

Abstract
Persons with the Lynch syndrome (LS) are at high risk for cancer, including cancers of the small bowel, stomach, upper urologic tract (renal pelvis and ureter), ovary, biliary tract, and brain tumors, in addition to the more commonly observed colorectal and endometrial cancers. Cancer prevention strategies for these less-common cancers require accurate, age-specific risk estimation. We pooled data from four LS research centers in a retrospective cohort study, to produce absolute incidence estimates for these cancer types, and to evaluate several potential risk modifiers. After elimination of 135 persons missing crucial information, cohort included 6041 members of 261 families with LS-associated MLH1 or MSH2 mutations. All were either mutation carriers by test, probable mutation carriers (endometrial/colorectal cancer-affected), or first-degree relatives of these. Among mutation carriers and probable carriers, urologic tract cancer (N=98) had an overall lifetime risk (to age 70) of 8.4% (95%CI: 6.6–10.8); risks were higher in males (p<0.02) and members of MSH2 families (p<0.0001). Ovarian cancer (N=72) had an lifetime risk of 6.7% (95%CI: 5.3–9.1); risks were higher in women born after the median year of birth (p<0.008) and in members of MSH2 families (p<0.006). Brain tumors and cancers of the small bowel, stomach, breast, and biliary tract were less common. Urologic tract cancer and ovarian cancer occur frequently enough in some LS subgroups to justify trials to evaluate promising prevention interventions. Other cancer types studied occur too infrequently to justify strenuous cancer control interventions.
Keywords: Lynch syndrome, cancer risk, MLH1, MSH2
Introduction
Persons with the Lynch syndrome (LS) are at high risk for cancer. Recommended cancer preventive measures in LS have focused on colorectal cancer (CRC) and endometrial cancer (EC), the two most commonly occurring cancers in this hereditary disorder. Several other cancer types, while less common than CRC and EC, occur more frequently in LS than in the general population: cancers of the small bowel, stomach, upper urologic tract (renal pelvis and ureter), ovary,1 biliary tract, and brain tumors.2 Breast cancer has also been observed in LS family members and, in some cases, the inherited mismatch repair gene mutation has been shown to have played a role in its development.3,4 However, LS-associated elevation of breast cancer risk has not been convincingly demonstrated.
Lack of primary data has hampered consensus development regarding prevention strategies for these less common LS tumors. Age-specific incidence rates are critical. Few published studies have enough cases to produce reliable rate estimates. A recent summary5 revealed large estimate ranges (e.g., for gastric cancer, estimates of lifetime risk from 2%–13%). We pooled data from four LS research centers, to produce reliable, age-specific incidence estimates for these cancer types. In addition, we evaluated the hypothesis that there are differences in incidence between MLH1 and MSH2 mutation carriers, and that, after adjustment for family size, there is significant inter-family variability in incidence rates.
Methods
Four LS registries collaborated in this study: the Danish hereditary colon cancer registry in Copenhagen, Denmark; the Foundation for the Detection of Hereditary Tumours in Leiden, Holland; the Lynch syndrome registry in Helsinki, Finland; and the Hereditary Cancer Center at Creighton University in Omaha, USA. Data were collected in protocols approved by the respective Institutional Review Boards.
Each center provided data on all eligible families, and on all eligible members of those families. Families were eligible if any member(s) had a known LS-associated mutation in MLH1 or MSH2. Cases were eligible if observed prior to the organization of the data in 1998.
In each family, progenitors were identified on the basis of CRC, EC, and mutation test results. Among the progenitors’ descendants we excluded all mutation noncarriers by test or inference (i.e. descendants of tested noncarriers). Among the remaining descendants, we identified as mutation carriers and probable mutation carriers (MC+PMC) all individuals with CRC/EC or a known mutation, and all individuals who had MC+PMCs among their descendants (MC+PMCs by inference). Among the remaining descendants, we identified as first-degree relatives (FDR), all individuals who were primary relatives of a MC+PMC. All FDRs and MC+PMCs were included in the study.
Information collected on all FDRs and MC+PMCs included the following: gender, year of birth (YOB), year of last follow-up, year of death, whether they had a history of CRC and/or EC, gene mutated in the family (MLH1 vs. MSH2), and whether they were a known mutation carrier by test or inference. Also recorded was whether the individual had a history of a primary brain tumor or a primary invasive cancer of the ovary (ovary or fallopian tube), urologic tract (UTC, kidney, renal pelvis, ureter, or bladder), small bowel (SBC), biliary tract and pancreatic cancer (BPC, intrahepatic and extrahepatic bile ducts, common bile duct, ampulla of Vater, liver, gallbladder or pancreas), stomach, and breast. For each of these cancer diagnoses, we recorded year of diagnosis and degree of verification (documented histologic verification, documented by clinical record or death certificate, or undocumented self/family report). Where patients had multiple primary tumors of the same type, only the first diagnosis was included.
Statistical analysis
Statistical analyses were carried out using SAS/STAT (SAS Institute, Cary NC). In all statistical testing, results with p<0.05 were considered to be significant.
Lifetime risk of each cancer type was calculated by the Kaplan-Meier (product-limit) method.6 Confidence intervals were calculated with the loglog transformation.6 Age-specific rates were estimated using proportional hazards modeling to produce a hazard function, plotted using the SMOOTH macro,7 which applies a kernel smoothing method. In both analyses, each family member was included from birth until death/last follow-up/diagnosis of the cancer type.
Sensitivity analyses were performed: gene mutated (MLH1 vs. MSH2), males vs. females, before vs. after midpoint year of birth, and registry. Hypotheses about differences between these groups were evaluated using Cox proportional hazards model regression, with the Wald chi-square test statistic. If significant differences were identified, risks were calculated separately for the subgroups of interest.
Permutation analysis was used to evaluate the hypothesis that there is significant heterogeneity among families which cannot be accounted for by known factors affecting incidence (identified in sensitivity analysis). This method has been previously described.1 An index, Z, of cancer frequency (relative to the expected frequency based on the pooled collection of families, adjusted for the number, ages and genders of family members, and other factors found to be significantly related to risk) was calculated for each family. The variance of the Z scores among families was calculated. A statistical test (permutation test) was performed to evaluate whether the variance was larger than expected. Because the sampling distribution of the variance is unknown, 1000 random permutations of the data were used to approximate the sampling distribution. In these permutations, each family was reconstructed at random from the pooled cohort (within restrictions for gender, age at end of follow-up, and other factors), Z scores were recalculated, and the Z score variance was recalculated. If the observed variance was larger than 95% of the variances found in the permutations, the hypothesis of homogeneity was rejected in favor of the alternative hypothesis that significant heterogeneity exists among families.
This study was not designed to provide an accurate idea of the relative risk of these cancers. However, to verify that the previously documented high risk was observed in this cohort, approximate expected numbers of cancers were calculated. Age-specific and sex-specific average annual incidence rates were calculated from the Surveillance, Epidemiology and End Results program public use data8 using the SEERStat software.9 Using the density method,10 risk up to the end of each age interval was calculated. Expected numbers of cancer cases were obtained by multiplying the number of persons in each age and sex category by these risks, and then summing across categories.
Results
The initial cohort included 6176 members of 261 families. After eliminating individuals who were missing crucial data (gender, year of birth, year of death), 6041 remained for analysis. Cohort members are described in Table 1.
Table 1.
Description of cohort: 6041 members of 261 families with hMLH1 or hMSH2 mutations. For extracolonic, extraendometrial cancers, expected number (E) based on general population incidence rates are also given.
| Characteristic | Number | Percent |
|---|---|---|
| Expected number of cases | ||
| All | 6041 | |
| Male Gender | 3115 | 51.6 |
| MLH1 Mutation | 3597 | 59.6 |
| Year of birth **** | ||
| <1924 | 1411 | 23.4 |
| 1924–46 | 1370 | 22.7 |
| 1947–62 | 1470 | 24.3 |
| 1963+ | 1790 | 29.6 |
| Registry***** | ||
| Denmark | 619 | 10.2 |
| Finland | 1452 | 24.0 |
| Netherlands | 1566 | 25.9 |
| USA | 2404 | 39.8 |
| PMC* | 2683 | 44.4 |
| Mutation carrier* | 1756 | 29.1 |
| 1° relative of mutation carrier | 2426 | 40.2 |
| Dead | 1959 | 32.4 |
| Cancer-affected | ||
| CRC ** | 1127 | 24.6 |
| EC ** *** | 233 | 10.5 |
| Urologic tract (E=38.50) | 98 | 1.62 |
| Ovary*** (E=11.87) | 72 | 2.46 |
| Gastric (E=11.64) | 85 | 1.41 |
| Breast*** (E=92.65) | 65 | 2.22 |
| Small Bowel (E=1.82) | 56 | 0.93 |
| Biliary-Pancreatic (E=22.26) | 66 | 1.09 |
| Brain Tumor (E=13.00) | 53 | 0.88 |
PMC (vs FDR) status is based on CRC status, EC status and mutation test results (see Methods). Mutation carrier status based on mutation test results only.
information missing on 1452 cases
females only included when calculating percentages
All participating centers seek documentation for all reported cancer diagnoses. Among the 493 diagnoses included in this study, documentation of histologic verification (generally a pathology report) was obtained in 70.6% of the cases. In 19.7% of the cases, other documentation (clinical report, death certificate) was obtained. In 48 (9.7%) of the cases, the diagnosis was based on family- or self-report only. Given this low percentage, and our experience that most reported cancers are verified when medical records are obtainable, the decision was taken to include all diagnoses in our analysis.
Table 1 provides the expected number of cases of each cancer type included in this study, based on USA general population incidence rates. All cancer types were found to be much more common than expected, except for breast cancer. Because of the lack of excessive numbers of breast cancers, this cancer type was dropped from further analysis.
The variable MC+PMC was designed to identify likely LS carriers, based on CRC/EC status and mutation test results. As such, MC+PMCs were expected to have a higher risk for LS-associated cancers and to provide the best model for LS carriers. As seen in Table 2, MC+PMCs consistently showed significantly elevated risk. The only exception to this finding was in the brain tumors, where MC+PMCs did not have higher risk than the cohort as a whole. So it was decided that we would base our risk estimates on MC+PMCs, except in the case of brain tumors, where we would use the entire cohort.
Table 2.
Multivariate proportional hazards modeling results: maximum likelihood parameter estimates, Wald chi-square test results, and hazard ratios in subgroups. Subgroup results are shown for significant factors only. Hazard rates for each registry are given relative to the Danish registry.
| Variable | DF | Parameter Estimate | Standard Error | Chi- Square | Pr>ChiSq | Hazard Ratio |
|---|---|---|---|---|---|---|
| UROLOGIC TRACT | ||||||
| Gender: male | 1 | 0.48 | 0.20 | 5.53 | 0.0186 | 1.618 |
| Gene mutated: MLH1 | 1 | −1.96 | 0.29 | 44.30 | <.0001 | 0.141 |
| PMC | 1 | 1.83 | 0.28 | 43.86 | <.0001 | 6.262 |
| Registry | 3 | 10.14 | 0.0174 | |||
| Netherlands | −0.63 | 0.30 | 0.530 | |||
| USA | −0.73 | 0.26 | 0.481 | |||
| Finland | −0.06 | 0.40 | 0.937 | |||
| OVARY | ||||||
| Gene mutated: MLH1 | 1 | −0.66 | 0.24 | 7.66 | 0.0057 | 0.517 |
| Year of birth: recent | 1 | 0.74 | 0.27 | 7.23 | 0.0072 | 2.087 |
| PMC | 1 | 1.08 | 0.29 | 13.88 | 0.0002 | 2.957 |
| GASTRIC | ||||||
| Gene mutated: MLH1 | 1 | −0.66 | 0.26 | 6.41 | 0.0114 | 0.519 |
| PMC | 1 | 1.12 | 0.25 | 19.20 | <.0001 | 3.052 |
| Registry | 3 | 11.63 | 0.0088 | |||
| Netherlands | 0.74 | 0.50 | 2.104 | |||
| USA | 0.54 | 0.48 | 1.712 | |||
| Finland | 1.42 | 0.51 | 4.120 | |||
| SMALL BOWEL | ||||||
| Gender: male | 1 | 1.14 | 0.30 | 14.20 | 0.0002 | 3.138 |
| PMC | 1 | 2.54 | 0.52 | 23.67 | <.0001 | 12.620 |
| Registry | 3 | 12.08 | 0.0071 | |||
| Netherlands | −0.54 | 0.38 | 0.582 | |||
| USA | −0.58 | 0.34 | 0.560 | |||
| Finland | −2.58 | 0.76 | 0.076 | |||
| BILIARY/PANCREATIC | ||||||
| PMC | 1 | 0.63 | 0.26 | 5.67 | 0.0173 | 1.869 |
| BRAIN | ||||||
| Gene mutated: MLH1 | 1 | −0.66 | 0.28 | 5.56 | 0.0184 | 0.514 |
| PMC | 1 | −0.77 | 0.30 | 6.69 | 0.0097 | 0.465 |
We next considered for which subgroups among the MC+PMCs to calculate separate risk estimates. To determine this, we used multivariate proportional hazards modeling, starting with all factors which had been included in bivariate analyses, and using backward elimination to simplify the model to significant factors. Results of these analyses are given in Table 2.
In the case of BPC, MC+PMC status was the only significant modifiers of risk. Gender was a significant modifier of UTC and SBC risk, with males having higher risk than females. Gene mutated (MLH1 vs. MSH2) was a significant modifier of brain tumor, ovarian cancer, UTC, and gastric cancer risk, and in all these cases members of MSH2 mutation families were at higher risk than members of MLH1 families. Year of birth was a significant modifier of ovarian cancer risk, with more recently born women at higher risk. Based on these findings, it was decided to calculate risk for MC+PMCs only in all cancer types except brain tumors; and separately for males and females, for MLH1 and MSH2 family members, and for YOB-recent and YOB-older, for types where these factors were significant.
Registry was also a significant independent risk modifier for UTC, SBC and gastric cancer. It was decided that separate calculations of risk by registry were unlikely to be useful, and so they were not done.
In a secondary analysis, the 30 bladder cancers were excluded from the UTCs. Proportional hazards modeling results were unchanged, except that inter-registry differences were no longer significant (p<0.11).
Risk estimates
Lifetime risk estimates are provided in Table 3. It will be noted that, in the case of gastric cancer, although gene mutated was found to be a significant risk modifier in multivariate proportional hazards modeling, with members of MLH1 families being at lower risk, life time risk estimates for MC+PMCs are similar between the MLH1 and MSH2 subgroups. The Wilcoxon test for equality of ranks between strata is not significant (p=0.77).
Table 3.
Kaplan-Meier analysis results: Lifetime cumulative cancer incidence rate (failure rate, 1-cumulative proportion surviving without disease). Cumulative incidence is to age 70 except in the case of ovarian cancer in subgroups with year of birth>1946, where risk is to age 50.
| GROUP | Cumulative Incidence | 95% Confidence Interval | Number of diagnoses |
|---|---|---|---|
| UROLOGIC TRACT: PMCs | 0.0843 | 0.0657–0.1079 | 82 |
| MLH1, FEMALE | 0.0113 | 0.0041–0.0312 | 6 |
| MLH1, MALE | 0.0373 | 0.0170–0.0810 | 9 |
| MSH2, FEMALE | 0.1186 | 0.0788–0.1763 | 29 |
| MSH2, MALE | 0.2777 | 0.1967–0.3830 | 38 |
| OVARY: PMC FEMALES | 0.0668 | 0.0535–0.0912 | 57 |
| MLH1, YOB<=1946 | 0.0388 | 0.0231–0.0648 | 15 |
| MLH1, YOB>1946 | 0.0630 | 0.0330–0.1183 | 10 |
| MSH2, YOB<=1946 | 0.0826 | 0.0536–0.1261 | 21 |
| MSH2, YOB>1946 | 0.1158 | 0.0621–0.2105 | 11 |
| GASTRIC: PMCs | 0.0581 | 0.0438–0.0768 | 64 |
| MLH1 | 0.0614 | 0.0431–0.0871 | 41 |
| MSH2 | 0.0521 | 0.0327–0.0824 | 23 |
| SMALL BOWEL: PMCs | 0.0427 | 0.0308–0.0590 | 51 |
| FEMALE | 0.0269 | 0.0150–0.0480 | 15 |
| MALE | 0.0609 | 0.0410–0.0901 | 36 |
| BILIARY/PANCREATIC: PMCs | 0.0407 | 0.0279–0.0592 | 44 |
| BRAIN: PMCs and FDRs | 0.0206 | 0.0147–0.0287 | 53 |
| MLH1 | 0.0173 | 0.0104–0.0288 | 21 |
| MSH2 | 0.0251 | 0.0163–0.0385 | 32 |
When bladder cancers were excluded from UTCs, overall lifetime risk dropped to 0.06; risk in MLH1 females, MLH1 males, MSH2 females, and MSH2 males were 0.004, 0.021, 0.091, and 0.203, respectively.
Figure 1 provides age-specific hazard rates for the two commonest cancer types, UTC and ovarian cancer.
Figure 1.
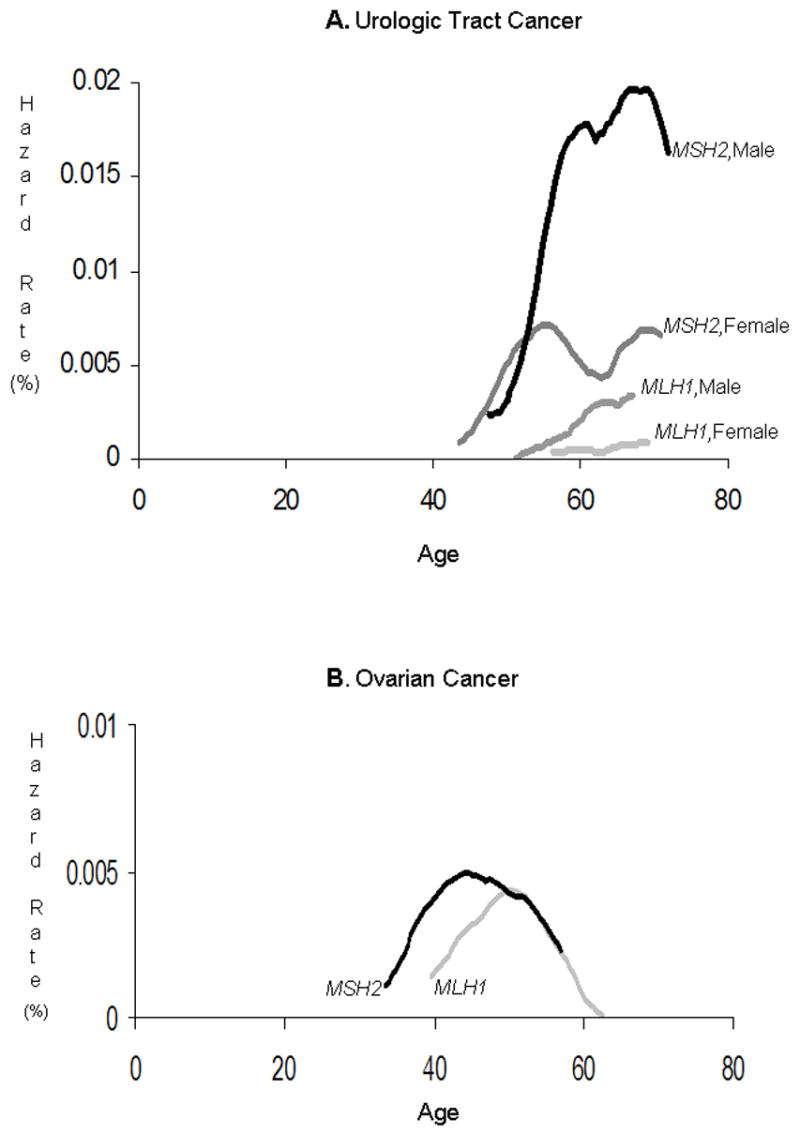
Smoothed hazard function for urologic tract cancer and ovarian cancer. Only MC+PMCs were included in this analysis.
Variation among families in cancer rates
Proportional hazards modeling, reported above, showed that there is significant variation among cohort members in risk for each of these cancer types. Some of this variation can be accounted for by gene mutated (MLH1/MSH2), gender, year of birth, group (MC+PMC vs. FDR) and registry. Permutation analysis was used to test the hypothesis of variation among families in risk, after adjustment for the age, gender, year of birth, and group membership structure of the family, and for the registry and gene mutated in the family, as appropriate for that cancer type. Results are shown in Table 4. Significant heterogeneity among families was observed in biliary/pancreatic cancers and brain tumors.
Table 4.
Permutation analysis results: Test for heterogeneity among families, after adjustment for ages of members and all factors found to be significant risk modifiers for the specific cancer type (see Table 2).
| Cancer type | Families (N)* | Variance of Z | Percentile Rank |
|---|---|---|---|
| UROLOGIC TRACT | 249 | 1.19 | 93.4 |
| OVARY | 255 | 1.11 | 73.8 |
| GASTRIC | 234 | 1.15 | 90.3 |
| SMALL BOWEL | 250 | 0.88 | 33.0 |
| BILIARY/PANCREATIC | 261 | 1.28 | 96.8 |
| BRAIN | 261 | 1.33 | 96.3 |
Note that families in which all members belonged to strata with a zero frequency of cancer cases were excluded from this analysis.
Discussion
A recent review of cancer risk and preventive strategies in the LS noted that research needs to define optimal cancer screening techniques, including age of initiation and surveillance frequency for CRC and EC, as well as for other LS-associated cancers.11 One critical step in this process is the estimation of overall and age-specific risk for each cancer type. To date, published estimates of the risk for the less-common LS cancers have limited reliability because of small sample size. We now provide estimates based on a cohort of 6041 high-risk members of families with known MLH1 and MSH2 mutations.
UTC was the most common of the cancers studied, and had the highest overall lifetime risk (8%). Rates were 1.6-fold higher in males than in females, and 7-fold higher in MSH2 than in MLH1 family members. The lifetime risk estimate for male carriers in MSH2 families was nearly 28%. The period of highest risk for urologic tract cancers was age 50–70. During this period, we estimate an annual hazard rate of around 0.5% for female members of MSH2 families; among male members of MSH2 families it exceeds 1% during this period. Prior to age 50, risk was low.
UTC risk observed in this cohort was markedly higher than that observed in a previous study of the Finnish families,2 and comparable to that observed in a previous study of the Dutch families.12 This may relate to the paucity of MSH2 family members in the Finnish study. The difference between males and females has not previously been reported to our knowledge.
Substantially and significantly different risks were seen among registries, with Finland and Denmark having higher incidence rates than the other registries. In the general population of these nations, Denmark and Finland have markedly lower rates of UTC than do the USA and the Netherlands.13 However, this is mainly due to the much higher incidence of bladder cancer in the USA and the Netherlands. Bladder cancer is by far the most frequent UTC in the general population. Epithelial upper urologic tract cancer is more common than bladder cancer in the LS,1 and upper urologic tract cancer rates are similar in all four countries.13 Environmental factors, diagnostic definitions,14 and screening practices may affect urologic tract cancer incidence rates, but we do not have the information necessary to determine if these might contribute to the inter-registry difference.
After adjustment for age distribution, gender, gene mutated, risk status and registry, no significant heterogeneity was seen among families in UTC risk.
Based on these findings, surveillance for early detection, and other strategies for prevention of UTC could be considered, especially among carriers of MSH2 mutations. Preventive management should focus on persons approaching or past the age of 50. Further studies are needed to identify the clinical features of UTC, so that an optimal prevention strategy can be developed and evaluated.
Ovarian cancer had the second highest incidence among the cancer types studied, with a lifetime risk of nearly 7% overall. MSH2 family members had nearly twice the incidence rate observed in MLH1 family members. There was a significant risk difference associated with year of birth: women born after the median year of birth had twice the risk of persons born before that year. These more recently born women were, at most, in their early 50s at the time of this study. The highest risk period for ovarian cancer was from age 40 to 55; in this period, ovarian cancer was diagnosed at annual rates exceeding 0.25%. Risk before age 50 was substantial: it was 2%, 6%, 5% and 12% in YOB-older MLH1, YOB-recent MLH1, YOB-older MSH2 and YOB-recent MSH2 strata. In the whole cohort, 8 of the 72 ovarian cancers were diagnosed before age 35; only two were observed after age 60. These risks are consistent with previously reported estimates,2,12,15 except that the birth year cohort difference has not been previously reported to our knowledge.
After adjustment for gene mutated, age, risk status and year of birth cohort distribution, no significant heterogeneity was seen among families in ovarian cancer risk.
Based on these findings, strategies for prevention of ovarian cancer should be considered, and if such strategies are available they should begin at or before age 40. Especially when potentially harmful interventions, such as early oophorectomy, are considered, differential recommendations for MLH1 and MSH2 carriers may be appropriate, since ovarian cancer risk is twice as high in the latter group.
After adjustment for significant risk modifiers, there was no significant heterogeneity among families in UTC or ovarian cancer risk. Thus, we do not support the practice of focusing prevention efforts on LS carriers with a positive family history of these cancers.
None of the other cancer types included in this study occurred frequently enough to justify our recommendation of special LS-associated cancer prevention measures.
A recent report proposed regular gastric surveillance in the LS starting at age 35.16 No age-specific rates were given to justify this recommendation, and our observations do not support it. Gastric cancer had the third highest incidence among the cancers we studied. MC+PMCs, overall, had a lifetime risk to age 70 of 5.8%. The greatest risk for gastric cancer in the LS occurs between 50 and 65. Risk before age 50 was low. We found no evidence that this risk differed markedly by gender or gene mutated, consistent with previous studies.12 We observed no significant heterogeneity among families in gastric cancer risk.
The risk we report here is consistent with some reports in the literature,12 but lower than the lifetime risk of 12% reported in a study of Korean LS families,17 and the risk of 13% reported in a study of Finnish families.2 The authors of the former study point out that the general population of Korea has a high incidence of gastric cancer, and hypothesize that the same environmental factors which increase gastric cancer risk in the general population act to increase it in LS. The high rate observed in Finland is more difficult to explain. Many cases from the Finnish study were included in this study, and our modeling also showed a significant difference among registries, with the highest rates in the Finnish registry and the lowest in the Danish registry. This is not a reflection of differential rates in the general population, where rates are similar among all four nations and the highest rates are seen in the Netherlands.13 If the findings of Park et al17 are shown to be reliable, LS patients who are members of populations with high background rates of gastric cancer may benefit from periodic screening for this cancer. More work is required to identify these populations.
Breast cancer was the fourth most common type of cancer among the cancer types studied; lifetime risk among female MC+PMCs was 5.4%. However, our results provide no evidence that LS cases are at higher risk for breast cancer than members of the general population. No special breast cancer surveillance is indicated for LS patients.
Based on our findings, we conclude that surveillance for early detection of SBC or BPC is unlikely to be cost-effective. However, the observation of significant inter-family heterogeneity in the case of BPC needs further study, in case some families have a sufficiently higher risk to justify special cancer screening for its members. In the case of small bowel cancer, it has been noted that new, less invasive technologies for evaluation of the small bowel are becoming available.18 If these are shown to be effective, the question of SBC screening for LS carriers over the age of 50 should be re-evaluated.
One recent study19 studied a large series of LS mutation carriers with SBC. They compared the frequency of mutations in various regions of the MSH2 and MLH1 genes with the expected frequency based on the distribution of mutations in a large registry of LS-associated mutations. They found no deviations from expectation in the MLH1 gene, but in the MSH2 gene, the region including Walker A (codon 626–733) was over-represented in the SBC group, while the MutL homologue interaction domain (codon 734–895) was underrepresented. We found no evidence of a higher rate in the former region or a lower rate in the latter, either when we evaluated crude numbers of SBCs or incidence rates (data not shown).
Brain tumors had the lowest incidence rate of the studied cancer types, with an overall lifetime risk of 2%. Risk was lower in MLH1 carriers than in MSH2 carriers. Brain tumors differed from the other cancer types studied, in that putative mutation carriers did not have a higher incidence of disease than their first-degree relatives. This may signal that these tumors are not associated with the LS, despite significant excesses which have been reported.2,20 An alternative explanation relates to the relatively early ages at which brain tumors occur (26% of diagnoses before age 25), and their high mortality (90% dead; 23% of these dead before age 25). Developing a brain tumor in childhood or early adulthood reduces one’s chance of being tested, of developing colorectal or endometrial cancer, and of having children. Based on this, we believe that mutation carriers with brain tumors are less likely be identified as MC+PMCs (vs. FDRs) by the method we used. This raises the possibility that the true risk of brain tumor may be underestimated.
Limitations
Some of these cases were used in previous studies.1,2,12,18–20 Where our results are similar to those of these earlier studies, it is not an independent confirmation of those earlier results but a recalculation, with improved reliability due to the current study’s larger cohort.
Only individuals from families with an MSH2 or MLH1 mutation were included in this study. Mutations in these genes account for most LS families, but mutations in MSH6 and PMS2 also occur21; numbers of MSH6 and PMS2 families in the participating centers were too small to effectively estimate cancer risks. Although studies to date are small, there are indications that the cancer risks associated with these less common mutations are different from the risks associated with MLH1 and MSH2. Our results cannot be generalized to such families.
The cancer types designated in this study combine several significantly different cancer subtypes, such as bladder cancer and ureteral cancer; and small bowel adenocarcinoma and small bowel carcinoid. In some cases, this was done because these sub-types might be screened together (e.g., bladder cancer and ureteral cancer), in other cases because many cancers cannot be precisely located in a specific organ (e.g., cancer of the pancreas vs. Ampulla of Vater). We concluded it was impractical and unnecessary (albeit desirable) to make estimates for all specific cancer sub-types. Secondary analyses excluding bladder cancers from the UTC diagnoses had little effect on differences among subgroups although, of course, lifetime risk estimates were reduced.
Our data are derived from LS families identified in high-risk registries. The majority of the families were identified because of a striking family history of cancer, especially colorectal cancer. Using such data retrospectively can be expected to bias cancer risk estimates.22 However, our focus in this study is on the less common LS cancers. None of the registries has recruited index cases from clinical centers that would favor recruitment of cases with these cancers (e.g., urological centers). Most recruitment has been done through gastroenterologists and gastrointestinal surgeons. Simulation23 supports our belief that, in LS, ascertainment is mainly affected by the family history of CRC and EC. Nevertheless, some ascertainment bias may exist in favor of index cases and families with the tumors included in the current study. It is noteworthy that in the largest registry, where it was possible to identify the index cases, the index cases were not significantly more likely than other MC+PMCs to be diagnosed with any of these cancers. Using prospective study data would avoid this type of ascertainment bias; however, given the rarity of LS families, and, in particular, of these tumor types within LS families, a prospective study will not yield reliable results for many years. Until that time, clinical decisions must be based on the best information available.
Another limitation to our conclusions is the use of cohorts identified in four specific locales (Finland, Denmark, the Netherlands, and the USA). Our cohort was a convenience sample which cannot be assumed to be representative of the LS as a whole. Among the included registries, significant differences were observed. Our estimates may not apply to other populations of LS carriers, especially those where the background population cancer incidence rates differ markedly from those in northern Europe and North America. The inclusion of very large families and multiple families with the same mutation, with no adjustment for the possibly resulting correlations, may have biased our estimates and affected our confidence intervals.
Our study included all reported cancer diagnoses among cohort members. All but 9.7% of these were confirmed by a medical record or death certificate. However, for the following cancer types the percentage of unconfirmed diagnoses was higher: brain tumors (29%); gastric cancers (21%). The relative numbers of misspecified diagnoses, which would overestimate incidence, and of missed diagnoses among cohort members, underestimating incidence, is unknown.
Conclusion
The decision to recommend a specific cancer prevention strategy, whether it is screening for early cancer detection, prophylactic surgery, or chemoprevention, depends on many factors. Primary among these is the incidence of the cancer, in the form of lifetime risk and age-specific annual risk. We studied the cancers most commonly seen in LS families, after colorectal and endometrial cancer. Annual hazard rates approaching or exceeding 0.5%, and lifetime risks approaching or exceeding 10%, were observed in urologic tract and ovarian cancer. Brain tumors, small bowel cancers, biliary-pancreatic and gastric cancers occurred less frequently.
Acknowledgments
This study was supported by revenue from Nebraska cigarette taxes awarded to Creighton University by the Nebraska Department of Health and Human Services. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the State of Nebraska or the Nebraska Department of Health and Human Services (P. Watson, H.T. Lynch).
Support was also given by the National Institutes of Health through grant #1U01 CA 86389 (P. Watson, H.T. Lynch), Creighton University through the Charles F. and Mary C. Heider Chair in Cancer Research (H.T. Lynch), The Dutch Collaborative Group on the Lynch Syndrome (J.H. Kleibeuker, F.M. Nagengast, A. Cats, F.H. Menko), the Helsinki University Hospital Research Fund (H.J. Järvinen), the clinical genetic departments in Copenhagen, Odense and Vejle (M.L. Bisgaard, M-M. Gerdes, D. Crüger), and the laboratories at Ålborg Hospital, Skejby Hospital, and Rigshospitalet.
Abbreviations used
- LS
Lynch syndrome
- CRC
colorectal cancer
- EC
endometrial cancer
- MC+PMC
mutation carriers and probable mutation carriers
- FDR
first degree relative of MC+PMC
- YOB
year of birth
- UTC
urologic tract cancer
- SBC
small bowel cancer
- BPC
biliary/hepatic/pancreatic cancer
- CI
cumulative incidence
Footnotes
Novelty and impact: This is the largest study to date on the less common but significantly elevated risk cancers in the LS, and provides markedly more accurate risk estimates. Our results should change guidelines for clinical management of LS patients.
References
- 1.Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71:677–685. doi: 10.1002/1097-0142(19930201)71:3<677::aid-cncr2820710305>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomäki P, Mecklin J-P, Järvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–218. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 3.Vasen HFA, Morreau H, Nortier JWR. Is breast cancer part of the tumor spectrum of hereditary nonpolyposis colorectal cancer? Am J Hum Genet. 2001;68:1533–1534. doi: 10.1086/320610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyd J, Rhei E, Federici MG, Borgen PI, Watson P, Franklin B, Karr B, Lynch J, Lemon SJ, Lynch HT. Male breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat. 1999;53:87–91. doi: 10.1023/a:1006030116357. [DOI] [PubMed] [Google Scholar]
- 5.Vasen HFA, Möslein G, Alonso A, Bernstein I, Bertario L, Blanco I, Burn J, Capella G, Engel C, Frayling I, Friedl W, Hes FJ, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer) J Med Genet. 2007;44:353–362. doi: 10.1136/jmg.2007.048991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. New York: John Wiley & Sons; 1980. [Google Scholar]
- 7.Allison P. Survival Analysis Using the SAS System. Cary, NC: SAS Institute Inc; 1995. [Google Scholar]
- 8.Surveillance EaERSPwscgSSD. Incidence - SEER 9 Regs Public-Use, Nov 2005 Sub (1973–2003) - Linked to County Attributes - Total U.S., 1969–2003 Counties. National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch. released April 2006, based on the November 2005 submission. Available from: URL: www.seer.cancer.gov.
- 9.Surveillance Research Program NCI. SEER*Stat software. Surveillance Research Program, National Cancer Institute 2007(version 6.2.4.) Available from: URL: www.seer.cancer.gov/seerstat.
- 10.Kleinbaum DG, Kupper LL, Morgenstern H. Epidemiologic Research: Principles and Quantitative Methods. New York: Van Nostrand Reinhold Company; 1982. [Google Scholar]
- 11.Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, Lynch P, Burke W, Press N. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: A systematic review. JAMA. 2006;296:1507–1517. doi: 10.1001/jama.296.12.1507. [DOI] [PubMed] [Google Scholar]
- 12.Vasen HFA, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, Taal BG, Moller P, Wijnen JT. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19:4074–4080. doi: 10.1200/JCO.2001.19.20.4074. [DOI] [PubMed] [Google Scholar]
- 13.Ferlay J, Bray F, Pisani P, Parkin DM. Cancer Incidence, Mortality and Prevalence Worldwide IARC CancerBase No. 5, version 2.0. Lyon: IARC Press; 2004. GLOBOCAN 2002. [Google Scholar]
- 14.Murphy WM. What’s the trouble with cytology? J Urol. 2006;176:2343–2346. doi: 10.1016/j.juro.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Brown GJ, St John DJ, Macrae FA, Aittomaki K. Cancer risk in young women at risk of hereditary nonpolyposis colorectal cancer: implications for gynecologic surveillance. Gynecol Oncol. 2001;80:346–349. doi: 10.1006/gyno.2000.6065. [DOI] [PubMed] [Google Scholar]
- 16.Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, Kloor M, Kuntsmann E, Vogelsang H, Keller G, Dietmaier W, Mangold E, et al. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: A report by the German HNPCC Consortium. J Clin Oncol. 2006;24:1–8. doi: 10.1200/JCO.2005.03.7333. [DOI] [PubMed] [Google Scholar]
- 17.Park YJ, Shin K-H, Park J-G. Risk of gastric cancer in hereditary nonpolyposis colorectal cancer in Korea. Clin Cancer Res. 2000;6:2994–2998. [PubMed] [Google Scholar]
- 18.ten Kate GL, Kleibeuker JH, Nagengast FM, Craanen M, Cats A, Menko FH, Vasen HFA. Is surveillance of the small bowel indicated for Lynch syndrome families? Gut. 2007;56:1198–1201. doi: 10.1136/gut.2006.118299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J-G, Kim D-W, Hong CW, Nam B-H, Shin Y-K, Hong S-H, Kim I-J, Lim S-B, Aronson M, Bisgaard ML, Brown GJ, Burn J, et al. Germ line mutations of mismatch repair genes in hereditary nonpolyposis colorectal cancer patients with small bowel cancer: International Society for Gastrointestinal Hereditary Tumours collaborative study. Clin Cancer Res. 2006;12:3389–3393. doi: 10.1158/1078-0432.CCR-05-2452. [DOI] [PubMed] [Google Scholar]
- 20.Vasen HFA, Sanders EACM, Taal BG, Nagengast FM, Griffioen G, Menko FH, Kleibeuker JH, Houwing-Duistermaat JS, Meera Khan P. The risk of brain tumours in hereditary non-polyposis colorectal cancer (HNPCC) Int J Cancer. 1996;65:422–425. doi: 10.1002/(SICI)1097-0215(19960208)65:4<422::AID-IJC4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 21.Lagerstedt Robinson K, Liu T, Vandrovcova J, Halvarsson B, Clendenning M, Frebourg T, Papadopoulos N, Kinzler KW, Vogelstein B, Peltomäki P, Kolodner RD, Nilbert M, et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst. 2007;99:291–299. doi: 10.1093/jnci/djk051. [DOI] [PubMed] [Google Scholar]
- 22.Bourne TH, Campbell S, Reynolds K, Hampson J, Bhatt L, Crayford TJ, Whitehead MI, Collins WP. The potential role of serum CA 125 in an ultrasound-based screening program for familial ovarian cancer. Gynecol Oncol. 1994;52:379–385. doi: 10.1006/gyno.1994.1065. [DOI] [PubMed] [Google Scholar]
- 23.Carayol J, Khlat M, Maccario J, Bonaiti-Pellie C. Hereditary non-polyposis colorectal cancer: current risks of colorectal cancer largely overestimated. J Med Genet. 2002;39:335–339. doi: 10.1136/jmg.39.5.335. [DOI] [PMC free article] [PubMed] [Google Scholar]