings reveal a slowing of normal thalamic rhythms (Gücer et al., 1978), not unlike the altered EEG seen in childhood absence epilepsy. More strikingly, whereas neurons in the VPL display regular 10 Hz action potential discharge in non-CPS patients, neurons in CPS patients fire brief bursts of high frequency action potentials (Lenz et al., 1987, 1989; Jeanmonod et al., 1993). This abnormal burst discharge is particularly prominent in regions corresponding to the parts of the patient's body with pain and loss of sensation. Finally, weak microstimulation in the thalamus is capable of eliciting painful sensations in CPS patients, particularly when the stimulation is delivered in regions in which excessive bursting is observed, but not in non-CPS patients (Lenz et al., 1987, 1998; Davis et al., 1996; Dostrovsky, 2000).
We produced lesions of the ventrolateral spinal white matter centered on the spinothalamic tract (STT), demonstrated pain hypersensitivity, and used this model to test the hypothesis that partial deafferentation of relay cells in the VPL, and subsequently a delayed increase in their excitability, accounts for the altered pain sensation. In addition, we tested the hypothesis that ethosuximide, an antiepileptic drug used successfully to treat childhood absence epilepsy, would restore normal thalamic excitability and alleviate the altered pain sensation in these rats.
Materials and Methods
Spinothalamic tract lesion.
Experiments were performed on adult male Sprague Dawley rats (Zivic-Miller) weighing 180–340 g. All experimental procedures were approved by the University of Maryland School of Medicine Animal Care and Use Committee and conform to the guidelines for use of experimental animals published by the International Association for the Study of Pain. Rats were kept on a 12 h light/dark cycle, housed 2–3/cage, and received food and water ad libitum. A total of 200 animals were used for all experiments: 102 for behavior, 52 for electrophysiology, 46 for anterograde labeling and degeneration visualization.
Animals were anesthetized with pentobarbital (50 mg/kg, i.p.). A laminectomy was performed on either the T8 or T9 vertebrae. The dura was lifted by a fine forceps and opened with iris scissors. The right spinothalamic pathway was lesioned using a tungsten microelectrode (1 MΩ) positioned 0.6–0.8 mm lateral to midline and 1.8–2.1 mm deep. In some cases (8 rats) two lesion sites were made at 0.6 mm lateral, 1.8 mm deep and 0.8 mm lateral, 2.10 mm deep. A brief current pulse (+460 μA, 90 s) was passed between the electrode and a ground electrode placed in the muscle beside the spine to lesion the nearby axons. Demyelinating lesions were produced by the same procedure as electrolytic lesions, except that a glass pipette filled with 0.2 μl of l-α-lysophosphatidylcholine (LPC, Sigma, 50 mg/ml) was inserted into the region of the spinothalamic tract. The pipette was allowed to remain in place for 3 min after injecting the LPC solution with pressure applied via an attached syringe. For sham control animals, laminectomy was performed without dural penetration or spinal injury.
Behavior assessment.
Mechanical nociception was assessed using the method described by Ren (Ren, 1999). Briefly, the rat was habituated to being held in the experimenter's hand with its eyes largely covered. After the habituation, rats remained still in that position for >10 min. To test forepaws, the animal stood with all four paws on the bench with its head in the experimenter's hand. To test hind paws, the animal stood on its hind paws with its forepaws lifted off the bench by the experimenter. Stimuli were delivered to the dorsal surface of the paw between the second and third toes by a set of calibrated Von Frey filaments (North Coast Medical). Each filament was tested five times at an interval of 5–15 s. The starting filament was 100 g (6.1 marking, hind paw) or 26 g (5.46 marking, forepaw). A descending or ascending series of the filaments was then used depending on the withdrawal response to the starting filament. If the five stimuli with the starting filament elicited withdrawal one or more times, then an ascending series was used until no withdrawal was elicited, then a descending series was used until all five stimuli elicited withdrawal. Stimulus-response function curves were prepared by plotting the paw withdrawal frequency as a function of the force delivered. The data were fit with a sigmoid function and a value, equal to the force that produced withdrawal in 50% of trials, was derived. This value will be referred to as the paw withdrawal response threshold.
Thermal nociception was assessed by placing rats under a clear plastic chamber on an elevated glass surface (Paw Thermal Stimulator System, University of California, San Diego, La Jolla, CA) and allowing them to become habituated to the test environment for 20–30 min. A radiant heat stimulus was applied to the plantar surface of each hind paw and forepaw from beneath a glass floor with a high-intensity projector lamp bulb. The paw withdrawal latency was automatically recorded when the rat withdrew its paw after the stimulus. A 20 s cutoff was used to prevent tissue damage. Three measures, each separated by a 5 min interval, were obtained for each hind paw and forepaw, with the mean designated the latency.
Open field behavior was monitored in a 50 × 30 inch arena, in which the floor was divided into a 5 × 6 grid of 18 outer quadrants and 12 inner quadrants, in a darkened room illuminated with low level red light. Animals were habituated in the room for 30 min and then put in one corner of the arena. The observer recorded the crossings into inner or outer quadrants within a 5 min test session. Every animal was tested once and had not been placed in the arena previously. The data were analyzed as total frequency of crossings from one quadrant to another inner or outer quadrant, from which the total number of crossings and the percentage of crossings into inner quadrants were calculated.
Drug application.
Ethosuximide (Sigma) was injected intraperitoneally, then thermal and mechanical nociceptive assessments were made at 15, 30, 60, and 90 min after drug application. The effect of ethosuximide on the behavior in the open field arena was assayed 30 min after peritoneal injection of ethosuximide (50 mg/kg). For the dose–response curves, ethosuximide was administered once per day starting at 1 mg/kg and was increased in succeeding trials until the maximal dose was delivered. Thermal paw withdrawal latency was tested at 30 min after ethosuximide was applied intraperitoneally.
Electrophysiology.
Animals were deeply anesthetized with pentobarbital (100 mg/kg), then decapitated. The brain was taken out quickly and transferred to a bath of ice-cold sucrose solution containing (in mm): 248 sucrose, 10 glucose, 26 NaHCO3, 1.25 NaH2PO4, 3 KCL, 5 MgSO4, and 1 CaCl2. This bath was bubbled continuously with a 95% O2-5% CO2 gas mixture (pH 7.2). The brain was blocked at the anterior surface at the level of the optic chiasm and at the posterior surface at the level of the midbrain, then bisected at the midline. The dorsal surface was then attached to the stage of a vibratome with cyanoacrylate glue and the brain was resubmerged in the iced, oxygenated sucrose solution. 400 μm-thick sections were cut with the ventral surface at a 10–15° angle from the horizontal plane, to retain the maximal number of thalamocortical and corticothalamic fibers. Slices were made from intact, sham, and lesioned animals in which unilateral spinothalamic tract lesions had been made 14 d earlier. After 1–2 h preincubation, slices were transferred to recording chamber with saline containing (in mm): 124 NaCl, 3 KCl, 1.3 MgSO4, 2 CaCl2, 15 glucose, 5 BES, and 25 NaHCO3. The saline was bubbled continuously with a 95% O2-5% CO2 gas mixture and had a pH = 7.2, as monitored with phenol red (0.01 mg/L). Extracellular field potentials, reflecting the extracellular current flow resulting from action potentials in populations of neurons located near the recording electrode, were recorded using patch pipettes containing extracellular saline (tip resistance = 1–4 MΩ). Responses were amplified 1000× and digitized, stored, and analyzed off-line using pClamp software (Molecular Devices). Field potentials were evoked by stimulating local thalamocortical or corticothalamic fibers within the VPL using a concentric bipolar metal electrode and pulses of 2.5 times threshold intensity with a duration of 0.1 ms, delivered at 0.05 Hz.
The degree of bursting was quantified using a modified coastline index (Korn et al., 1987). The coastline index was calculated as the sum of the point-to-point voltage differences during the length of a 100 ms long epoch of the evoked response starting at the peak positivity after the first population spike. For each slice, the coastline index was calculated for 10–20 responses and the average value for that slice was used for analysis. The coastline index is sensitive to changes in the number or amplitude of population spikes. It increases when the number of neurons in the vicinity of the recording electrode firing action potentials in response to the stimulus increases and when the number of cells firing multiple action potentials increases. In addition, we counted the average number of additional population spikes occurring after the first population spike.
Histology.
Myelin staining was used to assess the effects of LPC injection. Mounted sections were briefly hydrated in distilled water, then immersed in a 2.5% ferric ammonium sulfate solution overnight. After rinsing with distilled water (2×), sections were stained in fresh hematoxylin (1% alcoholic hematoxylin in an aqueous solution containing 70 ml/L saturated lithium carbonate) for 2 h. Slides were submerged into 80% alcohol to differentiate until background was clear and myelin stood out sharply, then dehydrated in ethanol and cleared in xylene. Slides then were coverslipped in Permount. All chemicals were purchased from Sigma-Aldrich
Fluorescence immunostaining was used for visualizing neurofilaments in the spinal cord after demyelinating lesions, using sections adjacent to those used for myelin staining. Rats were perfused as above. Fixed thoracic spinal cord segments were cut on a freezing cryostat at 25 μm. Sections were mounted on gelatin-coated slides, dried, and washed with 0.05 m potassium phosphate buffered saline (KPBS) for 10 min (3×). After blocking in 10% normal donkey serum for 1 h at room temperature, sections were incubated with a rabbit anti-neurofilament 200 antibody solution (1:80; Sigma) overnight at room temperature. Sections were washed with KPBS (3×) and then incubated with a solution of CY3-conjugated donkey anti-rabbit antibody (1:500; Jackson) for 1 h at room temperature. Sections were washed with KPBS, air dried, and coverslipped with aqueous mounting medium.
Statistics.
All data are expressed as mean ± SE. Various statistical tests were used (Sigmastat) depending on the experiment design, as described in the text. Statistical significance was taken as p < 0.05.
Results
To test the hypothesis that central pain syndrome after spinal cord injury results from chronic denervation of the somatosensory thalamus, we tested the effects of a unilateral electrolytic lesion of the ventrolateral spinal white matter, in the region containing the spinothalamic tract (eSTTx), at vertebral level T8 or T9 on pain sensation in rats. All animals displayed grossly normal locomotor function after the lesions and no signs of extreme pain, such as limb chewing, failure to groom, or hyperagitation in response to handling. In Nissl-stained sections prepared from all animals post hoc, the lesion site was characterized by evidence of gliosis extending approximately 100–300 μm in diameter (Fig. 1A) and for ∼200 μm in the rostrocaudal axis. Behavioral and electrophysiological data were excluded if there was evidence of the lesion site extending beyond the ventrolateral quadrant of the white matter or into the gray matter.
Figure 1.

Electrolytic and demyelinating lesions of the ventrolateral spinal white matter. a, A representative Nissl-stained section illustrating the focal gliosis induced 3 d after an electrolytic lesion of the ventrolateral spinal white matter (eSTTx), the region of the spinothalamic tract, at the T8 or T9 vertebral level. Drawings of the injury sites in 15 rats used for pain behavior assessment, as assessed post hoc at 31 d postlesion. Animals in which the lesion site extended beyond the ventrolateral quadrant white matter were not analyzed. b, Visualization of LPC-induced demyelination in ventrolateral spinal cord with myelin staining 14 d after injection (left). Immunostaining with an antibody against neurofilament protein 200 in an adjacent section, showing that the LPC injection does not damage spinothalamic tract axons (right).
Unilateral spinothalamic tract lesions induce bilateral above- and below-level hyperalgesia and allodynia
Human CPS after spinal cord injury is characterized by hyperalgesia and allodynia. We first assayed thermal sensation in both hind and forepaws after unilateral eSTTx or sham surgery at 3–31 d postsurgery (Fig. 2). We first detected a significant hyposensitivity to painful stimuli, apparent as an increase in the latency to paw withdrawal, in the contralateral hind paw at 3 d postlesion, consistent with a lesion-induced deficit in thermal sensation, with no concomitant decrease in thermal sensation in the ipsilateral hind paw or either forepaw. Thereafter, significant thermal hypersensitivity, apparent as a 20–30% decrease in the latency to paw withdrawal, was detected in both contralateral and ipsilateral forepaws by 7 d postlesion and in both hind paws by 14 d postlesion (Fig. 2A,B). For all paws, the hyperalgesia persisted for >31 d after the lesion.
Figure 2.

a–f, Hyperalgesia and allodynia to above- and below-level thermal and mechanical stimuli after electrolytic spinothalamic tract lesions. Paw withdrawal latencies are shown for ipsilateral (open symbols) and contralateral (closed symbols) thermal stimulation of hind paws (a) and forepaws (b) made at various times after spinothalamic tract lesions (eSTTx)(red) or sham surgeries (blue). After a transient increase in contralateral hind paw withdrawal latency at 3 d, lesioned rats respond to the thermal stimulus by withdrawing both ipsilateral and contralateral hind and forepaws more quickly at 7 d postlesion. This decrease in paw withdrawal latency to above- and below-level stimuli was maintained for at least 31 d postlesion (two-way RM ANOVA, Tukey's test, n = 15 lesioned, 16 sham, F = 79.3, p < 0.001 for lesioned vs sham contralateral hind paw; two-way RM ANOVA, Tukey's test, n = 5 lesioned, 8 sham, F = 110.58, p < 0.001 for lesioned vs sham ipsilateral forepaw). Stimulus-response curves were prepared by plotting the frequency of paw withdrawal elicited with repeated trials of mechanical stimuli of varying force delivered to the contralateral hind paw (c) and ipsilateral forepaw (d) at various times after spinothalamic tract lesions and in intact and sham-operated animals. A large leftward shift 7–31 d after spinothalamic lesions compared with either intact of sham animals was readily apparent. Data were fit with a sigmoid function and the force eliciting withdrawal in 50% of trials was taken as the paw withdrawal threshold (dashed lines). Plots of hind paw (e) and forepaw (f) withdraw thresholds for ipsilateral (open symbols) and contralateral (closed symbols) mechanical stimuli delivered at various times after electrolytic spinothalamic tract lesions (red) or sham surgery (blue). Paw withdrawal thresholds for both above- and below-level mechanical stimuli were decreased in lesioned animals beginning 3 d after lesion (two-way RM ANOVA, Tukey's test, n = 15 lesioned, 16 sham, F = 260.33 p < 0.001 for lesioned vs sham contralateral hind paw; two-way RM ANOVA, Tukey's test, n = 5 lesioned, 8 sham, F = 184.17, p < 0.01 for lesioned vs sham ipsilateral forepaw).
We next assayed nociception using a graded series of mechanical forces delivered to the dorsal surface of the hind and forepaws with von Frey's filaments. For each paw, we prepared a stimulus-response function with the frequency of paw withdrawal plotted as a function of the force used. Data were fit with a sigmoid function, and the threshold paw withdrawal force was defined as the force necessary to produce paw withdrawal in 50% of trials, according to the fit. In sham control animals, there was a small and transient decrease in the threshold withdrawal force for both hind and forepaws at 3 d postsurgery that recovered to control values by 7 d after the surgery. In lesioned animals in contrast, we observed that ∼80% less force was required to induce paw withdrawal for both ipsilateral and contralateral hind and forepaws, apparent as a leftward shift in the stimulus-response curves, by >3 d postlesion (Fig. 2C,D). Withdrawal thresholds remained decreased for >31 d after the lesion (Fig. 2E,F). We conclude that a physical transaction of the spinothalamic tract induces the delayed development of chronic alterations in objective pain sensation in rats.
Demyelinating lesions of the spinothalamic tract alter pain perception
CPS is also a complication of many demyelinating diseases, such as transverse myelitis (Bonica, 1991; Cannavero and Bonicalzi, 2007). To determine whether only physical interruption of spinothalamic tract axons can alter pain sensation, we triggered an autoimmune demyelinating lesion of the spinothalamic tract and action potential conduction failures by injecting lysophosphatidylcholine (LPC) into the right ventrolateral thoracic spinal cord (T8 or T9 level) (Foster et al., 1980). LPC injection induced long-lasting demyelination in spinothalamic tract, as apparent from the loss of normal dark-staining myelin (Fig. 1B). Neurofilaments in the demyelination site remained intact (Fig. 1B), however, indicating that the injection did not physically damage the spinothalamic tract axons themselves. As with the electrolytic lesions, demyelinating spinothalamic tract lesions (mSTTx) resulted in both mechanical and thermal hyperalgesia and allodynia bilaterally in both hind paws and forepaws, beginning three to 14 d postlesion and lasting for >31 d (Fig. 3). We conclude that altered input activity, rather than an interruption in the spinothalamic connection per se, is sufficient to alter pain sensation chronically.
Figure 3.

a–f, Hyperalgesia and allodynia to above- and below-level thermal and mechanical stimuli after demyelinating spinothalamic tract lesions. Paw withdrawal latencies are shown for ipsilateral (open symbols) and contralateral (closed symbols) thermal stimulation of hind paws (a) and forepaws (b) made at various times after demyelinating lesions (mSTTx)(red) or sham surgeries (blue). Lesioned rats respond to the thermal stimulus by withdrawing both ipsilateral and contralateral hind and forepaws more quickly at 14–31 d postlesion (two-way RM ANOVA, Tukey's test, n = 12 lesioned, 16 sham, F = 150.57, p < 0.001 for lesioned vs sham contralateral hind paw; two-way RM ANOVA, Tukey's test, n = 8 lesioned, 8 sham, F = 175.32, p < 0.01 for lesioned vs sham ipsilateral forepaw). Stimulus-response curves were prepared as in Figure 2 for mechanical stimuli delivered to the contralateral hind paw (c) and ipsilateral forepaw (d) at various times after demyelinating lesions and in intact and sham-operated animals. A large leftward shift 3–31 d after spinothalamic lesions compared with either intact of sham animals was readily apparent. Data were fit with a sigmoid function for calculation of the paw withdrawal threshold (dashed lines). Plots of hind paw (e) and forepaw (f) withdraw thresholds for ipsilateral (open symbols) and contralateral (closed symbols) mechanical stimuli delivered at various times after demyelinating lesions (red) or sham surgery (blue). Paw withdrawal thresholds for both above- and below-level mechanical stimuli were decreased in lesioned animals beginning 3 d after lesion (two-way RM ANOVA, Tukey's test, n = 12 lesioned, 16 sham, F = 278.48, p < 0.001 for lesioned vs sham contralateral hind paw; two-way RM ANOVA, Tukey's test, n = 8 lesioned, 8 sham, F = 142.30, p < 0.01 for lesioned vs sham ipsilateral forepaw).
Unilateral spinothalamic tract lesions induce subjective pain behavior
Thermal and mechanically induced responses provide objective measures of pain sensation in response to acute stimuli. We next wished to determine whether the lesioned rats experienced on-going, spontaneous subjective pain. We therefore examined spontaneous locomotor activity with an open field test (Hasnie et al., 2007; Suzuki et al., 2007). Rats in which the spinothalamic tract had been electrolytically lesioned 14 d earlier displayed 57% less spontaneous locomotion in the outer quadrants of an open field arena than sham operated control rats (Fig. 4A). This difference could not be attributed to locomotor deficits, because lesioned rats displayed no change in walking or rearing behavior, such as limping, foot dragging or uneven gait, nor did it result from anxiety, because there was no significant difference in the fraction of the crossings made by the lesioned and sham-operated animals from outer to inner quadrants (Fig. 4B).
Figure 4.
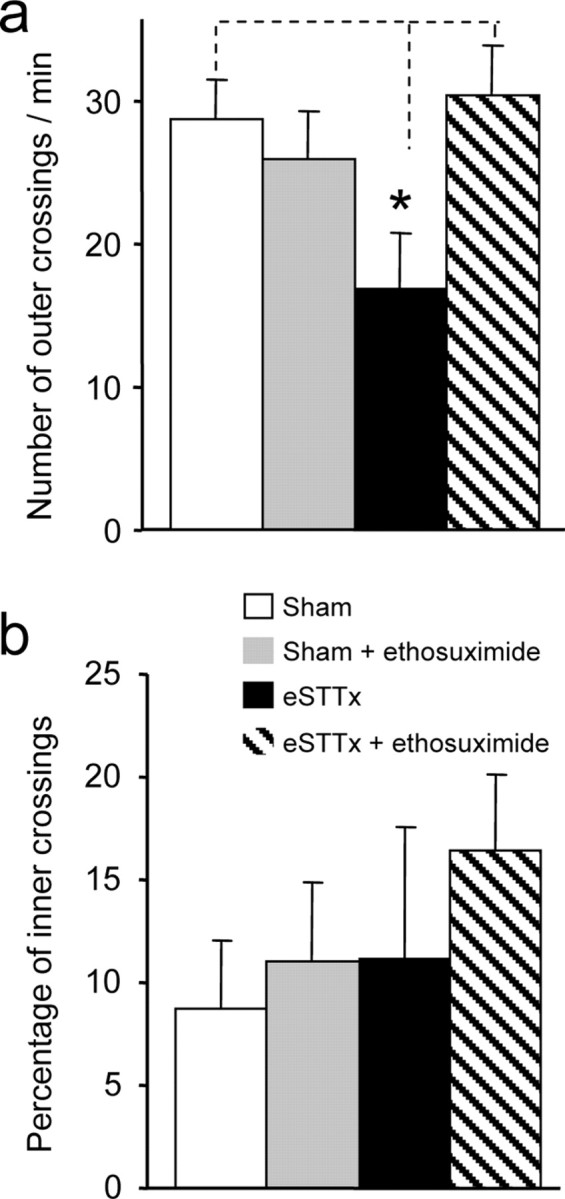
Spinothalamic tract lesions induce subjective pain that is reversed by ethosuximide. a, Rats receiving an electrolytic spinothalamic tract lesion (eSTTx) 14 d earlier (n = 5) displayed significantly reduced locomotion, quantified as the number of crossings of the outer quadrants during a 5 min trial, comparing with sham animals (one-way ANOVA, n = 5 lesioned, 8 sham, F = 6.27, *p < 0.05 for lesioned vs sham). Intraperitoneal administration of ethosuximide (50 mg/kg) had no effect on locomotion in sham animals (n = 8), but prevented the decrease in locomotion in lesioned animals (n = 7). b, There were no significant differences in the percentage of crossings into the inner quadrants, an assay of anxiety, between lesioned and sham animals, either with or without ethosuximide (one-way ANOVA, n = 5 lesioned, 8 sham, F = 1.26).
We conclude that interruption of the normal ascending activity carried by the spinothalamic tract leads to altered pain sensation, characterized by hypersensitivity, and reduced spontaneous locomotor activity. These changes were bilateral, occurred only after a delay of several days, and persisted for many weeks. These features resemble the allodynia and hyperalgesia experienced by humans suffering from central pain syndrome after spinal cord injury, suggesting that it may provide useful insights into the underlying biological mechanisms.
Spinothalamic tract lesions induce burst discharges in the VPL nucleus of the thalamus
We hypothesized that altered pain sensation might result from alterations in neuronal excitability in either neuronal populations in the dorsal horn of the spinal cord, giving rise to untransected ascending projections, or in the target neurons of the lesioned spinothalamic tract axons in the VPL thalamus. Furthermore, we predicted that if the intrinsic firing behavior of either population of cells was altered in response to lesions centered on the spinothalamic tract, then the pathological activity would be maintained in acutely prepared brain slices. We therefore recorded extracellular field potentials, to sample activity from a population of cells near the recording electrode, in the contralateral dorsal horn of spinal cord slices and ipsilateral thalamic brain slices prepared from intact animals and animals that had undergone a sham surgery or a unilateral electrolytic lesion 14–19 d earlier.
We found that the excitability of neurons was qualitatively altered in the VPL contralateral to the spinothalamic tract lesion site. In slices from intact animals or sham operated controls, field potentials elicited by single stimuli delivered at the edges of VPL ∼100–200 μm from the recording electrode were characterized by one or two negative-going deflections of 2–4 ms in duration at a latency of 3–5 ms from the stimulus, consistent with the discharge of one or two action potentials in nearby neurons. Field potentials in slices taken from lesioned animals, however, displayed numerous negative deflections at intervals of 10–20 ms, consistent with the discharge of bursts of action potentials at high frequency in the partially denervated VPL neurons (Fig. 5A). Bursting responses (>1 population spike) were elicited in 68% of slices from lesioned animals, but only 28% and 25% of slices from sham and intact animals (χ2 = 8.86, p < 0.05; n = 12 lesioned, 5 sham, 5 intact). For those slices displaying bursts, there was a significantly higher average number of population spikes per trial in lesioned slices than in slices from sham or intact animals (Fig. 5B). We quantified the degree of bursting activity using a modified version of the coastline index (Korn et al., 1987) and observed a three-fold increase in slices from lesioned animals (Fig. 5C). Similar bursting discharges were observed in the VPL in contralateral thalamic slices (n = 3), but not in the ipsilateral medial geniculate nucleus (n = 2). There was no difference in the amplitude of the first field potential between slices from lesioned, sham, and intact animals, indicating that the intensity of stimulation was comparable in all experiments (Fig. 5D). We conclude that neurons in the VPL exhibit hyperexcitability after spinothalamic tract lesion that is strikingly similar to the altered discharge displayed by neurons in the thalamus of human patients with central pain syndromes (Lenz et al., 1987, 1989; Jeanmonod et al., 1993). This altered excitability is intrinsic to the VPL because it is apparent in acutely isolated brain slices.
Figure 5.
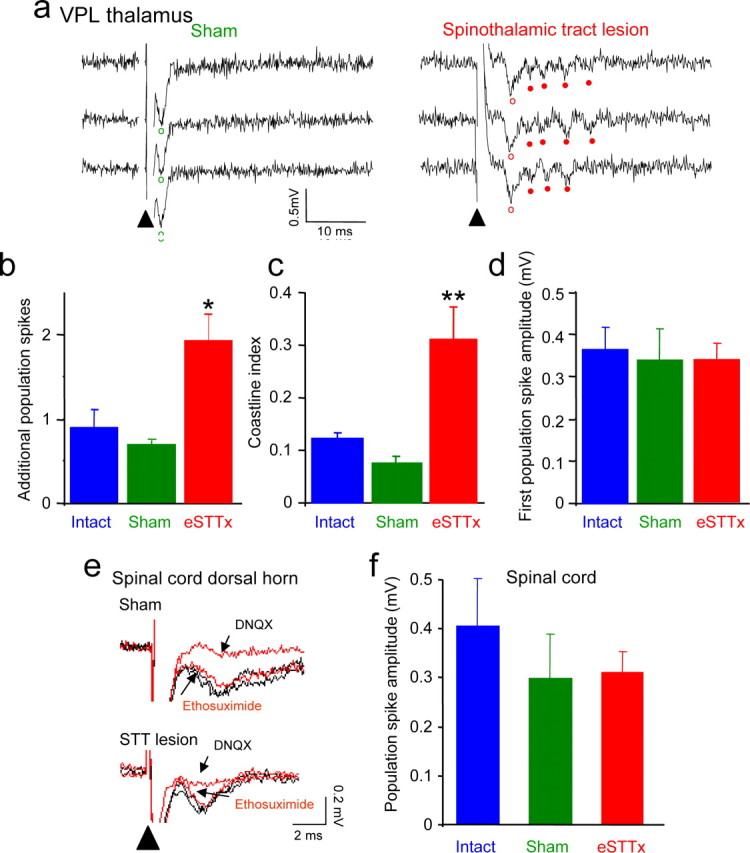
Abnormal burst discharges in the ventral posterior lateral nucleus (VPL) after spinothalamic tract lesions. a, Field potential recordings made in three consecutive trials in the VPL of thalamic slices from a sham rat and a rat lesioned 14 d earlier. Local stimulation elicits only a single population spike in the sham slice but bursts of 4 or 5 spikes in the slice from the lesioned animal. Open circles indicate first population spikes, closed circles indicate later spikes. b, The average number of additional spikes occurring after the first spike per sweep was greater in slices from lesioned animals (eSTTx) than in slices from sham or intact animals (one-way ANOVA, n = 12 lesioned, 5 sham, 5 intact, *p < 0.01). c, A modified coastline index, a quantitative measure of bursting, was calculated for 100 ms period starting after the first population spike. The coastline index was significantly greater for responses in slices from lesioned animals than in slices from sham or intact animals (one-way ANOVA, n = 10 lesioned, 8 sham, 10 intact, F = 13.58, **p < 0.001, lesioned vs sham and intact). d, There were no significant differences in the amplitudes of the first population spike, indicating that comparable levels of stimulation were used in all three sets of slices. e, Field potentials, consisting of single population spikes, were recorded in response to stimulation at the dorsal root entry zone in lamina III-V of the dorsal horn in spinal cord slices from a sham rat and a rat in which the spinothalamic tract was lesioned 14 d earlier. Population spikes were eliminated by the glutamate receptor antagonist DNQX (40 μm), but were unaffected by ethosuximide in slices from lesioned animals, shams, or controls. f, There was no difference in population spike amplitude in slices from lesioned, sham, or intact animals (n = 7 lesioned, 7 sham, 5 intact).
In spinal cord slices from lumbar levels, in contrast, field potentials recorded in laminas III-V in response to single stimuli delivered at the dorsal root entry zone displayed single negative going deflections of 2–3 ms in duration at a latency of 2–3 ms after stimulation, consistent with the firing of single action potentials by neurons in the vicinity of the recording electrode. In all cases, the field potentials were eliminated by the AMPA-type glutamate receptor antagonist DNQX (40 μm), indicating that they were the result of monosynaptic glutamatergic excitation (Fig. 5E). There was no significant difference in the amplitude of these field potentials in slices taken from lesioned, sham or intact animals (Fig. 5F).
Ethosuximide abolishes burst firing in thalamic brain slices from lesioned animals
T-type low threshold calcium channels are expressed at high levels in thalamic neurons (McKay et al., 2006) and are known to underlie bursting discharge in these neurons during sleep spindles and spike-and-wave discharges of childhood absence epilepsy (McCormick and Bal, 1997; Crunelli and Leresche, 2002; Steriade, 2005). We therefore tested the hypothesis that the irregular bursts observed in VPL neurons after spinothalamic tract lesions also require T-type calcium channels for their generation. We applied the widely used anti-absence epilepsy drug ethosuximide, which is known to be a selective blocker of T-type calcium channels (Coulter et al., 1989). We observed that burst firing was abolished when ethosuximide (0.7 mm) was applied to slices from lesioned animals, apparent as a 36 ± 3% decrease in the coastline index (Fig. 6A,B), so that responses in slices from lesioned animals were identical to those in slices from intact or sham operated control animals. In slices from control and sham operated rats, in contrast, ethosuximide had no significant effect on the coastline index. Ethosuximide had no effect on the amplitude of the first population spike in slices from control, sham or lesioned rats (Fig. 6C). Furthermore, ethosuximide had no effect on the amplitude of field potentials in laminas III-V of spinal cord slices from control, sham or lesioned animals (Fig. 5E). We conclude that bursting discharges in thalamic neurons experiencing chronic partial denervation after spinothalamic tract lesions require the participation of T-type voltage-dependent calcium channels and that ethosuximide can be used to restore normal physiological responses to the denervated cells.
Figure 6.

Ethosuximide abolished thalamic burst discharges induced by spinothalamic tract lesion. a, Field potential recordings made in three consecutive trials in the VPL in a thalamic slices from a rat lesioned 14 d earlier before, during, and after application of ethosuximide (700 μm). The bursts of 4–6 spikes were reversibly blocked by ethosuximide, leaving the first population spike unaffected. b, Group data indicating that ethosuximide produces a 40% decrease in the coastline index of slices from lesioned animals (eSTTx)(paired t test, *p < 0.05, before vs after; n = 10 lesioned, 8 sham and 10 intact), but has no effect on the coastline index of responses in slices from sham or intact rats. c, Grouped data indicating that ethosuximide has no significant effect on the amplitude of the first population spike in any condition.
Ethosuximide alleviates lesion-induced hyperalgesia and allodynia
Because ethosuximide alleviated the hyperexcitability of the thalamic neurons in lesioned animals, without affecting spinal cord responses, we next tested the hypothesis that this thalamic bursting activity underlies the altered pain sensation in these animals. We therefore determined the effects of an intraperitoneal injection of ethosuximide at a range of concentrations on paw withdrawal in response to thermal and mechanical stimuli in animals subjected to electrolytic or demyelinating lesions of the spinothalamic tract or sham surgery 14 d earlier.
We observed that ethosuximide significantly increased hind and forepaw withdrawal latency in response to thermal stimulation in lesioned animals within 15 min and that its analgesic action persisted for more than 1 h (Fig. 7A,B). Control intraperitoneal injections of saline had no effect on thermal paw withdrawal latencies in lesioned animals (Fig. 7A,B). Ethosuximide (15 mg/kg) was also effective in increasing paw withdrawal force thresholds in response to mechanical stimulation of hind and forepaws in lesioned animals (Fig. 7C,D). At 15 mg/kg, ethosuximide had no effect on paw withdrawal responses to mechanical or thermal stimuli in sham operated controls (Fig. 7A–D). By quantifying the effects of a range of doses of ethosuximide on thermal withdrawal latency, we derived an EC50 for this effect of 5.2 mg/kg, with a maximal effect at ∼50 mg/kg (Fig. 7E). Ethosuximide affected thermal pain sensation in sham operated control animals, but only at 100-fold higher doses (Fig. 7E), perhaps because of locomotor dysfunction as a consequence of actions at cerebellar T-type channels (McKay et al., 2006). In the open field test, too, ethosuximide (50 mg/kg) restored the level of locomotor activity displayed by the lesioned rats to levels that were not different from sham controls (Fig. 4A), but did not effect on the percentage of crossings into the inner zone (Fig. 4B). The ability of ethosuximide to reduce both pathological thalamic bursting activity and allodynia and hyperalgesia in animals subjected to spinothalamic tract lesions leads us to conclude that altered pain sensation after spinal cord injury results from a lesion-induced hyperexcitability of thalamic neurons.
Figure 7.
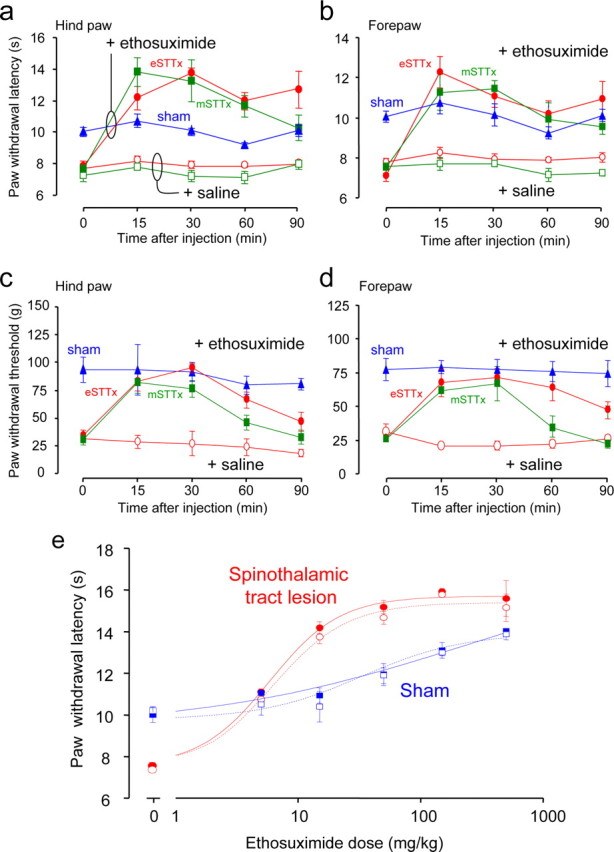
a–e, Ethosuximide eliminates hyperalgesia and allodynia induced by spinothalamic tract lesion. Contralateral hind (a) and forepaw (b) withdrawal latencies in response to thermal stimulation were measured at various times after administration of ethosuximide (15 mg/kg i.p., closed symbols) or saline (open symbols) in animals in which electrolytic (eSTTx, red, n = 6) or demyelinating (mSTTx, green, n = 4) spinothalamic lesions were made 14 d earlier, and sham-operated controls (blue, n = 5). Ethosuximide increased paw withdrawal latency significantly for 15–90 min for both hind and forepaws in eSTTx animals (two-way RM ANOVA, Tukey's test, F = 30.74; p < 0.001 in hind paw; F = 17, p < 0.01 in forepaw for ethosuximide vs saline), and also elevated the contralateral hind (c) and forepaw (d) paw withdrawal thresholds for mechanical stimuli in eSTTx animals (two-way RM ANOVA, Tukey's test, F = 30.74, in hind paw, F = 23.74 in forepaw, p < 0.001 for ethosuximide vs saline). Similarly, ethosuximide increased thermal paw withdrawal latency in both hind and forepaws in mSTTx animals (two-way RM ANOVA, Tukey's test, F = 30.03; p < 0.01 in hind paw; F = 35.29, p < 0.001 in forepaw for ethosuximide vs saline), but only significantly increased mechanical threshold at 30 min after injection (c, d) (two-way RM ANOVA, Tukey's test, intercomparison, q = 6.32, p < 0.001 in hind paw and q = 3.2, p < 0.05 in forepaw for ethosuximide vs saline). e, Dose–response curves were plotted for ipsilateral (open symbols) and contralateral (closed symbols) thermal paw withdrawal latency with various doses of ethosuximide. The ED50 of ethosuximide in increasing contralateral hind paw withdrawal latency was calculated at 5.2 mg/kg in lesioned animals (n = 4). Ethosuximide had some analgesic action in sham animals, but with a much higher ED50 of 67.5 mg/kg (n = 4).
Discussion
This study was designed to develop a rat model of central pain syndrome to reveal the biological bases of the condition, and to test novel therapeutic approaches for the treatment of CPS. The pain experienced by CPS patients is varied, but the most typical symptoms are hyperalgesia for evoked pain and allodynia, and this hyperesthesia is always delayed in onset (Nasreddine and Saver, 1997; Finnerup et al., 2004). We have observed several forms of altered pain sensation after unilateral lesions involving the ascending spinothalamic tract in rats, confirming previous observations in cats and primates (Vierck et al., 1990; Koyama et al., 1993; Weng et al., 2003). Spinal cord injury in rats induced hypersensitivity to thermal and mechanical stimuli delivered bilaterally at both above- and below-level sites, as well as a decrease in spontaneous locomotor activity in a novel environment. Changes in pain-related behaviors were apparent as early as 3 d after the injury and persisted for longer than 1 month. Furthermore, heightened pain sensitivity resulted from both electrolytic lesions, in which the ascending STT axons degenerate, and demyelinating lesions, in which the axons remain intact but their information transmitting properties are impaired.
There is considerable evidence that cats, primates, and humans experience central pain syndromes in response to lesions that involves the spinothalamic tract (Berić et al., 1988; Vierck et al., 1990; Koyama et al., 1993; Weng et al., 2003). Surgical interruption of the spinothalamic tract has been used for control of intractable pain in humans (White and Sweet, 1969), but the procedure induced the delayed occurrence of allodynia and CPS in many patients (Walker, 1943; King et al., 1957; Triggs and Beric, 1992). Our observations in rat thus seem applicable to the response of the human nervous system to injury. Altered pain-related behaviors in rats has been described after mechanical contusion (Hulsebosch et al., 2000) or local ischemia (Hao and Xu, 1996). Unfortunately, because degeneration is more widespread in those forms of injury, it is difficult to determine with these rat models, whether the pain symptoms result from interruption of ascending spinothalamic afferent pathways or rather from the severing of descending pathways that activate inhibitory intraspinal circuitry.
We observed that lesion-induced alterations in pain sensitivity were accompanied by abnormal neuronal excitability in populations of thalamic relay neurons in brain slices prepared 2 weeks after injury, without any evidence of altered excitability in the dorsal horn of the spinal cord. Furthermore, the maintenance of this pathological activity in acutely prepared brain slices indicates that the alterations are intrinsic to the thalamus and not projected on it from other afferent nuclei. There is abundant evidence of altered excitability of neurons in VPL of thalamus in human CPS (Lenz et al., 1987, 1989, 1993; Jeanmonod et al., 1993) (but see Radhakrishnan et al., 1999) and in primate CPS models (Weng et al., 2000, 2003), in the form of increased spontaneous activity, altered receptive field properties, and spike bursts in areas innervated by the spinothalamic tract. We demonstrated that the thalamic hyperexcitability in the denervated rat thalamus was due to an increase in bursting discharges in response to local stimuli mediated by T-type, low-threshold, voltage-dependent calcium channels, because it was eliminated by the T-type calcium channel blocker, ethosuximide.
We hypothesized that this alteration in thalamic excitability underlies the alteration in pain perception. Consistent with our hypothesis, treatment of lesioned rats with ethosuximide restored normal pain sensation in response to thermal and mechanical stimuli, as well as normal open field exploratory behavior. We suggest that ethosuximide may offer a simple and readily tested therapeutic approach for the treatment of human CPS. Ethosuximide is widely used to treat childhood absence epilepsy, and is safe and well tolerated in humans. It has not yet been tested in human CPS to our knowledge.
The molecular mechanisms underlying the increased bursting in denervated thalamic relay cells remain unknown and may be numerous. Thalamocortical oscillations are driven by reciprocal connections between excitatory relay neurons and inhibitory neurons in the reticular nucleus (Huguenard and McCormick, 2007) and zona incerta (Trageser et al., 2006). The T-type calcium channel CaV3.1 is the key component of this oscillation in relay neurons, whereas CaV3.2 and CaV3.3 predominate in reticular neurons (Talley, 1999). Altered channel expression levels or voltage dependence in either nuclei would increase bursting in relay cells, as would changes in the strength of GABAergic inhibition (Kim and McCormick, 1998a,b). Changes in the biophysical properties of voltage-dependent sodium channels, particularly NaV1.3, in VPL have also been implicated (Hains et al., 2003, 2006). Our results do not exclude the possibility of additional changes contributing to CPS, such as alterations in downstream pain-gating mechanisms.
Altered pain sensation has also been demonstrated previously in rodents after spinal hemisection or large contusions (Hulsebosch et al., 2000; Hains et al., 2003; Waxman and Hains, 2006). In these models, however, it is difficult to distinguish alterations in the descending control of spinal circuits from alterations in the sensory processing in the forebrain. For example, Hains et al. (2005, 2006) have suggested that contusion-induced potentiation of dorsal horn neuronal responsivity produces an increased afferent activation of VPL neurons, leading to upregulation of NaV1.3. Our finding that lesions involving primarily the spinothalamic tract are sufficient to induce abnormal thalamic burst discharges allows us to suggest an alternative model. We propose that it is a decrease in the level of afferent activation of VPL neurons that triggers an increase in their excitability. In this regard, the altered pain perception in CPS represents a form of maladaptive homeostatic plasticity, in which cells in the thalamus compensate for the loss of activity by increasing their excitability. We suggest that the increased gain of somasthetic sensory relay functions leads to an increase in the input received in primary sensory cortices in response to the uninjured peripheral inputs (Fig. 8), where it is mistakenly perceived as a painful peripheral stimulus.
Figure 8.

Working model of central pain syndrome after spinal cord injury. Top, In intact animals, non-noxious peripheral stimulation signals are relayed via the spinothalamic tract to VPL of thalamus. The thalamocortical neurons in VPL faithfully convey this signal to cortex to form normal perception. Bottom, After an injury to the spinothalamic tract, denervation of thalamocortical neurons in the VPL causes homeostatic changes that produce hyperexcitability and strong activation of T-type Ca2+ channels. As a result of these changes, VPL neurons amplify non-noxious peripheral inputs thereby increasing output to the cortex, where it is perceived as painful.
Footnotes
This work was supported by grants from University of Maryland, Baltimore and the National Institute of Neurological Disorders and Stroke (R21 NS055896), and the Center for Integrative Neuroscience Postdoctoral Training Grant (P32 NS07375). We thank Drs. Ronald Dubner, Joel Greenspan, and Asaf Keller for their advice and comments on this manuscript; Dr. Kun Yang for assistance with the preparation of spinal cord slices; Drs. Lixing Lao and Ruixin Zhang for the use of the thermal stimulation apparatus; and Leepeng Mok for superb technical assistance.
References
- Berić A, Dimitrijević MR, Lindblom U. Central dysesthesia syndrome in spinal cord injury patients. Pain. 1988;34:109–116. doi: 10.1016/0304-3959(88)90155-8. [DOI] [PubMed] [Google Scholar]
- Bonica JJ. History of pain concepts and pain therapy. Mt Sinai J Med. 1991;58:191–202. [PubMed] [Google Scholar]
- Cannavero S, Bonicalzi V. Central pain syndrome: pathophysiology, diagnosis and management. Cambridge, UK: Cambridge UP; 2007. [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol. 1989;25:582–593. doi: 10.1002/ana.410250610. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- Davis KD, Kiss ZH, Tasker RR, Dostrovsky JO. Thalamic stimulation-evoked sensations in chronic pain patients and in nonpain (movement disorder) patients. J Neurophysiol. 1996;75:1026–1037. doi: 10.1152/jn.1996.75.3.1026. [DOI] [PubMed] [Google Scholar]
- Dejerine JJ, Roussy G. Le syndrome thalamique. Revue Neurologique Paris. 1906;14:521–532. [Google Scholar]
- Dostrovsky JO. Role of thalamus in pain. Prog Brain Res. 2000;129:245–257. doi: 10.1016/S0079-6123(00)29018-3. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Gyldensted C, Fuglsang-Frederiksen A, Bach FW, Jensen TS. Sensory perception in complete spinal cord injury. Acta Neurol Scand. 2004;109:194–199. doi: 10.1034/j.1600-0404.2003.00219.x. [DOI] [PubMed] [Google Scholar]
- Foster RE, Kocsis JD, Malenka RC, Waxman SG. Lysophosphatidyl choline-induced focal demyelination in the rabbit corpus callosum. Electron-microscopic observation. J Neurol Sci. 1980;48:221–231. doi: 10.1016/0022-510x(80)90202-6. [DOI] [PubMed] [Google Scholar]
- Gonzales GR. Central pain: diagnosis and treatment strategies. Neurology. 1995;45:S11–S16. doi: 10.1212/wnl.45.12_suppl_9.s11. [DOI] [PubMed] [Google Scholar]
- Greenspan JD, Ohara S, Sarlani E, Lenz FA. Allodynia in patients with post-stroke central pain (CPSP) studied by statistical quantitative sensory testing within individuals. Pain. 2004;109:357–366. doi: 10.1016/j.pain.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Gücer G, Niedermeyer E, Long DM. Thalamic EEG recordings in patients with chronic pain. J Neurol. 1978;219:47–61. doi: 10.1007/BF00313368. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Waxman SG. Changes in electrophysiological properties and sodium channel Na(v)1.3 expression in thalamic neurons after spinal cord injury. Brain. 2005;128:2359–2371. doi: 10.1093/brain/awh623. [DOI] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Waxman SG. Alterations in burst firing of thalamic VPL neurons and reversal by Na(v) 1.3 antisense after spinal cord injury. J Neurophysiol. 2006;95:3343–3352. doi: 10.1152/jn.01009.2005. [DOI] [PubMed] [Google Scholar]
- Hao JX, Xu XJ. Treatment of a chronic allodynia-like response in spinally injured rats: effects of systemically administered excitatory amino acid receptor antagonists. Pain. 1996;66:279–285. [PubMed] [Google Scholar]
- Hasnie FS, Breuer J, Parker S, Wallace V, Blackbeard J, Lever I, Kinchington PR, Dickenson AH, Pheby T, Rice AS. Further characterization of a rat model of varicella zoster virus-associated pain: relationship between mechanical hypersensitivity and anxiety-related behavior and the influence of analgesic drugs. Neuroscience. 2007;144:1495–1508. doi: 10.1016/j.neuroscience.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE, Xu GY, Perez-Polo JR, Westlund KN, Taylor CP, McAdoo DJ. Rodent model of chronic central pain after spinal cord contusion injury. J Neurotrauma. 2000;17:1205–1217. doi: 10.1089/neu.2000.17.1205. [DOI] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A. Thalamus and neurogenic pain: physiological, anatomical and clinical data. Neuroreport. 1993;4:475–478. doi: 10.1097/00001756-199305000-00003. [DOI] [PubMed] [Google Scholar]
- Kim U, McCormick DA. Functional and ionic properties of a slow afterhyperpolarization in ferret perigeniculate neurons in vitro. J Neurophysiol. 1998a;80:1222–1235. doi: 10.1152/jn.1998.80.3.1222. [DOI] [PubMed] [Google Scholar]
- Kim U, McCormick DA. The functional influence of burst and tonic firing mode on synaptic interactions in the thalamus. J Neurosci. 1998b;18:9500–9516. doi: 10.1523/JNEUROSCI.18-22-09500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EE, Naquet R, Magoun HW. Alterations in somatic afferent transmission through the thalamus by central mechanisms and barbiturates. J Pharmacol Exp Ther. 1957;119:48–63. [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Koyama S, Katayama Y, Maejima S, Hirayama T, Fujii M, Tsubokawa T. Thalamic neuronal hyper-activity following transection of the spinothalamic tract in the cat: involvement of N-methyl-d-aspartate receptor. Brain Res. 1993;612:345–350. doi: 10.1016/0006-8993(93)91684-k. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Tasker RR, Dostrovsky JO, Kwan HC, Gorecki J, Hirayama T, Murphy JT. Abnormal single-unit activity recorded in the somatosensory thalamus of a quadriplegic patient with central pain. Pain. 1987;31:225–236. doi: 10.1016/0304-3959(87)90038-8. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Kwan HC, Dostrovsky JO, Tasker RR. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res. 1989;496:357–360. doi: 10.1016/0006-8993(89)91088-3. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Seike M, Richardson RT, Lin YC, Baker FH, Khoja I, Jaeger CJ, Gracely RH. Thermal and pain sensations evoked by microstimulation in the area of human ventrocaudal nucleus. J Neurophysiol. 1993;70:200–212. doi: 10.1152/jn.1993.70.1.200. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Garonzik IM, Baker FH, Richardson RT, Dougherty PM. Reorganization of sensory modalities evoked by microstimulation in region of the thalamic principal sensory nucleus in patients with pain due to nervous system injury. J Comp Neurol. 1998;14:125–138. [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW, Turner RW. Ca(v) 3 T-type calcium channel isoformes differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci. 2006;24:2581–2594. doi: 10.1111/j.1460-9568.2006.05136.x. [DOI] [PubMed] [Google Scholar]
- Nasreddine ZS, Saver JL. Pain after thalamic stroke: right diencephalic predominance and clinical features in 180 patients. Neurology. 1997;48:1196–1199. doi: 10.1212/wnl.48.5.1196. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan V, Tsoukatos J, Davis KD, Tasker RR, Lozano AM, Dostrovsky JO. A comparison of the burst activity of lateral thalamic neurons in chronic pain and non-pain patients. Pain. 1999;80:567–575. doi: 10.1016/S0304-3959(98)00248-6. [DOI] [PubMed] [Google Scholar]
- Ren K. An improved method for assessing mechanical allodynia in the rat. Physiol Behav. 1999;67:711–716. doi: 10.1016/s0031-9384(99)00136-5. [DOI] [PubMed] [Google Scholar]
- Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–324. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Amata M, Sakaue G, Nishimura S, Inoue T, Shibata M, Mashimo T. Experimental neuropathy in mice is associated with delayed behavioral changes related to anxiety and depression. Anesth Analg. 2007;104:1570–1577. doi: 10.1213/01.ane.0000261514.19946.66. [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker RR, Dostrovsky JO, Dolan EJ. Computerized tomography (CT) is just as accurate as ventriculography for functional stereotactic thalamotomy. Stereotact Funct Neurosurg. 1991;57:157–166. doi: 10.1159/000099568. [DOI] [PubMed] [Google Scholar]
- Tasker RR, DeCarvalho GT, Dolan EJ. Intractable pain of spinal cord origin: clinical features and implications for surgery. J Neurosurg. 1992;77:373–378. doi: 10.3171/jns.1992.77.3.0373. [DOI] [PubMed] [Google Scholar]
- Trageser JC, Burke KA, Masri R, Li Y, Sellers L, Keller A. State-dependent gating of sensory inputs by zona incerta. J Neurophysiol. 2006;96:1456–1463. doi: 10.1152/jn.00423.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triggs WJ, Berić A. Sensory abnormalities and dysaesthesias in the anterior spinal artery syndrome. Brain. 1992;115:189–198. doi: 10.1093/brain/115.1.189. [DOI] [PubMed] [Google Scholar]
- Vierck CJ, Jr, Greenspan JD, Ritz LA. Long-term changes in purposive and reflexive responses to nociceptive stimulation following anterolateral chordotomy. J Neurosci. 1990;10:2077–2095. doi: 10.1523/JNEUROSCI.10-07-02077.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AE. Central representation of pain. Res Publ Assoc Res Nerv Ment Dis. 1943;23:63–85. [Google Scholar]
- Waxman SG, Hains BC. Fire and phantoms after spinal cord injury: Na+ channels and central pain. Trends Neurosci. 2006;29:207–215. doi: 10.1016/j.tins.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Weng HR, Lee JI, Lenz FA, Schwartz A, Vierck C, Rowland L, Dougherty PM. Functional plasticity in primate somatosensory thalamus following chronic lesion of the ventral lateral spinal cord. Neurosci. 2000;101:393–401. doi: 10.1016/s0306-4522(00)00368-7. [DOI] [PubMed] [Google Scholar]
- Weng HR, Lenz FA, Vierck C, Dougherty PM. Physiological changes in primate somatosensory thalamus induced by deafferentation are dependent on the spinal funiculi that are sectioned and time following injury. Neuroscience. 2003;116:1149–1160. doi: 10.1016/s0306-4522(02)00796-0. [DOI] [PubMed] [Google Scholar]
- White JC, Sweet WH. Pain and the neurosurgeon: a forty-year experience. Springfield, IL: C.C. Thomas; 1969. [Google Scholar]
- Yezierski RP. Pain following spinal cord injury: pathophysiology and central mechanisms. Prog Brain Res. 2000;129:429–449. doi: 10.1016/S0079-6123(00)29033-X. [DOI] [PubMed] [Google Scholar]