

Abstract
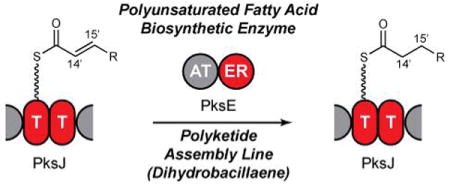
Polyketide biosynthesis is typically directed by cis-acting catalytic domains. In the case of the Bacillus subtilis secondary metabolite dihydrobacillaene, the cis-acting domains are not sufficient to generate the saturated C14′-C15′ bond. In this communication, we identify PksE as a trans-acting enoyl reductase utilized in the biosynthesis of this portion of dihydrobacillaene. PksE is homologous to the enzymes predicted to serve as enoyl reductases in polyunsaturated fatty acid (PUFA) biosynthesis, and we confirmed this functional assignment in vitro. These results suggest a general enoyl reduction pathway in polyketide biosynthesis and a means by which PUFA-like biosynthetic machinery can modulate small-molecule function.
Polyketide natural products are biosynthesized by an assembly line-like thiotemplated mechanism, wherein growing ketidyl intermediates are tethered by a thioester linkage to protein scaffolds known as thiolation (T) domains.1 These acyl-S-T intermediates are typically tailored by a series of cis-acting catalytic domains to establish the polyketide structure, although recently, several complementary trans-acting polyketide biosynthetic enzymes have been identified.2-6 One class of such trans-tailoring machinery is exemplified by PksE, an enzyme involved in the biosynthesis of the Bacillus subtilis signaling agent dihydrobacillaene (1).7-9 In this communication, we identify PksE as a trans-acting enoyl reductase (ER). We also confirm the activity of the PksE homolog PfaD, the predicted trans-enoyl reductase involved in polyunsaturated fatty acid (PUFA) biosynthesis.10 These results reveal a general enoyl reduction pathway in polyketide biosynthesis and suggest a strategy by which trans-enoyl reductases are utilized to modulate small-molecule function.
The saturated C14′-C15′ bond in dihydrobacillaene is incorporated while the growing intermediate is tethered to a non-standard doublet of T domains in the polyketide synthase PksJ (Figure 1B).11 However, the cis-acting catalytic domains in PksJ only possess ketoreduction and dehydration activities and as such are only able to generate the C14′-C15′ bond at the olefin oxidation state. We hypothesized that PksE acts as a trans-ER to reduce enoyl-S-PksJ and thereby saturate the C14′-C15′ bond in dihydrobacillaene. To test this, PksE and a construct bearing the T-domain doublet from PksJ [PksJ(T2)] were heterologously overexpressed in Escherichia coli.
Figure 1.
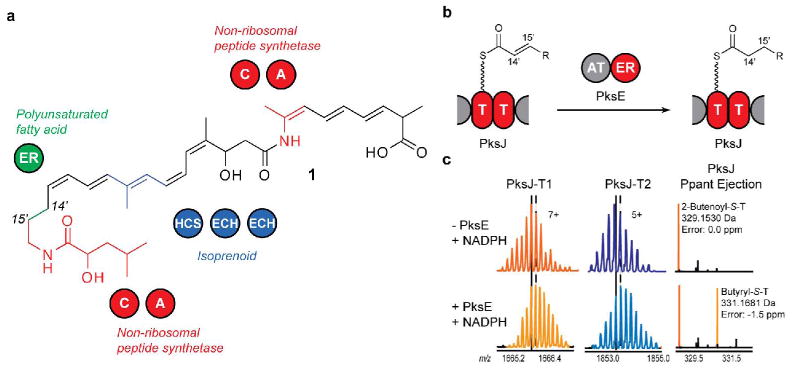
(a) Structure of dihydrobacillaene. C, condensation; A, adenylation; HCS, HMG-CoA synthase; ECH, enoyl-CoA hydratase. (b) PksE reduces an enoyl substrate tethered to the PksJ T-domain doublet in trans. (c) Confirmation of PksE enoyl reductase activity. (Left) PksJ-T1 active-site peptide. (Middle) PksJ-T2 active-site peptide. (Right) Ppant ejection ions. Vertical lines in (c) correspond to the most abundant isotopic peak (solid: absence of PksE; dash: presence of PksE).
Because enoyl reduction results in a mass increase of 2 Da, high-resolution Fourier-Transform Mass Spectrometry (FTMS)12 and the phosphopantetheinyl (Ppant) ejection assay13 were employed to monitor enoyl reduction. Proteolytic digestion of PksJ(T2) resulted in two active-site peptides of different mass, allowing analysis of mass shifts occurring at each T domain separately. Upon incubation of the model substrate 2-butenoyl-S-PksJ(T2) with PksE and NADPH, protein digestion, and analysis by liquid chromatography (LC)-FTMS, we observed a gain of 2 Da in approximately half of the substrate bound to each T-domain active-site peptide (Figure 1C), consistent with PksE's proposed activity as a C14′-C15′ enoyl reductase during dihydrobacillaene biosynthesis. Replacement of NADPH with NADH resulted in similar levels of reduction.
We next probed the role of the T-domain doublet in PksJ in facilitating in trans enoyl reduction. PksE is comprised of an acyltransferase (AT) domain along with the ER domain. Homologs of the PksE-AT domain load assembly-line T domains in trans with malonyl units during polyketide biosynthesis.14 Given the ability of these AT domains to interact with T domains, we tested the possibility that PksE-AT recognizes the PksJ T-domain doublet to recruit its fused ER domain to the correct region of PksJ. We heterologously overexpressed a PksE construct that harbors only the ER domain [PksE(ER)], and upon incubation of 2-butenoyl-S-PksJ(T2) with PksE(ER), formation of butyryl-S-PksJ was observed with no change in enoyl reduction relative to full-length PksE (Figure 2A). In parallel, we tested the ability of PksE-AT and PksC, a second dihydrobacillaene AT, to malonate a collection of T domains from the dihydrobacillaene assembly line, and both AT domains loaded every dihydrobacillaene T domain examined (Figure 2B). We therefore conclude that PksE-AT/PksJ interaction is not required for PksE-ER activity.
Figure 2.
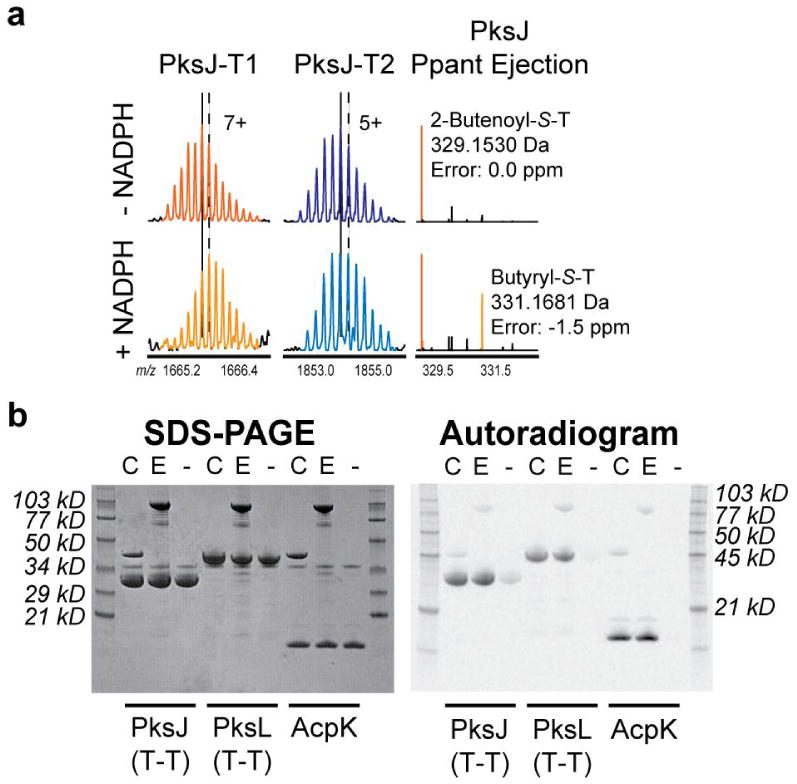
(a) Enoyl reduction is efficiently catalyzed by PksE(ER). (Left) PksJ-T1 active-site peptide. (Middle) PksJ-T2 active-site peptide. (Right) Ppant ejection ions. Vertical lines in (a) correspond to the most abundant isotopic peak (solid: absence of NADPH; dash: presence of NADPH). (b) PksC and PksE efficiently load several dihyrobacillaene-associated T domains (PksJ, PksL, AcpK) with 2-14C-malonate. C, including PksC; E, including PksE; -, no trans-AT. (Left) Coomassie-stained gel. (Right) Autoradiogram.
The tethering of substrates in proximity to the catalytic domains controls the selectivity of cis-tailoring reactions. In contrast, the selectivity of trans-tailoring reactions depends on recruitment of the catalytic machinery to only the appropriate T-domain scaffolds. Inspection of the dihydrobacillaene structure suggests that PksE-ER acts only on substrates tethered to the T-domain doublet in PksJ. A construct that contains a T domain from PksJ not predicted to serve as a scaffold for PksE-catalyzed enoyl reduction was expressed and purified in E. coli. PksE did not catalyze enoyl reduction of 2-butenoyl units tethered to this T domain, demonstrating that specific interactions of PksE with the dihydrobacillaene polyketide assembly line facilitate enoyl reduction and consequently the pattern of unsaturation in dihydrobacillane (Figure S1). The selective association of PksE with PksJ in vivo is in accord with fluorescence microscopy studies.8 Upon disruption of pksR, which encodes another multi-domain dihydrobacillaene synthase protein, a PksE-cyan fluorescent protein (CFP) fusion associated with a PksX megacomplex. In contrast, in a strain containing a disruption in pksJ, the PksE-CFP fusion did not co-localize with the megacomplex, demonstrating that PksE interacts selectively with PksJ.
Homologs of PksE are present in several polyketide biosynthetic clusters, including myxovirescin,15 leinamycin,4 and mupirocin,16 among others (Figure S2).9,17,18 Comparison of the structures of these polyketides and their associated domain architectures reveals that in each case at least one carbon-carbon bond can be identified that is saturated in the absence of a cis-ER, suggesting that trans-enoyl reduction by PksE-like ERs is a widely distributed biosynthetic strategy to incorporate saturated carbon-carbon bonds in polyketides. These PksE-like ER domains are phylogenetically distinct from ERs utilized in canonical polyketide and fatty acid biosynthesis. Instead, PksE-ER is highly homologous (47% identical/65% similar) to PfaD, the ER utilized in the biosynthesis of PUFAs. PUFAs are precursors to numerous essential cellular regulators (e.g. eicosanoids and arachidonic acid);19 however, humans do not possess the biosynthetic machinery necessary for their production. A thiotemplated assembly-line pathway to PUFAs in marine microorganisms was recently reported,10 and although it is a promising strategy to access large quantities of PUFAs, its mechanistic details remain enigmatic. In analogy to the PksJ T-domain doublet, runs of multiple T domains are a hallmark of this pathway.
To confirm the proposed activity of PfaD, we heterologously overexpressed the PfaD isoform from the model PUFA-synthesizing marine microbe Shewanella oneidensis MR-1,20 as well as a construct bearing four consecutive T domains [PfaA(T4)] in E. coli (Figure 3A). PfaA(T4) was loaded with 2-butenoyl-S-pantetheinyl units and subsequently treated with PfaD in the presence of NADPH. Upon proteolytic digestion and analysis by LC-FTMS, a gain of 2 Da was observed in the masses of substrates bound to T-domain active-site peptides corresponding to the acylated T1, T2, and T4 domains of PfaA(T4), consistent with PfaD-catalyzed enoyl reduction and production of butyryl-S-T thioesters (Figure 3B). As 2-butenoyl-S-PfaA is a predicted authentic intermediate during PUFA assembly, these observations show that PfaD is the ER used in PUFA assembly and acts on substrates tethered to the PUFA synthase T-domain multiplet.
Figure 3.
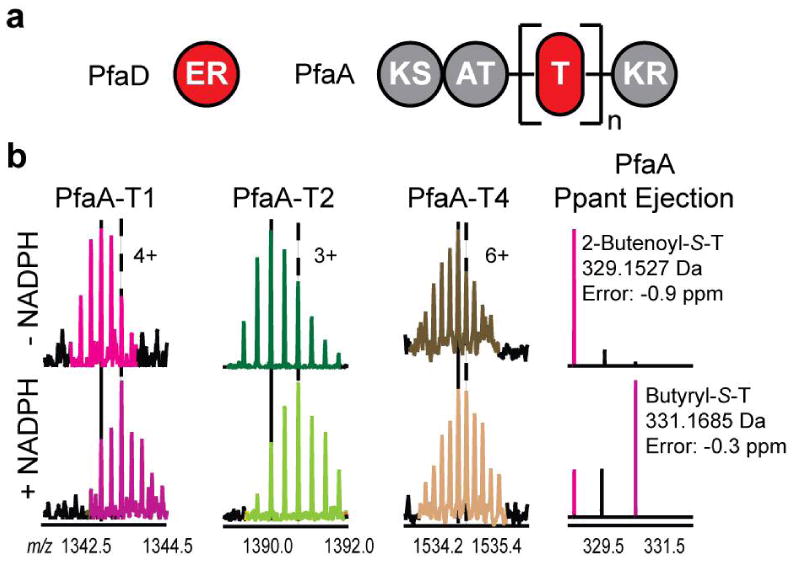
(a) Domain organization in a prototypical PUFA biosynthetic cluster. In S. oneidensis, n = 4. (b) Confirmation of PfaD enoyl reductase activity. (Left) PfaA-T1 active-site peptide. (Middle left) PfaA-T2 active-site peptide. (Middle right) PfaA-T4 active-site peptide. (Right) Ppant ejection ions. Vertical lines in (b) correspond to the most abundant isotopic peak (solid: absence of NADPH; dash: presence of NADPH).
Taken together, the above results suggest a benefit to the use of PUFA-like biosynthetic machinery in secondary metabolism. Variants of PUFA synthases are known which contain multiplets of 4 (as in the S. oneidensis PfaA), 6, or 9 consecutive T domains, all sharing a striking degree of homology (>95% identical).10 The role of the multiple tandem T domains is as-yet unknown, but this synthase architecture may ensure efficient tailoring of PUFA intermediates. Once an enoyl intermediate undergoes extension, the olefin can no longer be reduced. It is possible that the extra T domains make the extension step a biosynthetic bottleneck, increasing the chances that PfaD will reduce a given PUFA intermediate before extension. Under this model, the T-domain multiplets are scaffolds for parallel PUFA biosynthesis to ensure production only of the desired metabolite. Similar models have been previously proposed to explain the presence of T-domain multiplets in the mupirocin polyketide synthase,21 and in the present case, it is supported by recent work demonstrating that only a single T domain is required for PUFA biosynthesis.22
Though canonical polyketide synthases generally contain isolated T domains, several polyketide synthases harbor T-domain doublets and triplets. We propose that these T-domain multiplets mark insertion points of alternate biosynthetic logics. As shown here, the doublet in PksJ marks the insertion of PUFA logic, and the mupirocin16 and macrolactin23 polyketide synthases possess analogous T-domain multiplets at positions that correspond to predicted PUFA-like enoyl reductions. Similarly, elsewhere in the dihydrobacillaene assembly line, a second T-domain doublet marks the insertion of isoprenoid logic.3 Multiplets of T domains in polyketide assembly lines may be “scars” of the insertion of such multi-T-domain dependent machinery, and over the course of pathway evolution, the additional T domains could be lost. The myxovirescin biosynthetic machinery, for example, includes PUFA and isoprenoid-like enzymes to process substrates tethered to T-domain singlets, suggesting the myxovirescin assembly line has “sloughed off” the extra T domains resulting from the insertion.2,15
In many cases, the unsaturation pattern in the product small molecule set by the trans-ER has functional consequence. A striking example of the downstream effects of selective trans-enoyl reduction is observed in lovastatin biosynthesis, during which a trans-acting polyketide ER processes selected ketidyl-S-T domain intermediates to yield the specific unsaturation pattern necessary for conversion of a linear precursor to the polycyclic lovastatin skeleton.24 We propose that dihydrobacillaene represents a second example of an unsaturation pattern serving as a determinant of function. B. subtilis produces dihydrobacillaene as a mixture with its C14′-C15′ olefin congener bacillaene, generated by the post-synthetic C14′-C15′ desaturation of dihydrobacillaene catalyzed by the cytochrome P450 PksS, a formal reversal of PksE-catalyzed enoyl reduction.25 It is intriguing to speculate that the PksE/PksS reaction couple uses PUFA logic to control the function of the bacillaene/dihydrobacillaene ensemble by tuning the ratio of alkane and olefin congeners.
PksE is the first PUFA-like polyketide enzyme, and PfaD the first marine PUFA enzyme, whose enoyl reductase activities have been biochemically confirmed. The above observations reinforce the extreme hybridity of the dihydrobacillaene biosynthetic pathway, wherein multiple primary and secondary metabolic strategies converge, including polyketide, non-ribosomal peptide (red in 1), isoprenoid (blue), and PUFA logics (green).3,7 The opportunism of the dihydrobacillaene machinery in recruiting and cobbling together these disparate biosynthetic strategies in a single pathway is reflected in its chimeric structure. Indeed, as our understanding of secondary metabolism continues to grow, the use of multiple biosynthetic logics within a single pathway may be considered more of a norm than an exception.4,26 A key challenge in the future is to learn how we can insert multiple logics into pre-existing systems and bring the full force of Nature's biosynthetic ingenuity to access novel polyketides and other bioactive products.
Supplementary Material
Experimental protocols and supplemental data.
Acknowledgments
This work was supported by an NIH NRSA fellowship to C.T.C. (GM081743), an NIH Cell & Molecular Biology Training Grant to S.B.B. (T32 GM007283), and NIH grants to C.T.W. (GM49338) and N.L.K. (GM067725-05).
References
- 1.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–96. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Calderone CT, Iwig DF, Dorrestein PC, Kelleher NL, Walsh CT. Chem Biol. 2007;14:835–46. doi: 10.1016/j.chembiol.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Proc Natl Acad Sci USA. 2006;103:8977–82. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang GL, Cheng YQ, Shen B. Chem Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 5.Piel J. Proc Natl Acad Sci U S A. 2002;99:14002–7. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen T, Ishida K, Jenke-Kodama H, Dittmann E, Gurgui C, Hochmuth T, Taudien S, Platzer M, Hertweck C, Piel J. Nat Biotechnol. 2008;26:225–33. doi: 10.1038/nbt1379. [DOI] [PubMed] [Google Scholar]
- 7.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. Proc Natl Acad Sci USA. 2007;104:1506–9. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Straight PD, Fischbach MA, Walsh CT, Rudner DZ, Kolter R. Proc Natl Acad Sci USA. 2007;104:305–10. doi: 10.1073/pnas.0609073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen XH, Vater J, Piel J, Franke P, Scholz R, Schneider K, Koumoutsi A, Hitzeroth G, Grammel N, Strittmatter AW, Gottschalk G, Süssmuth RD, Borriss R. J Bacteriol. 2006;188:4024–36. doi: 10.1128/JB.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metz JG, Roessler P, Facciotti D, Levering C, Dittrich F, Lassner M, Valentine R, Lardizabal K, Domergue F, Yamada A, Yazawa K, Knauf V, Browse J. Science. 2001;293:290–3. doi: 10.1126/science.1059593. [DOI] [PubMed] [Google Scholar]
- 11.Moldenhauer J, Chen XH, Borriss R, Piel J. Angew Chem Int Ed Engl. 2007;46:8195–7. doi: 10.1002/anie.200703386. [DOI] [PubMed] [Google Scholar]
- 12.Dorrestein PC, Kelleher NL. Nat Prod Rep. 2006;23:893–918. doi: 10.1039/b511400b. [DOI] [PubMed] [Google Scholar]
- 13.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Biochemistry. 2006;45:12756–66. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aron ZD, Fortin PD, Calderone CT, Walsh CT. Chembiochem. 2007;8:613–6. doi: 10.1002/cbic.200600575. [DOI] [PubMed] [Google Scholar]
- 15.Simunovic V, Zapp J, Rachid S, Krug D, Meiser P, Müller R. Chembiochem. 2006;7:1206–20. doi: 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]
- 16.El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM. Chem Biol. 2003;10:419–30. doi: 10.1016/s1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 17.Carvalho R, Reid R, Viswanathan N, Gramajo H, Julien B. Gene. 2005;359:91–8. doi: 10.1016/j.gene.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, Matsunaga S. Proc Natl Acad Sci USA. 2004;101:16222–7. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serhan CN, Yacoubian S, Yang R. Annu Rev Pathol. 2008;3:279–312. doi: 10.1146/annurev.pathmechdis.3.121806.151409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong Y, Song S, Lee S, Hur B. Biotechnology and Bioprocess Engineering. 2006;11:127–133. [Google Scholar]
- 21.Rahman AS, Hothersall J, Crosby J, Simpson TJ, Thomas CM. J Biol Chem. 2005;280:6399–408. doi: 10.1074/jbc.M409814200. [DOI] [PubMed] [Google Scholar]
- 22.Jiang H, Zirkle R, Metz JG, Braun L, Richter L, Van Lanen SG, Shen B. J Am Chem Soc. 2008 doi: 10.1021/ja801911t. Articles ASAP. [DOI] [PubMed] [Google Scholar]
- 23.Schneider K, Chen XH, Vater J, Franke P, Nicholson G, Borriss R, Sussmuth RD. J Nat Prod. 2007;70:1417–23. doi: 10.1021/np070070k. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Science. 1999;284:1368–72. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- 25.Reddick JJ, Antolak SA, Raner GM. Biochem Biophys Res Commun. 2007;358:363–7. doi: 10.1016/j.bbrc.2007.04.151. [DOI] [PubMed] [Google Scholar]
- 26.Wenzel SC, Muller R. Curr Opin Chem Biol. 2005;9:447–58. doi: 10.1016/j.cbpa.2005.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental protocols and supplemental data.